
Abstract
The fungus Parastagonospora nodorum causes septoria nodorum blotch on wheat. The role of the fungal Velvet-family transcription factor VeA in P. nodorum development and virulence was investigated here. Deletion of the P. nodorum VeA ortholog, PnVeA, resulted in growth abnormalities including pigmentation, abolished asexual sporulation and highly reduced virulence on wheat. Comparative RNA-Seq and RT-PCR analyses revealed that the deletion of PnVeA also decoupled the expression of major necrotrophic effector genes. In addition, the deletion of PnVeA resulted in an up-regulation of four predicted secondary metabolite (SM) gene clusters. Using liquid-chromatography mass-spectrometry, it was observed that one of the SM gene clusters led to an accumulation of the mycotoxin alternariol. PnVeA is essential for asexual sporulation, full virulence, secondary metabolism and necrotrophic effector regulation.
Similar content being viewed by others

Introduction
Plant pathogenic microorganisms cause severe economic damage to the agricultural sector threatening food security [1]. Parastagonospora nodorum is a pathogenic fungus that causes Septoria nodorum blotch (SNB) of wheat and is considered a model necrotrophic fungal plant pathogen [2, 3]. The use of fungicides and the breeding of resistant cultivars have been successful in managing the spread of SNB [4]. However, continued yield losses caused by SNB prompt the investigation for the discovery of novel control methods [4].
P. nodorum is a necrotroph which infects susceptible wheat cultivars by causing necrosis of the plant tissue which it subsequently colonises [5]. P. nodorum spores germinate on the surface of wheat leaves [5]. During infection, the fungus produces proteinaceous necrotrophic effectors (NE) that interact with wheat dominant susceptibility receptors, in an inverse gene-for-gene manner, leading to host tissue necrosis [5]. The interaction between NEs and their corresponding susceptibility genes has been conceptualised as effector-triggered susceptibility and P. nodorum–wheat has been used as the model pathosystem to study this immune pathway in plants [6]. The infection cycle is completed by the production of asexual pycnidiospores and sexual ascospores which spread through rain splashes and wind, respectively, to infect other susceptible host plants [3]. For crop protection purposes, the removal of dominant susceptibility genes from wheats improves disease resistance [4].
Whilst transcription factors (TFs) are ubiquitous across all domains of life [7], the structural family Velvet is exclusive to the fungal kingdom [8]. Velvet TFs were first identified and characterised as important developmental regulators through pioneering studies in Aspergillus spp. [9, 10]. While all Velvet TFs share a conserved domain, four distinct classes have been described: Velvet A (VeA), Velvet-like B (VelB), Velvet-like C (VelC) and Viability of Spores A (VosA). Furthermore, interactions between the Velvet TFs have been reported, indicating that complex oligomeric regulatory units can form [11]. The Velvet complex can also interact with other cofactors, such as the methyltransferase LaeA, and has been associated with several regulatory functions including vegetative growth and development, stress tolerance, reproduction and secondary metabolite (SM) biosynthesis [9]. Orthologs of the canonical Velvet regulator VeA have been associated with virulence in both biotrophic and necrotrophic plant pathogens [12].
A number of P. nodorum TFs have been found to regulate the NE genes SnToxA and SnTox3 but little is known about the transcriptional regulation of the other two major NE genes SnTox1 and SnTox267 [12,13,14]. Previously, P. nodorum TFs such as PhmR and ElcR have been shown to regulate the production of phytotoxic SMs and be required for normal phytopathogenicity [15, 16].
Prior to this study, the role of the Velvet pathway of P. nodorum in NE and secondary metabolite regulation and host infection was not known. Therefore, this study aimed to identify and characterise the Velvet VeA ortholog in P. nodorum (PnVeA), and to investigate the genes regulated by this putative TF. PnVeA is essential for asexual reproduction, vegetative development and full phytopathogenicity. Furthermore, several NEs, as well as the mycotoxin alternariol are subjected to VeA regulatory control.
Results
Identification of Velvet TFs in P. nodorum and the deletion of PnVeA in P. nodorum
Four coding sequences were identified in the P. nodorum reference which harboured a putative velvet domain (InterPro ID: IPR037525) (Fig. 1A). A phylogenetic analysis incorporating characterised Velvet TFs from A. nidulans, A. flavus, Botrytis cinerea and Magnaporthe oryzae revealed four clades corresponding to the four distinct Velvet ortholog classes VeA, VelB, VelC and VosA (Fig. 1B). As such, the putative P. nodorum orthologues were designated PnVeA (SNOG_01807), PnVelB (SNOG_03679), PnVelC (SNOG_12917) and PnVosA (SNOG_06311). PnVeA was selected for further investigation and subsequent functional characterisation, given that it is highly expressed during P. nodorum infection, from necrotrophy to sporulation based on a microarray analysis [17], which aligns with the prominent role of VeA orthologs in other fungi, especially during host-pathogen interaction [9, 12].

