
Abstract
To improve the systematics and taxonomy of Patellogastropoda within the evolution of gastropods, we determined the complete mitochondrial genome sequences of Lottia goshimai and Nipponacmea fuscoviridis in the family Lottiidae, which presented sizes of 18,192 bp and 18,720 bp, respectively. In addition to 37 common genes among metazoa, we observed duplication of the trnM gene in L. goshimai and the trnM and trnW genes in N. fuscoviridis. The highest A + T contents of the two species were found within protein-coding genes (59.95% and 54.55%), followed by rRNAs (56.50% and 52.44%) and tRNAs (56.42% and 52.41%). trnS1 and trnS2 could not form the canonical cloverleaf secondary structure due to the lack of a dihydrouracil arm in both species. The gene arrangements in all Patellogastropoda compared with those of ancestral gastropods showed different levels of gene rearrangement, including the shuffling, translocation and inversion of single genes or gene fragments. This kind of irregular rearrangement is particularly obvious in the Lottiidae family. The results of phylogenetic and gene rearrangement analyses showed that L. goshimai and Lottia digitalis clustered into one group, which in turn clustered with N. fuscoviridis in Patellogastropoda. This study demonstrates the significance of complete mitogenomes for phylogenetic analysis and enhances our understanding of the evolution of Patellogastropoda.
Similar content being viewed by others

Introduction
The order Patellogastropoda (common name, true limpets) consists of the most primitive gastropod molluscs, which inhabit intertidal rocky shores worldwide, from tropical to polar regions1,2. Most species from this group feed by scraping a fine film of microalgae from what appears to be a bare rock surface. Their shells have the appearance of hats of different sizes, generally reaching no more than 20 cm3,4. These limpets, which are ecologically important in coastal regions, separated from other gastropods early in molluscan evolution5. They can be used in marine ecotoxicology research because of their wide distribution, and their gametes are available throughout the year6. This group is characterized by intraspecific polyphenism, the existence of cryptic species, and the intraspecific variation of characteristics such as shell morphology and colour. Therefore, it is difficult to identify species in this group by traditional methods alone7,8,9,10. The phylogenetic analysis of Patellogastropoda has generally focussed on Caenogastropoda, Neomphalina, Vetigastropoda, Neritimorpha and Heterobranchia. The phylogenetic position of the group and the evolutionary relationships among families of true limpets have been highly controversial11,12.
Nipponacmea fuscoviridis (Teramachi, 1949) and Lottia goshimai (Nakayama, Sasaki & T. Nakano, 2017) both belong to the family Lottiidae. N. fuscoviridis commonly appears in temperate areas around the Japanese islands and the southeastern coastal region of China13. Species of this genus are common along the Asian coast of the Pacific Ocean from Vietnam to Russia14. L. goshimai was previously thought to be an intraspecific variant of the northern population of N. fuscoviridis and was later proven to be a new species15; thus, there have been few studies on this species. Its developmental stages and gene expression were studied by Wang et al.16,17,18.
The complete mitochondrial genome provides more information than individual genes. It exhibits the characteristics of maternal inheritance, a high evolutionary rate and a relatively low intermolecular recombination rate, and it is becoming increasingly common for mitochondrial genomes to be used for phylogenetic reconstruction19,20,21,22. The circular mitochondrial genome of gastropods generally contains 37 genes (22 transfer RNA genes, two ribosomal RNA genes, 13 protein-coding genes) and a noncoding control region. Nevertheless, Lottiidae species seem to be an exception, exhibiting different numbers of tRNA genes23.
In the present study, two mitochondrial genomes (L. digitalis and N. fuscoviridis) from the Lottiidae family were sequenced, annotated and compared to the other available genomes from Patellogastropoda. We analysed the main characteristics of the newly generated mitogenomes, such as their nucleotide composition, codon usage and the secondary structure of their tRNAs. Complete mitogenome sequences from six subclasses of Gastropoda were downloaded from the GenBank database to reconstruct the phylogenetic tree. The results will help us to obtain further insight into the evolutionary relationships within Patellogastropoda.
