
Abstract
Background
Carbamoyl phosphate synthetase 1 (CPS1) deficiency (OMIM 237300), an autosomal recessive rare and severe urea cycle disorder, is associated with hyperammonemia and high mortality.
Methods
Herein we present 12 genetic variants identified in seven clinically well-characterized Chinese patients with CPS1 deficiency who were admitted to the Children’s Medical Center of Peking University First Hospital from September 2014 to August 2023.
Results
Seven patients (two male and five female patients including two sisters) experienced symptoms onset between 2 days and 13 years of age, and they were diagnosed with CPS1 deficiency between 2 months and 20 years. Peak blood ammonia levels ranged from 160 to 1,000 µmol/L. Three patients showed early-onset CPS1 deficiency, with only one surviving after treatment with sodium phenylbutyrate, N-carbamoyl-L-glutamate, and liver transplantation at 4 months, showing a favorable outcome. The remaining four patients had late-onset CPS1 deficiency, presenting with mental retardation, psychiatric symptoms, and self-selected low-protein diets. Among the 12 CPS1 variants identified in these patients, 10 were novel, with all patients exhibiting compound heterozygosity for CPS1 mutant alleles. Seven variants (c.149T > C, c.616 A > T, c.1145 C > T, c.1294G > A, c.3029 C > T, c.3503 A > T, and c.3793 C > T) resulted in single amino acid substitutions. Three frameshift variations (c.2493del, c.3067dup, and c.3241del) were identified, leading to enzyme truncation. One mutation (c.3506_3508del) caused an in-frame single amino acid deletion, while another (c.2895 + 2T > C) resulted in aberrant splicing.
Conclusions
Except for two known variants, all other variants were identified as novel. No hotspot variants were observed among the patients. Our data contribute to expanding the mutation spectrum of CPS1.
Similar content being viewed by others


Background
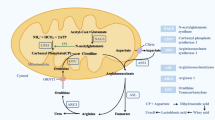
Carbamoyl phosphate synthetase 1 (CPS1) deficiency (OMIM 237300), an autosomal recessive inherited disease, is a rare and severe type of urea cycle disorder characterized by hyperammonemia [1,2,3]. CPS1 acts as the rate-limiting enzyme initiating ammonia detoxification in the urea cycle by synthesizing carbamoyl phosphate from bicarbonate, ammonia, and adenosine triphosphate with N-acetylglutamate. Mutations in CPS1 lead to enzyme activity deficiency, severely impeding the conversion of ammonia to carbamoyl phosphate. This results in hyperammonemic encephalopathy and liver damage, evident through symptoms such as vomiting, poor feeding, loss of consciousness, seizures, and rapid clinical deterioration, often leading to high mortality [4, 5]. The symptoms of CPS1 deficiency can vary widely, manifesting at any age. Typically, phenotypes are categorized into neonatal-onset and late-onset forms based on the age of onset [4]. Neonatal-onset forms are associated with a poorer prognosis and higher mortality rates compared to late-onset forms, which demonstrate a survival rate of > 90% [6]. The incidence of CPS1 deficiency is 1/62,000 to 1/1,300,000 worldwide [7, 8]. Only a few cases of CPS1 deficiency have been reported in China [9,10,11].
The severity of CPS1 deficiency, whether early- or late-onset, is influenced by residual enzyme activity related to the defect and the specific variant site and type. However, there has been limited analysis regarding the correlation between genotypes and phenotypes in patients with CPS1 deficiency. Our study aims to investigate the clinical features, metabolic profiles, and genotypes of seven cases of CPS1 deficiency, alongside discussing the potential relationship between genotypes and phenotypes.
Methods
Patients
Our study was approved by the Hospital Institution Committee in accordance with the Declaration of Helsinki. Written informed consent was obtained from the parents of each participant. Seven patients with CPS1 deficiency were from six non-consanguineous Chinese families. They were admitted to Peking University First Hospital between September 2014 and August 2023.
Routine examination
Clinical and laboratory data at onset and follow-up were collected. Data from routine physical examination and complete blood count; blood ammonia, glucose, lactic acid, and blood gas; liver and kidney function; and brain magnetic resonance imaging (MRI) were analyzed.
