
Abstract
Research in the last decade has shown that hematopoietic stem cells (HSCs) interact with and are modulated by a complex multicellular microenvironment in the bone marrow, which includes both the HSC progeny and multiple non-hematopoietic cell types. Intense work is gradually throwing light on the composition of the HSC niche and the molecular cues exchanged between its components, which has implications for HSC production, maintenance and expansion. In addition, it has become apparent that bidirectional interactions between leukemic cells and their niche play a previously unrecognized role in the initiation and development of hematological malignancies. Consequently, targeting of the malignant niche holds considerable promise for more specific antileukemic therapies. Here we summarize the latest insights into HSC niche biology and recent work showing multiple connections between hematological malignancy and alterations in the bone marrow microenvironment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The concept of stem cell niche, in the context of the hematopoietic system, implies that the behavior of hematopoietic stem cells (HSCs) is to a great extent modulated by their microenvironment in the bone marrow. Underlying the interest in the study of the HSC niche are two practical hypotheses: (1) precise characterization of the cell populations and factors produced by them, responsible for HSC maintenance in vivo, holds the clue for HSC expansion ex vivo; (2) knowledge of how the niche is modified in hematological diseases (such as dysplasias and leukemias) might allow selective therapeutic targeting that eliminates abnormal HSCs while restoring normal hematopoiesis. However, the complex cellular composition of the BM cavity, the multiple anatomical and functional interactions between its components—not yet fully characterized—and its apparent lack of physical compartmentalization into discrete, organized structures analogous to stem cell niches described in other tissues have complicated the cellular and molecular dissection of the HSC niche. As a consequence, significant progress in the two above mentioned directions has only begun to occur in very recent years. In this review, we discuss the most recent insights into the biology of HSCs and their niche, some of the technical difficulties that have been encountered, and recent work revealing the relationship between hematological disease and abnormalities of the HSC niche.
HSCs: current dogmas and evolving concepts
Our idea of the HSC has traditionally been dominated by a number of concepts:
-
1.
A static, long-term HSC with fixed specific features (including a characteristic immunophenotype, quiescent state, self-renewing properties), ultimately responsible for maintaining hematopoiesis through life.
-
2.
A convenient, straightforward, but simplistic model of progenitor hierarchy, in which a homogeneous population of HSC differentiates into progenitors and subsequently mature lineages along bifurcating, non-reversible, non-overlapping paths.
-
3.
A rigid idea of a “HSC niche”, in which—by analogy with certain non-hematopoietic tissues—the HSC is postulated to “reside” more or less statically in discrete anatomical microenvironments, the exact identity of which has remained elusive. The growing amount and complexity of information concerning the influence of many other bone marrow cells on HSC behavior has lead to postulate the existence of functionally different HSC niches, which is still not sufficiently substantiated by the experimental data available [1].
In the light of the results reported over the last few years, this image is gradually evolving toward a much more dynamic model:
(A) Heterogeneity in HSC proliferation and self-renewal Analyses of the mouse and human HSC compartments have revealed variability in the cell cycle status and self-renewal capacity of individual HSCs. In the mouse, populations of “dormant” and “activated” HSCs have been identified, which are to some extent interconvertible. Dormant HSCs divide at very slow rates in homeostasis (every 145 days), exhibit the highest self-renewal and multilineage repopulation activity and, although transiently activated by bone marrow injury or by granulocyte colony-stimulating factor (G-CSF), they revert to quiescence after reestablishment of homeostasis in a non-stochastic fashion [2]. HSCs switch from a proliferative to a quiescent status 3–4 weeks after birth [3] and are believed to undergo a similar transition after ex vivo manipulation requiring cytokine stimulation (e.g., retroviral transfer). Human HSCs also show heterogeneous self-renewal ability in xenotransplantation assays, with a limited number of clones providing long-term reconstitution and others exhibiting fluctuating contributions to hematopoiesis [4].