Results and discussion
Characteristics, structure and organization of the mitogenomes
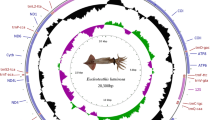
The gene arrangements found within Patellogastropoda mitochondrial genomes have been relatively conservative, but those of Lottiidae differ to some extent. The comparison of the two newly sequenced mitogenomes with a reported mitogenome from Lottiidae revealed the rearrangement of gene positions and structures. The complete mitochondrial genome sequences of L. goshimai and N. fuscoviridis were 18,192 bp and 18,720 bp, respectively (GenBank accessions MT248298 and MK395167) (Fig. 1, Table 1). Both circular mitochondrial genomes of the species contained 13 PCGs, 2 rRNA genes (12S rRNA and 16S rRNA), 22 putative tRNA genes and a control region (CR). Compared to the fragment of the genome previously published, we found an additional trnM gene in both species and additional trnW gene in N. fuscoviridis.

Maps of the mitochondrial genomes of two Lottiidae species. The direction of gene transcription is indicated by the arrows.
Overlapping and noncoding regions
Most of the genes identified in N. fuscoviridis are located on the heavy strand except for three PCGs and seven tRNAs. In addition, fourteen genes of L. goshimai (seven PCGs and seven tRNA genes) are located on the light strand, with the remaining genes being located on the heavy strand (Fig. 1 and Tables 2, 3). The mitochondrial genome of L. goshimai contains intergenic spacers with lengths ranging from 1 to 178 bp, and there are two genes showing overlapping nucleotides (6 and 20 bp). The longest intergenic spacer is located between trnY and nad5 (Table 2). The mitochondrial genome of N. fuscoviridis exhibits intergenic spacers with lengths ranging from 2 to 380 bp, and there are two genes with overlapping nucleotides (4 and 11 bp). The longest intergenic spacer is located between trnY and nad3 (Table 3). In conclusion, there are significant differences in the intergenic spacers and overlapping nucleotides of the two species, and these species of limpets also present large variations compared with other families (e.g., Nacellidae, Acmaeidae and Patellidae)24,25,26,27,28.
The control region (CR) is the largest non-coding region; it usually presents a high AT content and is therefore also known as the A + T rich region29. It is an essential element involved in mitochondrial genome replication and transcription initiation30. The mitogenomes of L. goshimai and N. fuscoviridis each contain one CR, and both CRs show relatively high AT contents of 61.61% and 53.43%, respectively. The CR is located between trnR and atp8 in L. goshimai, with a length of 1722 bp. In N. fuscoviridis, it is located between nad5 and atp8, with a length of 1561 bp. It also contains a replication origin for light-strand synthesis (OL), which is 21 bp (CCCTCCCCCCCAGGGGGAGGG) in length and folds into a hairpin secondary structure.
Base composition of mitogenomes
The A + T content of the whole mitogenome if 60.17% for L. goshimai (28.18% A, 32.00% T, 24.11% G and 15.71% C), and 54.15% for N. fuscoviridis (23.83% A, 30.32% T, 25.39% G and 20.46% C) (Table 4). The A + T contents of all PCGs in L. goshimai range from 55.65% (atp8) to 62.64% (cytb), and those in N. fuscoviridis range from 52.07% (nad4) to 57.25% (cox1) (Table 4). We observed the highest A + T contents of the two species in PCGs (59.95% and 54.55%), followed by rRNAs (56.50% and 52.44%) and tRNAs (56.42% and 52.41) (Table 4). The AT skew of the total PCGs is negative, and the GC skew is positive across the two species, indicating that they contain a slightly higher percentage of T and G bases than A and C bases. For each PCG of two Lottiidae species in addition to the cox2 gene of L. goshimai, most of the AT skew values are negative.
Protein-coding genes and codon usage
The total length of the all PCGs is 11,238 bp in L. goshimai and 11,154 bp N. fuscoviridis, accounting for 61.77% and 59.58% of the whole genome, respectively (Table 4). The comparison of the initiation and termination codons of all PCGs showed that most of the PGCs of the two Lottiidae species are initiated with an ATN codon and terminated with TAN. Only the cox1 gene of L. goshimai and nad3 of N. fuscoviridis start with GTG (Tables 2, 3). While the cox2, cox3 and cytb genes of N. fuscoviridis use an incomplete T stop codon, which is remarkably common in invertebrate mitogenomes.