Blood amino acid and acylcarnitine profiles in dried blood spots were analyzed by liquid chromatography–tandem mass spectrometry (API 3200, Triple Quad 4500; Applied Biosystems, CA, USA). ChemoView software was used to automatically calculate metabolite concentration. Urine organic acids were determined using gas chromatography-mass spectrometry (GC-MS, GCMS-QP2010plus; Shimadzu, Kyoto, Japan).
Genetic testing
Genomic DNA from peripheral blood samples was extracted and purified. Whole exon sequencing was conducted for seven patients and their parents. The DNA was sequenced on Illumina Hiseq2500 (Illumina, San Diego, USA). Each variant was compared with 1000 Genomics, the ExAC database (http://exac.broadinstitute.org/), and the gnomAD database (http://gnomad.broadinstitute.org/). Purified DNA samples were sent to Euler Genomics (Beijing, China), Berry Genomics Corporation (Beijing, China), and Running Gene Inc. (Beijing, China) for whole exon sequencing to screen variants in patients. Mutation Taster, PolyPhen-2, and SIFT were also referred to for in silico analysis of the variants to predict pathogenicity. The variants were evaluated according to the HGMD and Clinvar. The pathogenicity of the selected variants was assessed according to the American College of Medical Genetics and Genomics guidelines [12]. The variants were classified as pathogenic, likely pathogenic, variants of uncertain significance (VUS), likely benign, or benign.
Results
Clinical and biochemical features
Seven patients were included in this study, including two (28.6%) male and five (71.4%) female patients (Table 1). The age of onset ranged from 2 days to 13 years, and the patients were diagnosed at the age of 2 months to 20 years. Among the patients, three were diagnosed by postmortem examination.
Three patients (Patients 1 to 3) had neonatal-onset type. They exhibited reluctance to feed, convulsions, and loss of consciousness after breast milk feeding on days 2, 3, and 10 of life, respectively.
Patient 1 was treated with 250 mg/kg/day oral sodium phenylbutyrate and 130 mg/kg/day N-carbamoyl-L-glutamate intermittently upon the onset of symptoms, and a strict low-protein diet was started after diagnosis. Her condition stabilized with no vomiting; blood ammonia levels reduced from 514 µmol/L before treatment to 60 µmol/L after treatment, but anorexia and feeding difficulty did not improve. The physical growth and movement development were retarded. The infant underwent a liver transplant from her father at the age of 4 months. Subsequently, her blood ammonia levels returned to the normal range. She was fed with ordinary formula, and her general condition significantly improved. Her physical and psychomotor development gradually reached those of healthy infants. The girl, now 3 years old, shows normal physical and mental development.
Patient 2 experienced poor feeding, convulsions, and coma on the third day of life. His blood ammonia level was > 1,000 µmol/L. Patient 3 showed similar symptoms on day 10 of life, with a blood ammonia level of 489 µmol/L. Both patients underwent emergency management, including hemodialysis and a low-protein diet. Despite these interventions, they showed rapid deterioration and severe hyperammonemia and died on days 5 and 16 of life, respectively. Their blood and urine samples were taken for postmortem metabolic and gene analysis.
Four patients (Patients 4 to 7) were late-onset type with various non-specific symptoms. Patients 4 and 5 presented symptoms at the age of 2 and 4 months, respectively. Patients 6 and 7 presented with symptoms aged 3 years and 13 years, respectively. They had diverse degrees of mental retardation and psychotic behavioral disorders, such as apathy and short temper, and avoided high-protein foods. Patient 4 presented with non-specific symptoms, including crying, irritability, and developmental delay since 2 months of age, with a high blood ammonia level of 168 µmol/L. Patient 5 showed developmental delay since 4 months of age and later developed neuropsychiatric symptoms during puberty. She was diagnosed at the age of 20 years at our hospital. Patient 6 had recurrent severe symptoms such as vomiting, seizures, loss of consciousness, and coma following acute infections starting at 3 years of age. The definite diagnosis of CPS1 deficiency was made at the age of 6 years before death. Patient 5 and Patient 7 were sisters. Patient 7 had anorexia, altered temperament, and other neuropsychiatric disorders such as hallucinations since puberty. She experienced headaches, vomiting, seizures, coma after fever, and acute upper respiratory tract infection at the age of 26 years and died after 5 days. Patient 7 was diagnosed with CPS1 deficiency by postmortem gene analysis. All four patients received treatment involving intravenous and oral ammonia scavengers, including 250 mg/kg/day sodium phenylbutyrate, 250–300 mg/kg/day sodium benzoate, 300 mg/kg/day arginine, and 50–200 mg/kg/day levocarnitine.