These observations may suggest that HSC fate is initially unpredictable and occurs stochastically, but it may also in part reflect limitations in the methods currently used for the isolation and functional analysis of HSCs. Thus, despite significant improvements, the best combinations of phenotypic markers in the mouse reach about 50 % purity, measured by competitive transplant assays [1, 5], or exclude a substantial fraction of HSCs [6]. Recently, based on combined transcriptomic and functional analysis at the single cell level, Wilson et al. [7] have proposed an improved sorting strategy that increases purity up to 67 %. Strategies to isolate human HSCs still lag behind in terms of purity, and their functional validation is complicated by the relatively low engraftment frequency of xenotransplant assays. Only 9.5 % of lin- CD34+ CD38− CD45A− Thy1+ CD49f+ cells exhibit long-term repopulating activity in intrafemorally injected NOD-scid-IL2Rgc −/− mice, and a small fraction of HSCs is apparently Thy1- [8]. Therefore, even after selecting cell populations with the best combinations of markers available, studies at non-clonal levels must take into account that these cell populations are not pure but only enriched in the expected HSCs.
(B) Heterogeneity in multilineage differentiation capacity of HSCs Retrospective analyses of single cells or clonal transplant experiments have demonstrated different kinetics and patterns of multilineage haematopoietic reconstitution derived from individual murine HSCs. Up to 16 distinct differentiation patterns were identified, based on their relative lymphomyeloid output and kinetics [9–11]. In the absence of markers that would allow their prospective isolation, it remains unknown whether each HSC was deterministically imprinted with a differentiation program, whether cell fate choice occurred stochastically or whether it was imposed by the microenvironment upon transplantation. Moreover, there was variability in the stability or “memory” of such program: while sometimes the differentiation pattern was preserved upon serial transplantations, in other cases a switch was observed. In this regard, there is some evidence that spontaneous, stochastic gene expression “noise” in HSCs may affect lineage choice [12].
(C) Contribution of hematopoietic progenitor cells to hematopoiesis Traditionally, transplantation assays have served as the gold standard to assess HSC function. Under transplantation conditions, long-term reconstitution ability seems to be restricted to a small number of primitive LT-HSCs, and hematopoiesis is typically oligoclonal. However, recent studies using genetic labeling and clonal tracing of HSC have revealed a very different situation during adult steady-state hematopoiesis, in which blood cell production is highly polyclonal and predominantly maintained by “short-term HSCs” or progenitors downstream of LT-HSCs, with strong myeloid bias [13, 14]. Moreover, within myeloid progenitors, multiple subgroups with heterogeneous differentiation patterns have been identified [15].
Notwithstanding technical limitations, these data suggest that, contrasting the concept of stable and discrete HSPC populations, a more dynamic situation may exist in which there is some degree of plasticity in the proliferation and differentiation capacity of HSCs and their progeny. It is still unclear to what extent this is regulated through dynamic interactions with the microenvironment or via stochastic, cell-autonomous fluctuations in the HSC transcriptome.
The HSC niches: structurally organized or stochastic/variable entity?
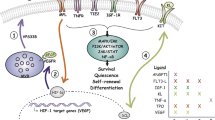
The growing amount of information concerning the HSC niche in recent years has paradoxically led to a situation of relative confusion, in which virtually every non-hematopoietic cell type in the bone marrow (osteoblasts, osteocytes, endothelial cells, stromal reticular cells, pericytes including MSC-like cells, adipocytes, non-myelinated Schwann cells, sympathetic neurons) plus several mature hematopoietic populations (macrophages, neutrophils, osteoclasts, megakaryocytes, Treg cells) have been proposed as niche components and/or critical regulators of HSC function (Fig. 1 left; Table 1). Here it would be worth attempting a clarification between those effects likely due to direct interaction between HSCs and anatomically close cellular components (thus properly called “niche” cells) and non-specific effects arising from the disruption of a very interconnected, complex multicellular system like the bone marrow, which requires a finely regulated homeostatic balance. Altering any major population in the bone, or the bone metabolism itself, will likely have indirect consequences on most bone marrow-resident cells due to profound structural and biochemical changes; on the other hand, experimental disruption of mature hematopoietic cells is bound to elicit a compensatory response in primitive HSPCs. However, neither of these are necessarily indicative of a “niche” function, and experimental dissection of direct and indirect effects may not be trivial.