The analysis of the two Lottiidae species indicated that the most frequently used amino acids are Gly, Ser1 and Val, while Gln and His are the least common amino acids (Fig. 2). In L. goshimai, the highest relative synonymous codon usage (RSCU) was found for UUA (Leu2), followed by AUU (Ile), GUU (Val) and UUU (Phe) (Fig. 2). In N. fuscoviridis, the highest relative synonymous codon usage (RSCU) was found for GGG (Gly), followed by GCU (Ala), UUU (Phe) and UUA (Leu2) (Fig. 2).

The codon distribution and relative synonymous codon usage (RSCU) in the mitogenomes of L. goshimai (Lg) and N. fuscoviridis (Nf). CDspT, codons per thousand codons.
Transfer RNA genes
We identified 23 tRNA genes from the mitochondrial genome of L. goshimai, including one more trnM gene than is common invertebrates, with lengths ranging from 65 (trnS2) to 72 bp (trnI). In addition, N. fuscoviridis exhibited one more trnW gene than L. goshimai, and 24 tRNA genes ranging from 64 (trnM1) to 72 bp (trnI) in length were identified. In both Lottiidae species, trnS1 and trnS2 cannot form a secondary structure due to the lack of dihydrouracil (DHU) arms, while other tRNAs are capable of folding into a typical clover-leaf secondary structure. The comparison of the tRNA genes of the two species showed that each corresponding amino acid is encoded by the same anticodon with the exception of the trnW1 gene of N. fuscoviridis, which is encoded by different anticodons (CCA). Moreover, methionine is encoded by two tRNAs with the same anticodons (CAT) (Tables 2, 3 and Figs. 3, 4).

Secondary structure of the tRNA genes of the L. goshimai mitochondrial genome.

Secondary structure of the tRNA genes of the N. fuscoviridis mitochondrial genome.
Nonsynonymous and synonymous substitutions.
We calculated the selection pressure (estimated by using Ka/Ks) on 13 PCGs in the two Lottiidae species (Fig. 5). Most of the Ka/Ks ratios are below 1 for these PCGs, indicating that they evolved under purifying selection. The remaining nad2, nad5, nad6 and cytb genes, with high Ka/Ks ratios, may have been affected by positive selection during evolution. Positive selection is influenced by the external environment for the self-regulation and transformation of genes, the elimination of genes that do not adapt to the environment, and the production of genes that can effectively adapt to the environment31. Therefore, advantageous genes are retained after non-synonymous mutations.

The Ka/Ks values of each PCG represent the ratios of nonsynonymous substitutions (Ka) to synonymous substitutions (Ks).
The substitution saturation index was analysed on the basis of the combined dataset of all PCGs of 60 Gastropoda mitogenomes, and the observed Iss value (Iss = 0.651) was significantly lower than that of the critical value (Iss.cSym = 0.859, p = 0.0000) (Fig. 6), indicating that sequence substitution is unsaturated; thus, the combined data are suitable for phylogenetic analysis.

Saturation plots for all PCGs. The plots show the uncorrected pairwise divergence in transitions (s) and transversions (v) against the divergence calculated using the GTR model.
Phylogenetic analysis
We used the Bayesian inference (BI) and maximum likelihood (ML) methods to reconstruct a phylogenetic tree based on 13 PCGs from the two new Lottiidae species and 58 other species within Gastropoda (i.e., 8 Patellogastropoda species, 11 Caenogastropoda species, 3 Neomphalina species, 17 Vetigastropoda species, 7 Neritimorpha species, and 12 Heterobranchia species), using two Mopaliidae species as outgroups.