The blood ammonia of the seven patients was elevated to 160 to 1,000 µmol/L. Their blood citrulline levels were either lower than or within the lower end of the normal range (2.3–6.5 µmol/L, normal range 5–40 µmol/L). Blood arginine levels were also lower than or within the lower end of the normal range (1.6–8.3 µmol/L, normal range 5–50 µmol/L). Urine organic acid analysis revealed normal orotic acid and uracil levels. All patients showed varied liver dysfunction.
Patients 1 and 6 underwent brain magnetic resonance imaging during the acute episode. Patient 1 showed short T1 and long T2 abnormal signals on both globus pallidus sites, in accordance with hyperammonemic encephalopathy (Fig. 1a and b). Patient 6 showed slight bilateral ventriculomegaly and an enlarged subarachnoid space in the temporal lobe.

a Brain magnetic resonance imaging (MRI) of Patient 1 at 10 days of age upon initial acceptance into our study before treatment. Brain MRI was performed at 3.0T at Peking University First Hospital. The axial T1 image revealed symmetrical diffuse high-signal shadows on both globus pallidus sites (indicated by the red arrow). b Brain MRI of Patient 1 at 10 days of age. The axial T2 image revealed symmetrical diffuse high-signal shadows on both globus pallidus sites (indicated by the red arrow)
Four patients (Patient 2, 3, 6, and 7) presented with severe hyperammonemic encephalopathy after acute infections or breast milk intake. The patients’ clinical conditions exacerbated rapidly, and all four patients died several days after symptom onset. Their blood ammonia level stabilized between 64 and 80 µmol/L with oral sodium phenylbutyrate and a protein-restricted diet. However, the psychological and behavioral abnormalities and mental retardation did not improve.
Gene analysis
Twelve CPS1 variants were identified (NM_001875.4) from the seven patients included in this study (Table 2). Only two mutations, c.1145 C > T (p.Pro382Leu) [13] and c.3029 C > T (p.Thr1010Met) [14], have been previously reported to cause CPS1 deficiency. The remaining 10 variants were novel. We assessed the pathogenicity of these novel variants using various prediction online tools, including Mutation Taster [15], PolyPhen-2 [16], and SIFT [17], and interpreted them according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology [12]. The five novel missense variations, namely c.149T > C, c.616 A > T, c.1294G > A, c.3503 A > T, and c.3793 C > T, were all located in the highly conserved regions of the translated protein. Three frameshift variations (c.2493del, c.3067dup, and c.3241del), one deletion (c.3506_3508del), and one splicing variation (c.2895 + 2T > C) were detected. Four (c.2493del, c.3067dup, c.3241del, and c.3506_3508del) were pathogenic variations. Three (c.149T > C, c.3793 C > T, and c.2895 + 2T > C) were likely pathogenic. Four (c.616 A > T, c.1294G > A, c.3029 C > T, and c.3503 A > T) were VUS. The data have been uploaded to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar, submission ID SUB12523111, ClinVar accession nos.: SCV003762244 to SCV003762254).
Genotype–phenotype correlations
Three of the 7 patients (Patients 1–3) had neonatal-onset type, and they carried three missense variants c.149T > C, c.3503 A > T and c.3793 C > t respectively. Most of the variants in our study were located in exon 25 (3/14) and exon 29 (2/14) and were in the carbamate phosphorylation domain (6/14). Notably, many of the variants in our study showed substitutions of Leu (2/14), Thr (2/14) and Pro (2/14) residues.