Overview of the main components of the HSC niche and their alterations in leukemia. Simplified schematic of the normal HSC niche (left) and its alterations in the context of malignancy. The diagram does not attempt to comprehensively include every cell population and molecule implicated in HSC regulation but to illustrate some of the best characterized candidate niche cells and factors, particularly those that have been found altered in leukemias. The right panel summarizes niche abnormalities observed in various experimental models representing different leukemia types. Therefore, it does not intend to propose a general model nor to describe the pathophysiology of any particular malignancy. HSC hematopoietic stem cell, LSC leukemia stem/initiating cell
One of the most controversial aspects in the last decade has been the role of osteolineage cells as HSC niche components. Although this was the first population in the BM linked to HSC regulation, and a fraction of HSCs tend to home to the vicinity of osteoblasts, some of the effects observed may be indirect, and deletion of key HSC-supporting factors from osteoblasts seems to have little effect on HSCs ([16, 17]; reviewed in [18]). One caveat of some of these studies is the difficulty to discriminate in vivo between osteoblasts/osteoprogenitors and more primitive mesenchymal progenitors, because genetic drivers routinely used to mark these populations show overlapping expression patterns (reviewed in [19]). Recent research suggests that the periosteal region may support more committed progenitors, particularly of the lymphoid lineages (see below).
Osteocytes have been reported to regulate hematopoiesis through both secretion [20] and responsiveness [21] to G-CSF. However, due to their anatomical location, effects on HSCs could be indirect. An emerging concept is that osteocytes communicate with endosteal osteoblasts through dendritic processes and may regulate their function via gap junctions [22, 23]. Mice deficient for Gsα in osteocytes exhibit loss of trabecular and cortical bone associated with expansion of myeloid cells, an effect partly mediated by G-CSF production by osteocytes [20]. In turn, osteocytes were shown to rapidly respond to G-CSF by downregulating bone-specific genes and undergoing morphological changes, with loss of the cellular processes that connect them with osteoblasts [21]. In this study, ablation or disruption of the osteocyte network preceded loss of osteoblasts, prevented G-CSF-induced HSPC mobilization, increased bone marrow Cxcl12 levels and induced HSC quiescence.
Non-myelinating Schwann cells have been reported to contribute to maintain HSCs in a quiescent state through TGF-β production [24]. Arterioles preferentially located near the bone surface were also found to provide niches for quiescent HSCs [25]. However, a recent study has found most quiescent and non-quiescent HSCs far from bone and arterioles and close to sinusoidal vessels, which are much more abundant and widely distributed in the BM [1]. Whereas most HSCs might reside in this larger region of the BM, it remains possible that a different microenvironment in the endosteum maintains a different HSC state/population. It must be kept in mind that studies reporting HSC localization in the BM have employed different marker combinations to identify non-identical HSC-enriched cellular pools, which may account for some of the discrepancies. Thus, the latter report utilized the newly described reporter α-catulin [1], while the arteriole study used SLAM markers [25].
One of the puzzling aspects when trying to extrapolate the concept of stem cell niche (as derived from evidence in solid tissues) to the bone marrow is the apparent absence of compartmentalization of the latter into discrete structures sheltering the HSC. The existence of such compartments, however, has been speculated. Structures termed “hemospheres” (containing mesenchymal, endothelial and hematopoietic cells) were shown to be enriched in CD150+ CD58− HSCs and seemed to represent sites of clonal hematopoietic cell proliferation. Hemospheres were found in sinusoidal vessels at the periphery of the BM cavity, associated with NG2+ Pdgfrb+ Nestin+ CD146+ pericytes, and required expression of VEGFR2 in the endothelium [26]. A recent study using high-resolution live imaging has suggested that this type of structures may not be permanent but dynamic. In zebrafish fetal hematopoietic tissue, the perivascular niche remodels upon arrival of a HSPC, forming a pocket-like compartment in which a group of endothelial cells surround an HSPC attached to a mesenchymal stromal cell. This structure seems to determine the polarity of HSC division [27]. Additional intravital imaging studies have revealed that HSCs also interact dynamically within the niche and, while their motility is restricted to small oscillations under resting conditions, they become more migratory upon activation (e.g., through acute infection), resulting in their contact with larger areas of the BM [28].
Recent evidence suggests that diverse bone marrow locations may function as niches for different hematopoietic progenitors. The evidence is stronger for lymphopoietic niches, which have been associated with IL-7-enriched bone regions [29, 30]. Also, specific deletion of Cxcl12 from osteoblasts reduces early lymphoid progenitors—a fraction of which was shown to localize at the endosteum—without affecting HSCs or myeloerythroid progenitors, suggesting a function of the so-called osteoblastic niche in regulating early lymphopoiesis [16]. Depletion of osteocalcin-expressing bone cells (mature osteoblasts and osteocytes), or deletion of the Notch ligand DLL4 from the same cells, impairs the production of T-lineage-competent lymphoid progenitors in the bone marrow, resulting in decreased T-cell generation in the thymus [31].