In addition, in the BI analysis, due to the high rearrangement rate of Lottiidae species, which exhibited a long branch compared to other species of Patellogastropoda, we encountered a long-branch attraction (LBA) artefact in the process of constructing phylogenetic trees. This is a common systemic error in phylogenetic reconstruction resulting from the clustering of fast-evolving taxa in the tree, instead of revealing their genuine phylogenetic positions32,33. Specifically, the three species of the Lottiidae family and Heterobranchia erroneously formed a clade, but this situation did not appear in the ML analysis. Finally, we combined these two methods and obtained a basically consistent evolutionary tree through reference to previous research on the phylogeny of gastropods34,35,36,37 (Fig. 7).

The phylogenetic tree for the two Lottiidae species and other gastropod species based on 13 PCGs. The purple dots indicate the two species sequenced in this study. The number at each node is the bootstrap probability.
The results showed a stable evolutionary tree topology in which each subclass formed a monophyletic clade. Most of the recovered clades were highly supported (Bayesian posterior probability (BPP) = 1, and Bootstrap (BS) = 100). The higher phylogenetic relationship of clade formed: (((Neomphalina + Vetigastropoda) + Neritimorpha) + Caenogastroopoda) + (Patellogastropoda + Heterobranchia). Patellogastropoda and Heterobranchia clustered together in the same clade, which was located on the outermost branch of the six subclasses. Lottiidae formed an independent branch as (N. fuscoviridis + (L. goshimai and Lottia digitalis)) within Patellogastropoda. L. goshimai was shown to be the closest extant relative of Lottia digitalis, and this clade clustered with N. fuscoviridis.
The significance of Lottiidae species in the evolution and development of gastropods was confirmed through this study. Further mitogenome sequencing work was carried out to provide more comprehensive taxon sampling for the future, thus improving the understanding of the Lottiidae phylogeny and evolution within Gastropoda.
Gene arrangement of Patellogastropoda
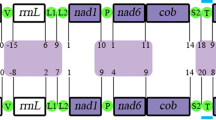
The gene arrangements in four subclasses were compared to the hypothetical ancestral gastropod gene order38 (Fig. 8). Among these subclasses, the fewest gene rearrangements are observed in Bathyacmaea nipponica of the Acmaeidae family, and only certain tRNA sequences exhibit shuffling (trnY and trnM), translocation (trnF, trnQ, trnF, trnC) and inversion (trnE)39. The gene order is closest to that of the family Nacellidae, with six tRNAs (trnT, trnR, trnN, trnA, trnK, trnI) and one PCG (nad3) exhibiting translocation. Recent studies of Nacellidae mitogenomes suggest that genome rearrangements are relatively conservative in this group11. The phylogenetic analyses showed that Nacellidae is the sister group of Acmaeidae, which confirmed that rearrangement may be helpful for phylogenetic analysis. Compared with the above two families, the gene order in Patellidae differs substantially, but the fragment from cytb to atp8 has been retained, with only a portion of this fragment exhibiting local inversion. However, the genome organization is almost the same in Patella ferruginea and Patella vulgate, indicating that they are conservative in the family Patellidae. The most noteworthy finding was that there are essential differences in gene arrangement among species of different Lottiidae families, but they share the common characteristic of rrnL and rrnS gene inversion. The mitogenomes of the Lottiidae family have retained a fraction of the clusters found in ancestral gastropods31. For instance, Lottia digitalis has retained nad4-nad4L, and L. goshimai has retained nad5-nad4-nad4l, with the nad4 and nad4l fragments inverted in both cases. In addition, an extremely high rate of gene rearrangement is found in N. fuscoviridis, and the irregular ordering may be caused by a high rate of sequence evolution40. We will need to conduct more research on the family to verify this in the future.

Mitochondrial genome organization of the two new Lottiidae species and available patellogastropod species.
Conclusion
In this study, the complete mitochondrial genome sequences of two new limpets, L. goshimai and N. fuscoviridis, belonging to Lottiidae, were characterized and compared. Duplications of tRNA genes are found in both species (trnM or trnW). In their tRNA secondary structures, both trnS1 and trnS2 are missing DHU stems, which is also observed in other species of the family. The phylogenetic relationships with other members of Gastropoda based on 13 mitochondrial PCGs were analysed. The results showed that the phylogeny was consistent with morphological observations and previous reports. In addition, a highly irregular rearrangement of mitochondrial genes was found within Lottiidae. Since there are currently few species in the family, it is impossible to determine whether this situation is associated with a single species or occurs throughout the family, which is worthy of further study.