Discussion
Herein we provide an account of the clinical presentations, biochemical features, genetic analysis, and outcomes of seven Chinese patients with CPS1 deficiency. Given the scarcity of research on this topic, our findings hold significant value for both clinicians and physicians dealing with such cases.
Our study included seven patients who exhibited a range of manifestations of CPS1 deficiency, spanning from the neonatal period to puberty. Among them, three were categorized as early-onset and four as late-onset type, each presenting with varied clinical symptoms. High-protein diets and acute infections emerged as the main triggers for onset in these patients. Notably, their blood ammonia levels were elevated, and liver dysfunction was evident. Four patients died, while three showed improvement following metabolic treatment comprising a low-protein diet, ammonia scavengers, and symptomatic drugs. One patient underwent N-carbamoyl-L-glutamate therapy and liver transplantation as part of their treatment regimen.
Patients with CPS1 deficiency usually present with nonspecific clinical manifestations in childhood. CPS1 deficiency can be divided into two types depending on the age of onset: the lethal early-onset (onset during the first month after birth) and the less severe late-onset (onset after the neonatal period) types [18, 19]. In this study, three cases (42.9%) were identified as having early-onset CPS1 deficiency. Only one infant recovered, while two died during the neonatal period. The results indicate that the prognosis of the early-onset type is extremely poor. Four cases (57.1%) were late-onset type, presenting symptoms from the age of 2 months to 13 years. The heterogeneity of the clinical manifestations was more significant, including developmental delay, mental and behavioral abnormalities, and anorexia. Among these four patients with late-onset CPS1 deficiency, two developed acute hyperammonemia and encephalopathy after infection. The disease progressed rapidly, and the patients died. These cases suggest that we should pay high attention to hyperammonemia and the underlying etiological disease. For late-onset types, despite stable clinical conditions, there remained a high risk of life-threatening metabolic crisis triggered by infections or high-protein diets.
The primary methods for detecting CPS1 deficiency include blood ammonia tests and gene analyses. No specific abnormalities are typically observed in blood amino acid and urine organic acid profiles. Notably, urinary orotic acid test results are negative, which helps distinguish CPS1 deficiency from other urea cycle disorders [20]. The definite diagnosis depends on detection of CPS1 pathogenic variants or liver CPS1 enzyme activity assay [1, 21]. In our study, blood ammonia levels of all patients were significantly elevated. The peak level of blood ammonia of Patient 2 was up to 1,000 µmol/L on Day 3, and the patient died on Day 5. Blood citrulline and arginine levels in all seven patients were below or within the lower limits of the normal range; however, glutamic acid levels in three patients were elevated, aligning with findings in existing literature [22]. These results suggested a definite diagnosis of CPS1 deficiency.
CPS1 is located on chromosome 2q35, contains 38 exons and 37 introns, and encodes the CPS1 protein of 1,462 amino acids [23, 24] (Fig. 2). More than 270 mutations have been reported on Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php). Up to now, no hot-spot variations of CPS1 has been reported. In this study, 12 CPS1 variations were detected among seven patients, of which only two variations have been previously reported [12, 13]. The other ten are novel. In addition to five missense variations, three were frameshift variations, one was a deletion, and one was a splicing variation. According to the ACMG pathogenicity grading, four were pathogenic, three were likely pathogenic, and three were VUS. The novel 10 variants further enrich the CPS1 pathogenic gene variation database.

Exon and domain structure of CPS1, along with the localization of CPS1 variants from this study. Human CPS1 comprises 1,462 amino acids. The blue box represents exons, connected by dark blue lines, which represent introns. Colored blocks denote specific protein domains as indicated. Dashed lines connect reported disease-causing variants with their respective exons and protein domains
The existing literature does not reveal any obvious genotype–phenotype correlations among patients with CPS1 deficiency. As we did not perform enzyme activity analysis in this study, we attempted to analyze genotype–phenotype correlations based on the clinical manifestations of our patients. Most variants identified in this study were located in the CPS1 domain (6/14, 42.9%), indicating that the CPS1 domain plays a vital role in enzyme activity and clinical presentation.