Regulation of the HSC niche by the mature hematopoietic progeny
Recent work has highlighted the influence of different mature hematopoietic cell subsets on the regulation of the HSC niche. Among mature bone marrow cells, the greatest body of evidence implicates monocytes/macrophages as potential HSC regulators, usually indirectly through interactions with other components of the niche [32–36]. Different studies have highlighted a role of monocytes/macrophages in HSPC mobilization through regulation of osteoblastic cells. G-CSF was shown to deplete bone marrow monocytic cells, including endosteal macrophages (osteomacs). In vivo depletion of macrophages caused loss of osteoblasts and mobilization of HSPCs. Notably, expression of G-CSF receptor in monocytic cells was sufficient to induce HSPC mobilization, osteoblast suppression and Cxcl12 downregulation, suggesting that G-CSF signaling in macrophages represses some osteoblast-supporting factors [33, 36]. A population of CD169+ macrophages was shown to promote HSPC retention in the bone marrow. Experimental depletion of macrophages enhances physiological or enforced HSPC mobilization, associated with a decrease in Cxcl12 levels and downregulation of HSC maintenance genes in nestin+ cells (but not in osteoblasts) [34]. In addition, CD169+ macrophages were implicated in regulating erythropoiesis by mediating erythroblast retention in the bone marrow. Although dispensable under homeostasis, macrophages were required for a proficient erythropoietic stress response, an effect mediated by VCAM1 expression by macrophages [35]. A different study identified a population of α-SMA+ activated monocytes and macrophages that localized adjacent to HSPCs and contributed to protect HSPC from exhaustion under stress [32]. Again, the effect was partially dependent on regulation of Cxcl12 expression by stromal cells, although macrophages were also proposed to directly protect HSCs from oxidative damage by PGE2 production.
On the other hand, the influence of osteoclasts—which also differentiate from the monocyte/macrophage lineage—on HSCs through bone remodeling has remained controversial; different studies have reported that osteoclasts are either necessary, dispensable or even inhibitory for HSC maintenance and mobilization. Thus, some studies have suggested that osteoclasts promote the formation of BM niches [37]. Also, osteoclast inhibition with calcitonin decreased homeostatic and G-CSF-enforced HPSC mobilization [38], but their suppression by zoledronate actually enhanced G-CSF mobilization of HSPCs [36]. However, different strains of osteoclast-deficient (osteopetrotic) mice had normal or increased HSPC mobilization [39]. Also, the BM of osteoclast-deficient, osteopetrotic mice (oc/oc or RANK knockout mice) contained normal HSC activity [40].
It has been recently shown that HSCs frequently localize in the vicinity of megakaryocytes, and ablation of megakaryocytes leads to cell cycle entry of quiescent HSC, suggesting that this cellular population contributes to maintaining HSC quiescence. This interaction was reported as being mediated by soluble factors such as TGF-β1, Cxcl4 and CLEC-2-mediated thrombopoietin production [41–44]. Megakaryocytes were also shown to participate in HSC niche remodeling after radioablation. Irradiation induces megakaryocyte migration to the endosteum mediated by thrombopoietin and CD41, and PDGF-BB secreted by megakaryocytes mediates osteoblast expansion. Stimulation of megakaryocyte function by thrombopoietin administration improves HSC engraftment after transplantation [45].
Neutrophil clearance has been proposed as an additional regulatory mechanism of the HSC niche [46]. Infiltration and elimination of aged CD62LO CXCR4HI neutrophils in the bone marrow was proposed to cause a reduction in the size and function of the HSC niche by decreasing the number of CXCL12-abundant reticular (CAR) cells and the levels of Cxcl12, thus promoting the circadian egress of HSPCs into circulation. However, circadian oscillations of Cxcl12 expression, directly induced in bone marrow stromal cells by oscillating sympathetic activity [47], might also contribute to some of the observed changes. The reported effects of neutrophils on the HSC niche were ultimately mediated by bone marrow macrophages—which attract and phagocytose aged neutrophils—and required activation of LXR in these cells.
Treg lymphocytes colocalize with HSCs near the endosteal surface in the bone marrow and contribute to protecting transplanted allogeneic HSCs from immune clearance, suggesting that the niche does not only nurture HSCs but also provides them with immune privilege [48].