Materials and methods
Sample collection and DNA extraction
Specimens of L. goshimai and N. fuscoviridis were collected from the coastal area of Xiangshan County (29.48°N, 121.98°E), Zhejiang Province, in the East China Sea and Jinzhou City (40.88°N, 121.13°E), Liaoning Province, in the Bohai Sea, respectively. Morphological identification of these samples was carried out by using published taxonomic books/available taxonomic keys, and we consulted with a taxonomist from the Museum of Marine Biology of Zhejiang Ocean University41,42. The samples were preserved in absolute ethyl alcohol before DNA extraction. Total genomic DNA was extracted from the operculum using the salting-out method43 and was then stored at − 20 °C before sequencing.
Mitochondrial genome sequencing, assembly and annotation
The whole mitogenomes of the two limpets were sequenced using the Illumina HiSeq X Ten platform (Shanghai Origingene Bio-pharm Technology Co., Ltd. China). An Illumina PE library with an insert size of 400 bp was generated. The original sequencing data have been stored in the sequence read archive (SRA, https://trace.ncbi.nlm.nih.gov/Traces/sra/) of the National Center for Biotechnology Information (NCBI). NOVOPlasty software (https://github.com/ndierckx/NOVOPlasty) was used for the de novo assembly of the clean data without sequencing adapters to obtain the optimal assembly result44. Two newly assembled mitochondrial genomes were annotated on the MITOS web server (https://mitos2.bioinf.uni-leipzig.de/index.py) using the invertebrate genetic code, and start and stop codons were confirmed by comparing the obtained nucleotide sequences with those from closely related limpets24,45,46.
Genome visualization, secondary structure prediction and comparative studies
Circular genome visualization was conducted with the CGView Server (https://stothard.afns.ualberta.ca/cgview_server/index.html)47. The secondary structure of the tRNA genes was predicted using the software ARWEN (https://130.235.244.92/ARWEN/) and the tRNAscan-SE v.2.0 web server (https://lowelab.ucsc.edu/tRNAscan-SE/), as implemented on the MITOS web server45,48,49. The nucleotide composition and relative synonymous codon usage (RSCU) of each PCG were calculated using MEGA 7.050. AT and GC skew values were calculated with the following formula: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)51. The ratio of nonsynonymous substitutions (Ka) to synonymous (Ks) substitutions was estimated with DnaSP6.052.
Preparation of datasets, model selection, phylogenetic analyses
For the phylogenetic analysis, DAMBE 5.3.19 was used to adjust the nucleotide sequences of 13 protein-coding genes (PCGs) of each species, and the nucleotide substitution saturation was analysed to determine whether these sequences were suitable for constructing phylogenetic trees53. Sixty published mitochondrial genomes were downloaded from NCBI as references, including those of 58 other marine gastropods and two outgroups (Cryptochiton stelleri and Katharina tunicata of Polyplacophora), and were analysed along with the mitogenome sequence of the two new Lottiidae species (Table 1). Then, the sequences of each of 62 species were aligned using ClustalW with the default parameters in MEGA 7.0. The phylogenetic analyses incorporated Bayesian inference (BI) methods using the program MrBayes v3.2 and maximum likelihood (ML) using IQ-TREE54,55. MrMTgui was used to combine the results of PAUP 4.0, Modeltest 3.7 and MrModeltest 2.3 to find the best substitution models (GTR + I + G) with the AIC for Bayesian inference (BI)56,57,58. BI analyses were conducted with two Markov chain Monte Carlo (MCMC) runs, each with four chains (three heated and one cold) run for 2,000,000 generations, with tree sampling every 1000 steps and a burn-in of 25%. ML analysis was performed with the best-fit substitution model automatically selected by ModelFinder, and the number of bootstrap replicates was set to 1000 in ultrafast likelihood bootstrapping to reconstruct a consensus tree59. The phylogenetic trees were visualized and edited using FigTree v1.4.360.