Protein sequence analysis of Velvet transcription factors. (A) Domain architecture of putative Velvet-domain (IPR037525) proteins in Parastagonospora nodorum showing the location of the Velvet domain in each ortholog. (B) A neighbour-joining phylogenetic tree showing characterised Velvet-domain transcription factors from four different species compared to putative orthologues in P. nodorum. Orange, green, grey, and blue boxes indicate the clustered VeA, VosA, VelB and VelC orthologs respectively. UniProt IDs are indicated right of protein names. Branch lengths denote the dissimilarities in amino acid sequences between proteins. Bootstrap values are indicated at node branches
PnVeA is required for vegetative development, asexual sporulation and full virulence
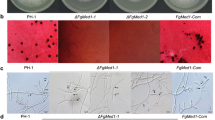
Two independent P. nodorum mutant strains lacking a functional PnVeA (SNOG_01807, NCBI accession: XM_001792381.1) were generated via homologous recombination (pnvea-23 and pnvea-25) in the P. nodorum SN15 background (Additional file 1). Mutant strains pnvea-23 and pnvea-25 were phenotypically compared to SN15 to assess the role of PnVeA in the development of the fungus. Strains lacking PnVeA had produced a dark secretion when cultivated on agar medium, which was lacking in SN15, and were unable to produce pycnidia and pycnidiospores (Fig. 2A and B).

Comparative examination of vegetative morphology and virulence of P. nodorum pnvea mutants with SN15. (A) Vegetative morphology of P. nodorum strains on V8PDA (top row) highlighting differences in pycnidial development (orange arrows) (bottom row). (B) Pycnidiospore count of P. nodorum strains on V8PDA media after 12 days of growth showing the abolishment of pycnidiospore production in pnvea-23 and pnvea-25. (C) Wheat leaf assay infection at day 6 post-inoculation showing reduced virulence of knockout mutants on wheat cultivar Axe. Tween was used as a negative control. (D) Pycnidiospore count of P. nodorum strains normalized to the length of infected leaf tissue after 10 days, showing the abolishment of pycnidiospore production in pnvea-23 and pnvea-25. For (B) and (D), ANOVA with the Tukey-Kramer test was used to compare the means of pycnidiospore counts (p ≤ 0.05, n = 3). Different letters above the bars indicate statistical significance between strains. Strains without detectable pycnidiospores are indicated by “n.d.” for “not detected”
To determine whether PnVeA affected virulence in-planta, pnvea-23 and pnvea-25 were compared to SN15 using a wheat leaf infection assay. Both pnvea-23 and pnvea-25 were highly reduced in virulence and unable to produce pycnidia (Fig. 2C and D; Additional file 2). Microscopic analysis revealed pnvea-25 was capable of host penetration attempts via the stomata and epidermis comparable to SN15 (Additional file 3).
To confirm that the phenotype of mutants is the result of the deletion of PnVeA, a gene complementation mutant was generated using the pnvea-25 background (PnVeA-25C). Phenotypic analyses indicated that the P. nodorum PnVeA-25C is comparable to SN15 (Fig. 2).
RNA Sequencing revealed that PnVeA regulates necrotrophic effectors, transcription factors and secondary metabolite genes in vitro
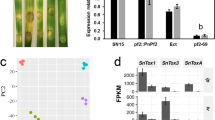
As the deletion of PnVeA affects vegetative morphology and virulence, we then used a comparative RNA-Seq approach to investigate the expression profile in SN15 and pnvea-25 under in vitro growth to identify genes responsible for the observed phenotypes. A principal component (PC) analysis of the SN15 and pnvea-25 RNA-Seq data revealed a clear segregation between SN15 and pnvea-25 consistent across replicates (Fig. 3A). PC1 accounted for 85% of the total variation while PC2 accounted for 12% and this discriminated SN15 from pnvea-25 (Fig. 3A). The in vitro RNA-Seq analysis revealed 5052 differentially expressed (DE) genes between pnvea-25 and the SN15 with 2898 up-regulated and 2154 down-regulated genes (Additional file 4). The putative orthologs PnVelB and PnVosA were significantly down-regulated with a Log2 fold change (LFC) of -1.35 and -5.54, respectively, while PnVelC expression remained unaltered. Previously characterised TFs involved in effector regulation, PnPf2 (-1.70 LFC) and PnCon7 (-1.05 LFC), were both significantly down-regulated [18, 19].

Comparative RNA-Seq analysis between SN15 and pnvea-25 under in vitro condition. (A) Principal component analysis (PCA) of transcriptomes of SN15 and pnvea25. Three biological replicates were used. Two SN15 biological replicates overlapped on the plot resulting in only two visual data points. (B) A ridge plot illustrating the enriched gene ontology (GO) terms between SN15 and pnvea-25 plotted against the LFC of genes within the GO terms. The shading represents the percentage of genes in the GO term that was enriched. (C) The expression profile of necrotrophic effectors SnToxA, SnTox1, SnTox267 and SnTox3 in the RNA-Seq analysis. Horizontal dashed grey lines at one fragment per kilobase of transcript per million mapped reads (FPKM) indicate the lowest threshold at which a gene is considered expressed. Asterisks indicate significant difference while n.s. is not significant according to Wald test with the Benjamini-Hoschberg adjustment, p < 0.0001 (****)
The enrichment of gene ontology (GO) terms within DE genes was analysed to assess the nature of the gene expression changes between SN15 and pnvea-25. This revealed that overall, PnVeA down-regulates transport within the cell and up-regulates processes related to carbohydrates (Fig. 3B). Major enriched genes up-regulated in pnvea-25 encoded for oxidoreductase activity (GO:0016491, 138 out of 502 genes), transmembrane transport (GO:0055085, 142 out of 561 genes) and transmembrane transporter activity (GO:0022857, 111 out of 420 genes). Notably, 21 out of 142 down-regulated transmembrane transport genes were predicted to be transporters associated with nitrogen assimilation (Additional file 5). A similar number of genes in catalytic activity (GO:0003824) and various GO terms related to binding (GO:0020037, GO:0005506, GO:0071949, GO:0008061, GO:0030246, GO:0050660) were equally both up and down-regulated between SN15 and pnvea-25. Genes that are associated with carbohydrate metabolic processes (GO:0005975 – 58 out of 229 genes) and hydrolase activity (GO:0004553 – 46 out of 140 genes) were enriched and predominantly down-regulated in pnvea-25. Notably, 35 of the 46 down-regulated genes in the GO term hydrolase activity (GO:0005975) were putative plant cell wall depolymerases (Additional file 6).