Notably, five of the novel mutations were missense changes. Among them, c.149T > C and c.616 A > T were observed in the N-terminal domain. This domain is hypothesized to activate catalytic activity to stabilize the enzyme [13]. c.149T > C was found in very severe neonatal patients, while c.616 A > T was found in late-onset patients, indicating that the variants in the same domain influence residual enzyme activity differently in patients. c.2493delA introduces a stop codon in the integrating domain, which is vital for proper enzyme folding and regulatory cross-talk between NAG and phosphorylation sites [25]. This may explain the neonatal type observed in Patient 1 in this study. The variant c.3029 C > T, on the other hand, in late-onset patients, may account for attenuated clinical manifestations and partial residual enzyme activity. In the third severe neonatal patient, c.3503 A > T was found to coexist with a second allele where a conserved valine residue is deleted (c.3506_3508del), resulting in severe effects from these two single amino acid variations.
On the opposite side of the clinical spectrum, data pertaining to late-onset patients also shed light on the lack of severity for the novel missense mutation c.1294G > A, as it coexists with a null allele in two sisters with late-onset CPS1 deficiency (Patients 5 and 7). In addition, the frequently reported c.1145 C > T variant was found in a late-onset patient (Patient 6), further supporting that this missense mutation does not completely abolish CPS1 activity.
At present, promptly reducing blood ammonia production is key to the treatment of CPS1 deficiency, as patient prognosis is strongly influenced by peak blood ammonia levels. Commonly used methods for managing this condition include the administration of phenylbutyric acid, benzoate, L-arginine, and L-citrulline, as well as liver transplantation [26, 27]. L-carnitine was also supplied to avoid secondary carnitine deficiency. N-carbamoyl-L-glutamate is an analog of N-acetylglutamic acid, which can activate CPS1 enzyme activity and promote the synthesis of carbamoyl phosphate. The drug is approved for the treatment of N-acetylglutamic acid synthetase deficiency. Earlier studies [28,29,30] have reported good outcomes on treating patients with CPS1 deficiency with N-carbamoyl glutamic acid. In our study, the clinical symptoms of Patient 1 were significantly relieved after N-carbamoyl glutamic acid was administered with increased tolerance to natural protein, and the blood ammonia stabilized, which allowed for liver transplantation. The child underwent paternal liver transplantation at 4 months of age and became the youngest child with CPS1 deficiency in the world to have successfully undergone liver transplantation.
Conclusions
In conclusion, CPS1 deficiency represents a serious form of urea cycle disorder, with a low incidence, high mortality and disability, and poor prognosis. This condition manifests as devastating acute attacks with severe consequences impacting multiple organs. Analyzing the genotype–phenotype relationship revealed that the variants c.149T > C, c.3503 A > T, c.3793 C > T, and c.3506_3508del markedly reduce CPS1 protein activity, while c.1294G > A and c.1145 C > T cause partial deficiency. Notably, no hotspot variants were identified among the patients in our study. These results further expand the mutation spectrum of CPS1.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the ClinVar repository(https://www.ncbi.nlm.nih.gov/clinvar). Submission ID is SUB12523111, and the ClinVar accession numbers are SCV003762244 to SCV003762254.
References
Nitzahn M, Lipshutz GS. CPS1: looking at an ancient enzyme in a modern light. Mol Genet Metab. 2020;131:289–98. https://doi.org/10.1016/j.ymgme.2020.10.003.
Lopes FF, Sitta A, de Moura Coelho D, Ribas GS, Faverzani JL, Dos Reis BG, et al. Clinical findings of patients with hyperammonemia affected by urea cycle disorders with hepatic encephalopathy. Int J Dev Neurosci. 2022;82:772–88. https://doi.org/10.1002/jdn.10229.
Ribas GS, Lopes FF, Deon M, Vargas CR. Hyperammonemia in inherited metabolic diseases. Cell Mol Neurobiol. 2022;42:2593–610. https://doi.org/10.1007/s10571-021-01156-6.