Data availability
The mitochondrial genome data has been submitted to NCBI GenBank under the following accession numbers: Lottia goshimai (MT248298), Nipponacmea fuscoviridis (MK395167).
References
Kawashima, H., Ohnishi, M. & Ogawa, S. Differences in Sterol Composition of Gonads of the Lottiid Limpets Nipponacmea concinna and Nipponacmea fuscoviridis from Northeastern Japan. J. Oleo Sci. 60, 501–504 (2011).
Stimson, J. The role of the territory in the ecology of the intertidal limpet Lottia gigantea (Gray). Ecology 54, 1020–1030 (1973).
Kristof, A., de Oliveira, A. L., Kolbin, K. G. & Wanninger, A. A putative species complex in the Sea of Japan revealed by DNA sequence data: A study on Lottia cf. kogamogai (Gastropoda: Patellogastropoda). J. Zool. Syst. Evol. Res. 54, 177–181 (2016).
Niu, C. J., Nakao, S. & Goshima, S. Energetics of the limpet Lottia kogamogai (Gastropoda: Acmaeidae) in an intertidal rocky shore in southern Hokkaido Japan. J. Exp. Mar. Biol. Ecol. 224, 167–181 (1998).
Nakano, T. & Sasaki, T. Recent advances in molecular phylogeny, systematics and evolution of Patellogastropod limpets. J. Molluscan Stud. 77, 203–217 (2011).
Perez, S., Fernandez, N. & Ribeiro, P. A. Standardization of a Patella spp. (Mollusca, Gastropoda) embryo-larval bioassay and advantages of its use in marine ecotoxicology. Ecotoxicol. Environ. Saf. 127, 175–186 (2016).
Nakano, T. & Spencer, H. G. Simultaneous polyphenism and cryptic species in an intertidal limpet from New Zealand. Mol. Phylogenet. Evol. 45, 470–479 (2007).
Nakano, T., Marshall, B. A., Kennedy, M. & Spencer, H. G. The phylogeny and taxonomy of New Zealand Notoacmea and Patelloida species (Mollusca: Patellogastropoda: Lottiidae) inferred from DNA sequences. Molluscan Res. 29, 33–59 (2009).
Brian, S. W. & Lindberg, D. R. Morphological and molecular resolution of a putative cryptic species complex: a case study of Notoacmea fascicularis (menke, 1851) (Gastropoda: Patellogastropoda). J. Molluscan. Stud. 65, 99–109 (1999).
Yu, S. S., Wang, J., Wang, Q. L., Huang, X. W. & Dong, Y. W. DNA barcoding and phylogeographic analysis of Nipponacmea limpets (Gastropoda: Lottiidae) in China. J. Molluscan. Stud. 80, 420–429 (2014).
Gaitan-Espitia, J. D., Gonzalez-Wevar, C. A., Poulin, E. & Cardenas, L. Antarctic and sub-Antarctic Nacella limpets reveal novel evolutionary characteristics of mitochondrial genomes in Patellogastropoda. Mol. Phylogenet. Evol. 131, 1–7 (2019).
Zapata, F. et al. Phylogenomic analyses of deep gastropod relationships reject Orthogastropoda. Proc. R. Soc. B Biol. Sci. 281, 20141739 (2014).
Sasaki, T. & Nakano, T. The southernmost record of Nipponacmea fuscoviridis (Patellogastropoda: Lottiidae) from Iriomote Islands, Okinawa (Japan). Venus 66, 99–102 (2007).
Sharina, S. N., Chernyshev, A. V. & Zaslavskaya, N. I. Genetic diversity and phylogeny of limpets of the genus Nipponacmea (Patellogastropoda: Lottiidae) based on mitochondrial DNA sequences. Mitochondrial DNA Part A 28, 703–710 (2017).
Nakayama, R., Sasaki, T. & Nakano, T. Molecular analysis reveals a new cryptic species in a limpet Lottia kogamogai (Patellogastropoda: Lottiidae) from Japan. Zootaxa 4277, 237–251 (2017).