SnToxA, SnTox1, SnTox267 and SnTox3 are well-characterised necrotrophic effector-coding genes present in SN15 [13, 20,21,22]. Another effector SnTox5 has been characterised in P. nodorum, but is absent in the reference isolate studied here [23]. In addition, 37 uncharacterised candidate effector genes were previously identified in SN15 (Additional file 7) [24]. These candidate effector genes and known effector genes were then examined in the RNA-Seq analysis between SN15 and pnvea-25 to explore a connection with known virulence factors. SnTox1 and SnTox267 were significantly down-regulated while the expression of SnToxA and SnTox3 was not altered during in vitro growth (Fig. 3C). Of the 37 candidate effector genes, 12 genes had significant dysregulation of expression in pnvea-25 compared to SN15 (Additional file 7).
PnVeA deletion leads to upregulation of necrotrophic effectors during late infection
The dysregulation of effector genes in the RNA-Seq prompted an analysis of their expression during host infection as it could be a factor involved in the observed reduction in virulence for the PnVeA deletion mutants. Digital droplet PCR (ddPCR) demonstrated that pnvea-25 had a significantly different profile of effector gene expression compared to the SN15 and PnVeA-25C (Fig. 4). The expression SnToxA did not significantly change without PnVeA (Fig. 4A). In contrast, SnTox1 expression in pnvea-25 was higher than in SN15 and PnVeA-25C at 10 days post infection (Fig. 4B). Interestingly, pnvea-25 expression of SnTox3 was increased from six days to 10 days post inoculation in-planta compared to SN15 and PnVeA-25C (Fig. 4C). Furthermore, SnTox267 expression in pnvea-25 was significantly higher than in SN15 and PnVeA-25C in-planta at 10 days post inoculation (Fig. 4D). Overall, pnvea-25 expressed the effector genes at higher levels than SN15 and PnVeA-25C at 10 days post-inoculation, excluding SnToxA where expression remained unchanged in-planta. This corresponds to pycnidiation in the infection cycle of P. nodorum, which was abolished in pnvea-25 and also where effector expression is typically reduced [25].

Digital droplet PCR of necrotrophic effector genes revealed that PnVeA coordinates SnTox1, SnTox3 and SnTox267 expression in-planta. The in-planta gene expression profiles of effector genes, (A) SnToxA, (B) SnTox1, (C) SnTox267 and (D) SnTox3, for the wildtype strain (SN15), PnVeA deleted mutant (pnvea-25) and complemented mutant (PnVeA-25C) were measured at three-, six-, eight- and 10-days post-infection. Asterisks indicate a significant difference in mean within a time point between strains according to a one-way ANOVA with the Tukey-Kramer test post-hoc (p ≤ 0.05, n = 3), p < 0.05 (*), < 0.01 (**), < 0.001 (***) and < 0.0001 (****)
PnVeA is associated with the regulation of putative secondary metabolite gene clusters
The identification and characterisation of PnVeA presents an opportunity to further investigate SMs in P. nodorum at a global regulatory level based on the current understanding of Velvet regulation of SMs in other fungi. In A. flavus and the corn pathogen Fusarium verticillioides, the VeA orthologs are regulators of secondary metabolism [26, 27]. Since 38 SM gene clusters have been previously discovered in P. nodorum [28], the expression of these clusters was analysed. Five putative polyketide synthase (PKS) gene clusters showed differential expression patterns between SN15 and pnvea-25 in the RNA-seq analysis (Fig. 5).

Differential gene expression of predicted SM gene clusters in P. nodorum [28]. In vitro RNA-Seq analysis compared the expression between wildtype SN15 and PnVeA deleted strain (pnvea-25). Log2 fold change (LFC) is coloured according to its significance: green is significantly up-regulated in pnvea-25, red is significantly down-regulated in pnvea-25 and grey is not significant. SNOG is the gene locus names. “PKS” are polyketide synthase genes, “TF” are transcription factors, “Other” are genes of various miscellaneous functions and “Previously not annotated” are genes within a cluster but only present in the genome by Bertazzoni et al. [48]
Cluster 17 contains the highly reducing PKS (HR-PKS) gene SNOG_08274. Out of the 18 predicted genes in this cluster, 13 were significantly down-regulated, including the backbone gene. antiSMASH comparative gene cluster analysis suggests that Cluster 17 can produce a SM similar to the naphthopyrone YWA1, a conidial pigment intermediate in the opportunistic human pathogenic fungus A. fumigatus [29]. Furthermore, SNOG_08274 has been previously associated with pigment production [28].
Two adjacent SM gene clusters (Cluster 25 and Cluster 26) containing putative HR-PKS genes (SNOG_11066 and SNOG_11076, respectively) were up-regulated in pnvea-25 mutant. However, it should be noted that SNOG_11066 is not expressed in P. nodorum SN15 [17]. While Cluster 25 has been associated with pyranonigrin E production [30], antiSMASH analysis has linked Cluster 26 to the macrolide AKML [31]. Genes in Cluster 28, which contains a NR-PKS gene SNOG_11981, were also up-regulated. Of the six genes predicted within Cluster 28, five were up-regulated. SNOG_11981 has been linked to melanin production [32], this may be a link to the observed darker pigmentation in pnvea-23 and pnvea-25.