Isler J, Rüfenacht V, Gemperle C, Allegri G, Haberle J. Improvement of diagnostic yield in carbamoylphosphate synthetase 1 (CPS1) molecular genetic investigation by RNA sequencing. JIMD Rep. 2020;52:28–34. https://doi.org/10.1002/jmd2.12091.
Nettesheim S, Kolker S, Karall D, Haberle J, Posset R, Hoffmann GF, et al. Incidence, disease onset and short-term outcome in urea cycle disorders -cross-border surveillance in Germany, Austria and Switzerland. Orphanet J Rare Dis. 2017;12:111–8. https://doi.org/10.1186/s13023-017-0661-x.
Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. 2007;356:2282–92. https://doi.org/10.1056/NEJMoa066596.
Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110:179–80. https://doi.org/10.1016/j.ymgme.2013.07.008.
Martinez AI, Perez-Arellano I, Pekkala S, Barcelona B, Cervera J. Genetic, structural and biochemical basis of carbamoyl phosphate synthetase 1 deficiency. Mol Genet Metab. 2010;101(4):311–23. https://doi.org/10.1016/j.ymgme.2010.08.002.
Zhang G, Chen Y, Ju H, Li X, Liu Y, Bei F, Li J, Wang J, et al. Carbamoyl phosphate synthetase 1 deficiency diagnosed by whole exome sequencing. J Clin Lab Anal. 2018;32: e22241. https://doi.org/10.1002/jcla.22241.
Chen X, Yuan L, Sun M, Liu Q, Wu Y. Two novel CPS1 mutations in a case of carbamoyl phosphate synthetase 1 deficiency causing hyperammonemia and leukodystrophy. J Clin Lab Anal. 2018;32: e22375. https://doi.org/10.1002/jcla.22375.
Bai R, He A, Guo J, Li Z, Yu X, Zeng J, et al. Novel pathogenic variant (c.2947C > T) of the carbamoyl phosphate synthetase 1 gene in neonatal-onset deficiency. Front Neurosci. 2022;16:1025572. https://doi.org/10.3389/fnins.2022.1025572.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.
Fan L, Zhao J, Jiang L, Xie L, Ma J, Li X, et al. Molecular, biochemical, and clinical analyses of five patients with carbamoyl phosphate synthetase 1 deficiency. J Clin Lab Anal. 2020;34: e23124. https://doi.org/10.1002/jcla.23124.
Rodriguez-Flores JL, Fakhro K, Hackett NR, Salit J, Fuller J, Agosto-Perez F, et al. Exome sequencing identifies potential risk variants for mendelian disorders at high prevalence in Qatar. Hum Mutat. 2014;35:105–16. https://doi.org/10.1002/humu.22460.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6. https://doi.org/10.1038/nmeth0810-575.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. https://doi.org/10.1038/nmeth0410-248.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. https://doi.org/10.1038/nprot.2009.86.
Imataka G, Ishii J, Ando Y, Yoshihara S, Takagi Y, Nitta A, Arisaka O, Yoshihara S. Long-term survival of a patient with acute neonatal-onset metabolic encephalopathy with carbamoyl phosphate synthetase 1 deficiency. Eur Rev Med Pharmacol Sci. 2020;24(19):10051–3. https://doi.org/10.26355/eurrev_202010_23220.
Choi Y, Oh A, Lee Y, Kim GH, Choi JH, Yoo HW, et al. Unfavorable clinical outcomes in patients with carbamoyl phosphate synthetase 1 deficiency. Clin Chim Acta. 2022;526:55–61. https://doi.org/10.1016/j.cca.2021.11.029.
Matsumoto S, Haberle J, Kido J, Mitsubuchi H, Endo F, Nakamura K. Urea cycle disorders-update. J Hum Genet. 2019;64:833–47. https://doi.org/10.1038/s10038-019-0614-4.
Haberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42:1192–230. https://doi.org/10.1002/jimd.12100.