Huan, P., Wang, Q., Tan, S. J. & Liu, B. Z. Dorsoventral decoupling of Hox gene expression underpins the diversification of molluscs. PNAS 117, 503–512 (2020).
Tan, S. J., Huan, P. & Liu, B. Z. The expression pattern of a tyrosinase gene potentially involved in early larval shell biogenesis of the limpet Lottia goshimai. Mar. Sci. 09, 1000–3096 (2018).
Wang, Q., Huan, P. & Liu, B. Z. Expression patterns of soxb and mox genes in Lottia goshimai during the formation of molluscan foot. Oceanolog. Limnolog. Sin. 05, 1091–1097 (2019).
Shi, G. et al. The complete mitochondrial genomes of Umalia orientalis and Lyreidus brevifrons: the phylogenetic position of the family Raninidae within Brachyuran crabs. Mar. Genomics 21, 53–61 (2015).
Tyagi, K. et al. Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta). Sci. Rep. 10, 695 (2020).
Li, T. et al. A mitochondrial genome of rhyparochromidae (Hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci. Rep. 6, 35175 (2016).
Kumar, V. et al. The Complete Mitochondrial Genome of endemic giant tarantula, Lyrognathus crotalus (Araneae: Theraphosidae) and comparative analysis. Sci. Rep. 10, 74 (2020).
Simison, W. B., Lindberg, D. R. & Boore, J. L. Rolling circle amplification of metazoan mitochondrial genomes. Mol. Phylogenet. Evol. 39, 562–567 (2006).
Fuenzalida, G., Poulin, E., Gonzalez-Wevar, C., Molina, C. & Cardenas, L. Next-generation transcriptome characterization in three Nacella species (Patellogastropoda: Nacellidae) from south america and antarctica. Mar. Genomics 18, 89–91 (2014).
Goldstien, S. J., Gemmell, N. J. & Schiel, D. R. Molecular phylogenetics and biogeography of the nacellid limpets of New Zealand (Mollusca: Patellogastropoda). Mol. Phylogenet. Evol. 38, 261–265 (2006).
González-Wevar, C. A., Nakano, T., Cañete, J. I. & Poulin, E. Molecular phylogeny and historical biogeography of Nacella (Patellogastropoda: Nacellidae) in the Southern Ocean. Mol. Phylogenet. Evol. 56, 115–124 (2010).
Sun, J. et al. The mitochondrial genome of the deep-sea limpet Bathyacmaea nipponica (Patellogastropoda: Pectinodontidae). Mitochondrial DNA Part B 4, 3175–3176 (2019).
Sousa, R., Delgado, J., Pinto, A. R. & Henriques, P. Growth and reproduction of the north-eastern Atlantic keystone species Patella aspera (Mollusca: Patellogastropoda). Helgol. Mar. Res. 71, 8 (2017).
Meng, X., Shen, X., Zhao, N., Tian, M. & Zhu, X. The complete mitochondrial genome of the clam Mactra veneriformis (Bivalvia: Mactridae): Has a unique non-coding region, missing atp8 and typical tRNASer. Mitochondrial DNA 24, 613–615 (2013).
Fernández-Silva, P., Enriquez, J. A. & Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 88, 41–56 (2003).
Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573 (1998).
Uribe, J. E., Irisarri, I., Templado, J. & Zardoya, R. New patellogastropod mitogenomes help counteracting long-branch attraction in the deep phylogeny of gastropod mollusks. Mol. Phylogenet. Evol. 133, 12–23 (2019).
Williams, S. T., Foster, P. G. & Littlewood, D. T. J. The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny. Gene 533, 38–47 (2014).
Sevigny, J. L. et al. The Mitochondrial Genomes of the Nudibranch Mollusks, Melibe leonina and Tritonia diomedea, and Their Impact on Gastropod Phylogeny. PLoS ONE 10, e0127519 (2015).
Nakajima, Y. et al. The mitochondrial genome sequence of a deep-sea, hydrothermal vent limpet, Lepetodrilus nux, presents a novel vetigastropod gene arrangement. Mar. Genomics 28, 121–126 (2016).