Notably, of the five DE PKS gene clusters, one (NR-PKS; SnPKS19) is involved in the production of alternariol, a previously characterized mycotoxin in P. nodorum [33]. To assess the correlation between RNA-seq results and SM production, particularly the abundance of alternariol, the SM profiles of all strains were analysed through LC-DAD-MS. PnVeA deleted strains (pnvea-23 and pnvea-25) displayed a new peak 1 (m/z 257 [M-H]-) [33], also present in lower quantities in PnVeA-25C, but undetectable in the control strain SN15 (Fig. 6A). Besides having a similar mass, the compound UV-vis spectrum and molecular formula were consistent with alternariol (Fig. 6B and C, Additional file 8).

The deletion of PnVeA resulted in a modified metabolic profile. (A) LC-DAD (DAD 254 nm) chromatograms showing the metabolomic profiles of P. nodorum SN15, pnvea-23, pnvea-25 and PnVeA-25C strains grown on V8PDA. Deletion of PnVeA led to increased production of 1 (alternariol), the structure of which is depicted in (B). (C) Alternariol UV-vis spectrum
Additionally, two uncharacterized compounds, 2 (m/z 378 [M + H]+) and 3 (m/z 392 [M + H]+), without apparent UV absorption were detected in SN15 and PnVeA-25C, but were absent in pnvea-23 and pnvea-25 (Additional file 9).
Discussion
The Velvet TFs are known to function as global regulators involved in diverse functions in filamentous fungi [9, 12]. Until now, their regulatory role had not been investigated in the NE-producing pathogen P. nodorum. Using a gene deletion approach, we highlight several key processes controlled by the VeA ortholog. Phenotypic analysis and RNA sequencing data suggest that PnVeA is required for full virulence, asexual sporulation, normal effector regulation, SM production and pigmentation.
Regarding sporulation, changes are also reported following VeA deletion in other fungi [9], yet we observed the complete abolishment of pycnidia and pycnidiospore production both in-vitro and during infection. This indicates PnVeA has a pivotal role in asexual sporulation in P. nodorum relative to orthologs in other characterised fungi, such as the pathogens: A. flavus, B. cinerea and M. oryzae [34,35,36,37].
The comparative RNA-Seq analysis conducted in this study revealed that PnVeA has a global role in gene expression, including significant changes in genes associated with carbohydrate/chitin interactions and carbohydrate metabolism, as well as iron/FAD metabolism and secondary metabolite biosynthesis. Previous transcriptomic investigations of the VeA ortholog in the plant pathogen A. flavus showed that oxidoreductase genes and genes related to carbohydrate metabolism were shown to be differentially regulated similarly to P. nodorum [38].
Leaf infection assays using the pnvea mutants demonstrated reduced virulence, despite retaining the capacity to attempt to penetrate the host tissue [39]. This indicates PnVeA may be required for biochemical pathways and other factors paramount for normal virulence. A key TF involved in virulence and NE expression in P. nodorum is PnPf2 [19, 40]. Gene silencing of another P. nodorum TF PnCon7 also led to reduced NE expression [18]. Interestingly, both these TFs, as well as SnTox1 and SnTox267 were downregulated in pnvea-25. SnToxA was not expressed in either SN15 nor pnvea-25 in-vitro which is consistent with previous literature [19, 41], yet 12 additional NE candidates identified by Jones et al. [24] were also altered in this analysis (Additional file 7). We therefore sought to investigate NE expression further during infection with ddPCR in order to provide insight into the reduced virulence observed for pnvea-23 and pnvea-25. Surprisingly, of the characterised NE genes, only Tox267 exhibited significantly reduced expression during early infection (3 days post inoculation). However, we did observe late necrotic symptoms induced by the pnvea mutants at 10 days post inoculation where the expression of effector genes was also greater than SN15 or PnVeA-25C (Fig. 4 and Additional file 2). There may be an undiscovered role of PnVeA related to the detection of wheat environmental cues to enable infection, reflected by the delayed expression of NE genes and the subsequent appearance of necrotic symptoms.
This investigation also revealed a significant role for PnVeA in SM regulation, including the mycotoxin alternariol, found in P. nodorum and in the members of the fungal genus Alternaria including the broad host phytopathogen Alternaria alternata [42, 43]. We demonstrated using LC-DAD-MS that PnVeA negatively regulates the production of alternariol. This is in direct contrast to what has been observed in A. alternata, where VeA was a positive regulator of alternariol production [44]. The previously characterised PKS gene, SnPKS19 is required to produce alternariol in P. nodorum [33], and this gene was significantly upregulated in the RNA-seq dataset. Interestingly, expression of the gene encoding a short-chain dehydrogenase Sch1, which is negatively correlated with alternariol production [43], was unchanged in pnvea-25. This suggests that the synthesis of alternariol is directly attributed by SnPKS19 and overcomes any suppressive effects previously attributed to Sch1. Alternariol was not detected in SN15 in this study but that may be due to the variation in the growth medium compared to previous studies [33, 43].
Three interesting uncharacterised PKS genes which were found to be upregulated in pnvea-25, include SNOG_08274 (Cluster 17), SNOG_11076 (Cluster 26) and SNOG_11981 (Cluster 28). However, the identity of the metabolite products is yet to be determined. Two compounds without UV absorption absent in pnvea-25 may be associated with Cluster 17 as the sole cluster to be down-regulated in pnvea-25 or the two compounds may be from unknown biosynthetic origins. Although tools like antiSMASH can predict SM-related genes for various classes of molecules, the biosynthetic origin of many compounds remains to be determined [45].