Lin HT, Enchautegui-Colon Y, Huang YR, Zimmerman C, DeMarzo D, Tsai AC. Novel compound heterozygote variants: c.4193_4206delinsG (p.Leu1398Argfs*25), c.793C > A (p.Pro265Thr), in the CPS1 gene (NM_001875.4) causing late onset carbamoyl phosphate synthetase 1 deficiency-lessons learned. Mol Genet Metab Rep. 2022;33:100942. https://doi.org/10.1016/j.ymgmr.2022.100942.
Ali EZ, Khalid MK, Yunus ZM, Yakob Y, Chin CB, Abd Latif K, et al. Carbamoylphosphate synthetase 1 (CPS1) deficiency: clinical, biochemical, and molecular characterization in Malaysian patients. Eur J Pediatr. 2016;175(3):339–46.
Yan B, Wang C, Zhang K, Zhang H, Gao M, Lv Y, et al. Novel neonatal variants of the carbamoyl phosphate synthetase 1 deficiency: two case reports and review of literature. Front Genet. 2019;10:718–28. https://doi.org/10.3389/fgene.2019.00718.
Diez-Fernandez C, Hu L, Cervera J, Haberle J, Rubio V. Understanding carbamoyl phosphate synthetase (CPS1) deficiency by using the recombinantly purified human enzyme: effects of CPS1 mutations that concentrate in a central domain of unknown function. Mol Genet Metab. 2014;112(2):123–32. https://doi.org/10.1016/j.ymgme.2014.04.003.
Kido J, Matsumoto S, Momosaki K, Sakamoto R, Mitsubuchi H, Endo F, et al. Liver transplantation may prevent neurodevelopmental deterioration in high-risk patients with urea cycle disorders. Pediatr Transpl. 2017;21: e12987. https://doi.org/10.1111/petr.12987.
Kido J, Nakamura K, Mitsubuchi H, Ohura T, Takayanagi M, Matsuo M, et al. Long-term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis. 2012;35:777–85. https://doi.org/10.1007/s10545-011-9427-0.
Yap S, Gougeard N, Hart AR, Barcelona B, Rubio V. N-carbamoylglutamate-responsive carbamoyl phosphate synthetase 1 (CPS1) deficiency: a patient with a novel CPS1 mutation and an experimental study on the mutation’s effects. JIMD Rep. 2019;48:36–44. https://doi.org/10.1002/jmd2.12034.
Sugiyama Y, Shimura M, Ogawa-Tominaga M, Ebihara T, Kinouchi Y, Isozaki K, et al. Therapeutic effect of N-carbamylglutamate in CPS1 deficiency. Mol Genet Metab Rep. 2020;24:100622. https://doi.org/10.1016/j.ymgmr.2020.100622.
Gragnaniello V, Gueraldi D, Puma A, Commone A, Loro C, Cazzorla C, et al. Variant in the allosteric domain of CPS1 protein associated with effectiveness of N-carbamoyl glutamate therapy in neonatal onset CPS1 deficiency. J Pediatr Endocrinol Metab. 2023;36(9):873–8.
Acknowledgements
We would like to thank all the patients and their families for their participation. We are grateful to the Euler Genomics (Beijing, China), Berry Genomics Corporation (Beijing, China), and Running Gene Inc. (Beijing, China) for their professional genetic sequencing and analysis services.
Funding
This work was supported by the National Key Research and Development Program of China [grant numbers 2021YFC2700903, 2022YFC2703401].
Author information
Authors and Affiliations
Contributions
HD, TS, and XM wrote the original draft of the manuscript. HD and YY participated in the study design and study conception. YZ, XM, HZ, DD, ZC, LS, and ZZ collected the clinical data and followed up with the patients. JS, YJ, and ML performed the metabolic assays. YZ and YY designed the study and supervised the clinical work. All the authors read and approved of the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was reviewed and approved by the Institutional Ethics Committee of Peking University First Hospital and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from the guardians of all patients in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, H., Sang, T., Ma, X. et al. Clinical features and CPS1 variants in Chinese patients with carbamoyl phosphate synthetase 1 deficiency. BMC Pediatr 24, 539 (2024). https://doi.org/10.1186/s12887-024-05005-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-05005-5