Romero, P. E., Weigand, A. M. & Pfenninger, M. Positive selection on panpulmonate mitogenomes provide new clues on adaptations to terrestrial life. BMC Evol. Biol. 16, 164 (2016).
Lee, H. et al. Incorporation of deep-sea and small-sized species provides new insights into gastropods phylogeny. Mol. Phylogenet. Evol. 135, 136–147 (2019).
Osca, D., Irisarri, I., Todt, C., Grande, C. & Zardoya, R. The complete mitochondrial genome of Scutopus ventrolineatus (Mollusca: Chaetodermomorpha) supports the Aculifera hypothesis. BMC Evol. Biol. 14, 197 (2014).
Dowton, M., Castro, L. R. & Austin, A. D. Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome “morphology”. Invertebr. Syst. 16, 345–356 (2002).
Xu, W., Jameson, D., Tang, B. & Higgs, P. G. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J. Mol. Evol. 63, 375–392 (2006).
Fuchigami, T. & Sasaki, T. The shell structure of the Recent Patellogastropoda (Mollusca: Gastropoda). Paleontolog. Res. 9, 143–168 (2005).
Vortsepneva, E., Ivanov, D., Purschke, G. & Tzetlin, A. Morphology of the jaw apparatus in 8 species of Patellogastropoda (Mollusca, Gastropoda) with special reference to Testudinalia tesulata (Lottiidae). Zoomorphology 132, 359–377 (2013).
Aljanabi, S. M. & Martinez, I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic. Acids. Res. 25, 4692–4693 (1997).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic. Acids. Res. 45, e18 (2016).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Simison, W. B., Lindberg, D. R. & Boore, J. L. Rolling circle amplification of metazoan mitochondrial genomes. Mol. Phylogenet. Evol. 39, 562–567 (2006).
Grant, J. R. & Stothard, P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic. Acids. Res. 36, 181–184 (2008).
Laslett, D. & Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic. Acids. Res. 25, 955–964 (1997).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Hassanin, A., Léger, N. & Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298 (2005).
Rozas, J. & Rozas, R. DnaSP, DNA sequence polymorphism: an interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 11, 621–625 (1995).
Xia, X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30, 1720–1728 (2013).
Ronquist, F. et al. Mrbayes 3.2: Efficient Bayesian phylogenetic inference and model selection across a large model space. Syst. Biol. 61, 539–542 (2012).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Swofford, D. L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods) Version 4. (Sinauer Associates, Sunderland, Massachusetts, 1998).
Posada, D. & Crandall, K. A. Modeltest: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Nylander, J. A., Ronquist, F., Huelsenbeck, J. P. & Nieves-Aldrey, J. Bayesian phylogenetic analysis of combined data. Syst. Biol. 53, 47–67 (2004).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587 (2017).
Rambaut A. FigTree, version 1.4.3, https://tree.bio.ed.ac.uk/software/figtree/ (accessed 1 July 2016).
Acknowledgements
This work was financially supported by the Fundamental Research Funds for Zhejiang Provincial Universities and Research Institutes (No. 2019J00021), the Natural Science Foundation of Zhejiang Province (Grant No. LQ18D060004), the Open Foundation from Marine Sciences in the First-Class Subjects of Zhejiang Province and the Project of Bureau of Science and Technology of Zhoushan (2020C21026).
Author information
Authors and Affiliations
Contributions
F.J.T. analyzed the data, wrote the paper, and prepared the figures and tables. G.Y.H. and Y.C.R. collected field material and processed the samples. Y.Y.Y. conceived and designed the experiments, reviewed drafts of the paper. L.J.J. contributed analysis tools, reviewed drafts of the paper. G.B.Y and L.Z.M supervised and directed the work, all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feng, Jt., Guo, Yh., Yan, Cr. et al. Comparative analysis of the complete mitochondrial genomes in two limpets from Lottiidae (Gastropoda: Patellogastropoda): rare irregular gene rearrangement within Gastropoda. Sci Rep 10, 19277 (2020). https://doi.org/10.1038/s41598-020-76410-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76410-w
- Springer Nature Limited