This study has characterised PnVeA, the ortholog of a fungal-specific global regulator VeA. It was discovered that PnVeA regulates growth, pigmentation and sporulation, as well as both effector expression and SM production either directly or indirectly. Uniquely, in P. nodorum, PnVeA is essential for sporulation. Furthermore, PnVeA is a negative regulator of alternariol production and is the first TF in P. nodorum observed to putatively regulate the NE gene SnTox267. Four uncharacterised SM gene clusters identified to be regulated by PnVeA were observed in this study. Efforts are underway to investigate SMs attributed to these canonical SM gene clusters. In addition, we were unable to find an enriched PnVeA binding motif in the differentially expressed genes using a previous method [40], likely due to indirect regulation of some differentially expressed genes. Future studies to include chromatin immunoprecipitation [46] could be used to uncover the binding motif and direct gene targets of PnVeA.
Materials and methods
Strains and cultures
P. nodorum strains used in this study are listed in Table 1. All fungal strains were maintained on V8PDA (150 mL/L Campbell’s V8 Juice, 3 g/L CaCO3, 10 g/L Difco PDA and 10 g/L Agar) [47]. Fungal strains were grown in a 12: 12 h light: dark cycle at 22 °C for 7 to 12 days.
Identification of Velvet orthologs in P. nodorum and phylogenetic tree construction
Velvet proteins VeA (NCBI accession: XP_658656.1), VelB (XP_657967.1), VelC (XP_659663.1) and VosA (XP_659563.1) from A. nidulans were used to search for the ortholog in P. nodorum reference strain SN15 [48] using the Basic Local Alignment Search Tool BlastP [49]. The probable orthologs identified were analysed using InterPro [50] to confirm the presence of the Velvet domains. Characterised velvet proteins from A. nidulans and three plant pathogenic fungi (A. flavus, B. cinerea and M. oryzae) were retrieved from UniProt [51] and selected to compare against the identified orthologs in P. nodorum [34,35,36,37]. An alignment of full length proteins was performed on MEGA11 using the ClustalW algorithm with default settings [52]. A neighbour-joining phylogenetic tree was constructed using default settings except with a bootstrap analysis set at 3000 repetitions [52].
Generation of PnVeA P. nodorum mutants
The primers used in this study are listed in Additional file 10. The gene deletion construct of PnVeA including a phleomycin selectable marker was assembled using Golden Gate cloning system [53] using a modified pUC19 (New England Biolabs, Ipswich, Massachusetts, USA) as the backbone vector [54]. The PnVeA complementation construct that included a 1,021 bp promoter region, the full intact gene and 290 bp of the terminator sequence was fused to a hygromycin selectable marker via fusion PCR [55].
The P. nodorum wild-type strain, SN15, was transformed via polyethylene glycol-mediated transformation as previously described [47]. PnVeA-deleted P. nodorum mutant strain, pnvea-25, was similarly transformed but the hyphae were used for the inoculation of the starter culture instead of spores due to its non-sporulating phenotype.
Deletion of the gene and subsequent complementation were confirmed using PCR (Additional file 1). Single copy integration of the constructs was confirmed using a previously developed quantitative PCR method (Additional file 1) [56].
Phenotypic assay
Agar plugs were cut out from the growing edge of each strain with the back of a 200 µL pipette tip and inoculated onto another V8PDA plate and left to grow under the conditions outlined before. Spores were then counted for each strain after 12 days.
Infection assay
Hyphae from each strain were homogenised in 0.02% Tween solution using metal beads and a ball mill shaken at 30 Hz for 10 s using a Mixer Mill MM400 (Retsch, Haan, Germany). The hyphal mixture was diluted to 20 mg/mL by wet weight in 0.02% Tween 20. This hyphal mixture was painted using a paintbrush on 10-day-old wheat seedlings (cv. Axe) embedded in 0.5% benzimidazole water agar and grown as a detached leaf assay under a 12: 12 h light: dark cycle at 22 °C [47]. After 10 days, the infected leaves were put into 500 µL of water and shaken at 30 Hz for 10 s to dislodge the pycnidiospores. The pycnidiospores were then counted and normalised to pycnidiospores per cm of infected leaf tissue.
The staining of infected leaves with Trypan Blue was as previously described [47].
RNA extraction, sequencing and quality control
SN15 and pnvea-25 were grown on V8PDA for 12 days and RNA extraction was performed using an extraction involving TRIzol™ reagent (Invitrogen, La Jolla, USA) and DNase treatment using DNase I (New England Biolabs, Ipswich, USA) both following manufacturers’ protocols. Library preparation and sequencing were performed by the Australian Genome Research Facility (https://www.agrf.org.au/). Following the library preparation, 150 bp paired-end stranded sequences were generated on a NovaSeq 6000 S4 (Illumina, San Diego, USA). The experiment was performed with three biological replicates. The quality of the raw RNA-Seq reads was assessed in FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The sequencing adapters were trimmed using Trimmomatic v0.39 [57] and the trimmed paired reads were mapped to the P. nodorum SN15 genome [48] with known splice sites using STAR v2.7.3 using default settings [58].
Determination and functional enrichment of differentially expressed genes in RNA-Seq
The counts of the reads overlapping the annotated gene features in the genome were determined using the featureCount v2.0.0 package in SubRead [59]. Differentially expressed (DE) genes were determined using the R package DESeq2 v1.36.0 [60]. The criterion for a DE gene was the LFC against the null hypothesis -1 ≤ LFC ≤ 1 (Ha = |LFC| > 1) with the Benjamini-Hoschberg adjusted P-value significance threshold of 0.05.
Gene ontology (GO) terms in DE genes were analysed in Goseq v1.26.0 to determine its overrepresentation [61]. GO terms were assigned using InterProScan where possible [62]. The predicted function of genes in the top five most dysregulated GO terms were added using predictions from UniProt [51]. SM cluster genes described by Chooi et al. [28] that were DE were analysed further on antiSmash 7 fungal version [63] with default settings using the SN15 genome [48] as the query.
Gene expression analysis of effector genes in in-planta condition
RNA from wheat leaves (cv. Axe) three-, six-, eight- and 10-days post inoculation with all P. nodorum strains in this study was extracted as described above. Reverse transcription PCR was performed on the sample to generate cDNA using iScript™ cDNA Synthesis Kit (BioRad, Hercules, USA) according to the manufacturer’s protocol. A QXDx AutoDG ddPCR system (BioRad) was used to perform ddPCR on the samples in biological triplicates. The expression of SnToxA, SnTox1, SnTox267 and SnTox3 was normalised to the expression of Act1 in the form of absolute quantification [18, 54].
Secondary metabolite assay
Three plates of each P. nodorum strains were grown on V8PDA were cut into approximately 1 cm x 1 cm pieces and immersed in 25 mL of methanol. This immersion was kept on ice and sonicated using a UW3100 sonicator with a VS70 tip (Bandelin, Berlin, Germany) for 2 min at 10 s: 10 s on: off bursts at 50% amplitude to disrupt the cells. This was then centrifuged at 3900 g for 5 min, the supernatant filtered through a 0.2 μm filter, and then methanol was evaporated off in a vacuum to retain the solid organic extracts. The crude extracts were re-dissolved in methanol for LC-DAD-MS analysis. Chromatographic separation was achieved with a linear gradient of 5–95% acetonitrile-H2O (0.1% (v/v) formic acid) over 10 min, followed by 95% acetonitrile for 3 min, with a flow rate of 0.6 mL/min. The MS data were collected in the m/z range of 100–1000. The experiments were performed using an Agilent 1260 liquid chromatography (LC) system, coupled to a diode array detector (DAD) and an Agilent 6130 Quadrupole mass spectrometer (MS) with an electrospray ionisation source. Chromatographic separation was carried out at 40 °C, employing a Kinetex C18 column (2.6 μm, 2.1 × 100 mm; Phenomenex).
Data availability
The raw RNA-Seq reads generated in this study are available in the National Center for Biotechnology Information Sequence Read Archive database under the BioProject ID: PRJNA1044467.
References
Savary S, Willocquet L, Pethybridge SJ, Esker P, McRoberts N, Nelson A. The global burden of pathogens and pests on major food crops. Nat Ecol Evol. 2019;3(3):430–9.
Quaedvlieg W, Verkley GJM, Shin HD, Barreto RW, Alfenas AC, Swart WJ, et al. Sizing up Septoria. Stud Mycol. 2013;75(1):307–90.
Solomon PS, Lowe RGT, Tan K-C, Waters ODC, Oliver RP. Stagonospora nodorum: cause of stagonospora nodorum blotch of wheat. Mol Plant Pathol. 2006;7(3):147–56.
Downie RC, Lin M, Corsi B, Ficke A, Lillemo M, Oliver RP, et al. Septoria nodorum blotch of wheat: disease management and resistance breeding in the face of shifting disease dynamics and a changing environment. Phytopathology®. 2020;111(6):906–20.
Oliver RP, Friesen TL, Faris JD, Solomon PS. Stagonospora nodorum: from pathology to genomics and host resistance. Annu Rev Phytopathol. 2012;50:23–43.
Faris JD, Friesen TL. Plant genes hijacked by necrotrophic fungal pathogens. Curr Opin Plant Biol. 2020;56:74–80.
Charoensawan V, Wilson D, Teichmann SA. Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res. 2010;38(21):7364–77.
Bayram Ö, Braus GH. Coordination of secondary metabolism and development in fungi: the velvet family of regulatory proteins. FEMS Microbiol Rev. 2012;36(1):1–24.
Calvo AM, Lohmar JM, Ibarra B, Satterlee T. 18 velvet regulation of Fungal Development. In: Wendland J, editor. Growth, differentiation and sexuality. Cham: Springer International Publishing; 2016. pp. 475–97.
Käfer E. Origins of translocations in aspergillus nidulans. Genetics. 1965;52(1):217–32.
Ahmed YL, Gerke J, Park H-S, Bayram Ö, Neumann P, Ni M, et al. The velvet family of fungal regulators contains a DNA-Binding domain structurally similar to NF-κB. PLoS Biol. 2014;11(12):e1001750.
John E, Singh KB, Oliver RP, Tan K-C. Transcription factor control of virulence in phytopathogenic fungi. Mol Plant Pathol. 2021;22(7):858–81.
Richards JK, Kariyawasam GK, Seneviratne S, Wyatt NA, Xu SS, Liu Z, et al. A triple threat: the Parastagonospora nodorum SnTox267 effector exploits three distinct host genetic factors to cause disease in wheat. New Phytol. 2022;233(1):427–42.
Tan K-C, Oliver RP. Regulation of proteinaceous effector expression in phytopathogenic fungi. PLoS Pathog. 2017;13(4):e1006241.
Chooi Y-H, Zhang G, Hu J, Muria-Gonzalez MJ, Tran PN, Pettitt A, et al. Functional genomics-guided discovery of a light-activated phytotoxin in the wheat pathogen parastagonospora nodorum via pathway activation. Environ Microbiol. 2017;19(5):1975–86.
Li H, Wei H, Hu J, Lacey E, Sobolev AN, Stubbs KA, et al. Genomics-driven discovery of phytotoxic cytochalasans involved in the virulence of the wheat pathogen parastagonospora nodorum. ACS Chem Biol. 2020;15(1):226–33.
Ipcho SV, Hane JK, Antoni EA, Ahren D, Henrissat B, Friesen TL, et al. Transcriptome analysis of Stagonospora nodorum: gene models, effectors, metabolism and pantothenate dispensability. Mol Plant Pathol. 2012;13(6):531–45.
Lin SY, Chooi YH, Solomon PS. The global regulator of pathogenesis PnCon7 positively regulates Tox3 effector gene expression through direct interaction in the wheat pathogen parastagonospora nodorum. Mol Microbiol. 2018.
Rybak K, See PT, Phan HT, Syme RA, Moffat CS, Oliver RP, et al. A functionally conserved Zn(2) cys(6) binuclear cluster transcription factor class regulates necrotrophic effector gene expression and host-specific virulence of two major Pleosporales fungal pathogens of wheat. Mol Plant Pathol. 2017;18(3):420–34.
Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, Ling H, Faris JD, et al. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet. 2006;38(8):953–6.
Friesen TL, Zhang Z, Solomon PS, Oliver RP, Faris JD. Characterization of the interaction of a novel Stagonospora nodorum host-selective toxin with a wheat susceptibility gene. Plant Physiol. 2008;146(2):682–93.
Liu ZH, Faris JD, Meinhardt SW, Ali S, Rasmussen JB, Friesen TL. Genetic and physical mapping of a gene conditioning sensitivity in wheat to a partially purified host-selective toxin produced by Stagonospora nodorum. Phytopathology. 2004;94(10):1056–60.
Kariyawasam GK, Richards JK, Wyatt NA, Running KLD, Xu SS, Liu Z, et al. The Parastagonospora nodorum necrotrophic effector SnTox5 targets the wheat gene Snn5 and facilitates entry into the leaf mesophyll. New Phytol. 2022;233(1):409–26.
Jones DAB, Rybak K, Bertazzoni S, Tan K-C, Phan HTT, Hane JK. Pathogenicity effector candidates and accessory genome revealed by pan-genomic analysis of Parastagonospora nodorum. bioRxiv. 2021:2021.09.01.458590.
IpCho SV, Tan KC, Koh G, Gummer J, Oliver RP, Trengove RD, et al. The transcription factor StuA regulates central carbon metabolism, mycotoxin production, and effector gene expression in the wheat pathogen Stagonospora nodorum. Eukaryot Cell. 2010;9(7):1100–8.
Duran RM, Cary JW, Calvo AM. Production of cyclopiazonic acid, aflatrem, and aflatoxin by aspergillus flavus is regulated by veA, a gene necessary for sclerotial formation. Appl Microbiol Biotechnol. 2007;73(5):1158–68.
Myung K, Li S, Butchko RAE, Busman M, Proctor RH, Abbas HK, et al. FvVE1 regulates biosynthesis of the mycotoxins fumonisins and fusarins in Fusarium verticillioides. J Agric Food Chem. 2009;57(11):5089–94.
Chooi YH, Muria-Gonzalez MJ, Solomon PS. A genome-wide survey of the secondary metabolite biosynthesis genes in the wheat pathogen parastagonospora nodorum. Mycology. 2014;5(3):192–206.
Piras M, Patruno I, Nikolakopoulou C, Willment JA, Sloan NL, Zanato C, et al. Synthesis of the fungal metabolite YWA1 and related constructs as tools to study MelLec-mediated immune response to aspergillus infections. J Org Chem. 2021;86(9):6044–55.
Awakawa T, Yang X-L, Wakimoto T, Abe I, Pyranonigrin E. A PKS-NRPS hybrid metabolite from Aspergillus Niger identified by genome mining. ChemBioChem. 2013;14(16):2095–9.
Morishita Y, Aoki Y, Ito M, Hagiwara D, Torimaru K, Morita D, et al. Genome mining-based discovery of fungal macrolides modified by glycosylphosphatidylinositol (GPI)–ethanolamine phosphate transferase homologues. Org Lett. 2020;22(15):5876–9.
Fulton TR, Ibrahim N, Losada MC, Grzegorski D, Tkacz JS. A melanin polyketide synthase (PKS) gene from Nodulisporium sp. that shows homology to the pks1 gene of Colletotrichum lagenarium. Mol Gen Genet MGG. 1999;262(4):714–20.
Chooi Y-H, Muria-Gonzalez Mariano J, Mead Oliver L, Solomon Peter S. SnPKS19 encodes the polyketide synthase for alternariol mycotoxin biosynthesis in the wheat pathogen parastagonospora nodorum. Appl Environ Microbiol. 2015;81(16):5309–17.
Eom TJ, Moon H, Yu JH, Park HS. Characterization of the velvet regulators in aspergillus flavus. J Microbiol. 2018;56(12):893–901.
Kim H-J, Han J-H, Kim KS, Lee Y-H. Comparative functional analysis of the velvet gene family reveals unique roles in fungal development and pathogenicity in Magnaporthe oryzae. Fungal Genet Biol. 2014;66:33–43.
Müller N, Leroch M, Schumacher J, Zimmer D, Könnel A, Klug K, et al. Investigations on VELVET regulatory mutants confirm the role of host tissue acidification and secretion of proteins in the pathogenesis of Botrytis Cinerea. New Phytol. 2018;219(3):1062–74.
Schumacher J, Pradier J-M, Simon A, Traeger S, Moraga J, Collado IG, et al. Natural variation in the VELVET gene bcvel1 affects virulence and light-dependent differentiation in Botrytis Cinerea. PLoS ONE. 2012;7(10):e47840.
Cary JW, Han Z, Yin Y, Lohmar JM, Shantappa S, Harris-Coward PY, et al. Transcriptome analysis of aspergillus flavus reveals vea-dependent regulation of secondary metabolite gene clusters, including the novel aflavarin cluster. Eukaryot Cell. 2015;14(10):983–97.
Solomon PS, Tan K-C, Sanchez P, Cooper RM, Oliver RP. The disruption of a Gα subunit sheds new light on the pathogenicity of Stagonospora nodorum on wheat. Mol Plant-Microbe Interactions®. 2004;17(5):456–66.
Jones DAB, John E, Rybak K, Phan HTT, Singh KB, Lin S-Y, et al. A specific fungal transcription factor controls effector gene expression and orchestrates the establishment of the necrotrophic pathogen lifestyle on wheat. Sci Rep. 2019;9(1):15884.
Tan KC, Phan HT, Rybak K, John E, Chooi YH, Solomon PS, et al. Functional redundancy of necrotrophic effectors - consequences for exploitation for breeding. Front Plant Sci. 2015;6:501.
Ostry V. Alternaria mycotoxins: an overview of chemical characterization, producers, toxicity, analysis and occurrence in foodstuffs. World Mycotoxin J. 2008;1(2):175–88.
Tan K-C, Trengove RD, Maker GL, Oliver RP, Solomon PS. Metabolite profiling identifies the mycotoxin alternariol in the pathogen Stagonospora nodorum. Metabolomics. 2009;5(3):330–5.
Wang L, Wang M, Jiao J, Liu H. Roles of AaVeA on mycotoxin production via light in Alternaria alternata. Front Microbiol. 2022;13.
Yee DA, Niwa K, Perlatti B, Chen M, Li Y, Tang Y. Genome mining for unknown-unknown natural products. Nat Chem Biol. 2023;19(5):633–40.
Park PJ. ChIP–Seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–80.
Solomon PS, Lee RC, Wilson TJ, Oliver RP. Pathogenicity of Stagonospora nodorum requires malate synthase. Mol Microbiol. 2004;53(4):1065–73.
Bertazzoni S, Jones DAB, Phan HT, Tan KC, Hane JK. Chromosome-level genome assembly and manually-curated proteome of model necrotroph parastagonospora nodorum Sn15 reveals a genome-wide trove of candidate effector homologs, and redundancy of virulence-related functions within an accessory chromosome. BMC Genomics. 2021;22(1):382.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402.
Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar Gustavo A, et al. InterPro in 2022. Nucleic Acids Res. 2022;51(D1):D418–27.
Consortium TU. UniProt: the Universal protein knowledgebase in 2023. Nucleic Acids Res. 2022;51(D1):D523–31.
Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol Biol Evol. 2021;38(7):3022–7.
Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE. 2008;3(11):e3647.
John E, Jacques S, Phan HTT, Liu L, Pereira D, Croll D, et al. Variability in an effector gene promoter of a necrotrophic fungal pathogen dictates epistasis and effector-triggered susceptibility in wheat. PLoS Pathog. 2022;18(1):e1010149.
Hilgarth RS, Lanigan TM. Optimization of overlap extension PCR for efficient transgene construction. MethodsX. 2020;7:100759.
Solomon PS, Ipcho SVS, Hane JK, Tan KC, Oliver RP. A quantitative PCR approach to determine gene copy number. Fungal Genet Rep. 2008;55:5–8.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-Seq aligner. Bioinformatics. 2013;29(1):15–21.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014;15(12):550.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-Seq: accounting for selection bias. Genome Biol. 2010;11(2):R14.
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33(Web Server issue):W116–20.
Blin K, Shaw S, Augustijn HE, Reitz ZL, Biermann F, Alanjary M, et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023;51(W1):W46–50.
Acknowledgements
We would like to acknowledge Dr Huyen T. T. Phan for providing the SnTox267 qPCR primers.
Funding
This study was supported by the Centre for Crop and Disease Management, a joint initiative of Curtin University and the Grains Research and Development Corporation under the research grant CUR00023 Project F3.4 awarded to KCT. SM was supported by an Australian Government Research Training Program Scholarship.
Author information
Authors and Affiliations
Contributions
KCT and EJ conceptualised the project. KCT acquired funding. SM, DJ and NS carried out experimental investigations. KCT, SM, CV, EJ, YCH and NS analysed the data. KCT, CV, LL, CT and EJ provided project supervision. SM, EJ, CV and KCT wrote the original manuscript draft. All authors reviewed and edited the manuscript draft.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Morikawa, S., Verdonk, C., John, E. et al. The Velvet transcription factor PnVeA regulates necrotrophic effectors and secondary metabolism in the wheat pathogen Parastagonospora nodorum. BMC Microbiol 24, 299 (2024). https://doi.org/10.1186/s12866-024-03454-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-024-03454-7