The malignant niche: a few knowns and many unknowns
Although it has long been recognized that leukemia/lymphoma development is associated with an abnormal tissue microenvironment in the affected organs, only in recent years has experimental work begun to dissect specific roles of the HSC niche in leukemogenesis (Fig. 1 right; Table 1). Progress in this direction so far has lagged behind the gradual understanding of the physiological HSC niche and shared similar technical limitations. On one hand there are uncertainties derived from the incomplete specificity, efficiency and characterization of genetic drivers used to target stromal cell populations (reviewed in [19]). These problems add to the intrinsic limitations of murine cancer models, e.g., how faithfully they recapitulate the human diseases (regarding both their hematopoietic and microenvironmental abnormalities) or how much the experimental procedure itself perturbs the niche (particularly in assays where myeloconditioning is necessary, such as xenotransplantation).
Two major ideas have emerged, supported by growing evidence: (1) genetic alterations of the niche, rather than the hematopoietic cell, may represent driving mutations during malignant transformation; (2) cancer cells remodel their niche into an abnormal environment that provides preferential support of malignant cells—in detriment of normal hematopoiesis—protects cancer cells from therapy-induced cell death, and/or drives disease progression. From these concepts immediately follows the idea that manipulating the leukemic niche might represent an advantageous therapeutic strategy, particularly in malignancies for which targeting the hematopoietic cells has proven inefficient.
Niche abnormalities as potential initiating events
Initial studies showed that certain genetic alterations that affected broadly the BM microenvironment also promoted the development of abnormal hematopoiesis. Mice usually developed a myeloproliferative-like disease characterized by hypergranulopoiesis, extramedullary hematopoiesis, increased myeloid progenitors and eventually bone marrow failure and anemia. These phenotypes were observed upon deletion of retinoic acid receptor gamma (microenvironment dependent) [49], deletion of retinoblastoma protein (simultaneously required in microenvironment and myeloid cells) [50], ubiquitous deletion of IkBα (non-hematopoietic cell autonomous, but it is unclear whether the alteration in the microenvironment was sufficient) [51], deletion of the Notch ligand endocytosis regulator Mib1 (causing defective Notch activation in the microenvironment) [52] and haploinsufficiency of Crebbp in the stromal cells [53]. These phenotypes were associated with histological changes in the BM, such as loss of trabecular bone [49, 50, 53] or decreased number of adipocytes [52]. Among the molecular pathways implicated in these processes were increased TNFα/NFkB and aberrant Notch signaling. In these studies—more extensively discussed in recent reviews [54]—widespread deletion of the gene of interest did not allow dissection of the cell population(s) responsible for the phenotype.
Subsequent studies refined this concept by modifying specific stromal cell subsets. Specific deletion of the RNA processing enzyme Dicer1 under the regulatory elements of Osterix, a gene required for osteoblastic differentiation—but not in mature osteoblasts, targeted by the regulatory elements of Osteocalcin promoter—did not only impair osteoblast differentiation, but was also found to cause a myelodysplastic syndrome characterized by dysplastic morphology, lymphoid-deficient hematopoiesis and increased HSPC proliferation and apoptosis. Key aspects of the phenotype were recapitulated by specific Osterix-cre-mediated deletion of Sbds, a Dicer-regulated gene that is mutated in some leukemias [55]. However, it should be noted that, although Osterix was originally reported to label only cells committed to become osteoblasts [56], a much wider recombination has been observed more recently after the development of stronger reporter lines [57–59] (see [19] for a review). Therefore, some of these phenotypes might be derived from gene excision in other cells, besides osteoprogenitors.
VEGF overexpression in Col2.3-cre+ osteochondroprogenitors and their progeny constitutively activated β-catenin and caused hematological abnormalities, extramedullary hematopoiesis and HSPC mobilization, likely related to the dramatic bone mass increase and the aberrant vascularization [60]. More dramatically, direct expression of a constitutively active β-catenin form in osteoblasts (Ctnnb1 CAosb) impaired the differentiation of hematopoietic progenitors and caused accumulation of immature myeloid cells and dysplastic neutrophils in hematopoietic tissues, resembling acute myeloid leukemia (AML). These changes were caused by expression of jagged1 in osteoblasts and consequent excessive Notch signaling in HSPCs [61].
These studies demonstrated that genetic alterations in the microenvironment can trigger a pre-leukemic condition (analogous to an oncogenic “first hit”), but at the same time suggested that additional mutations are required in the hematopoietic cells to induce malignancy and/or overt leukemia. Evidence of malignant transformation was limited in most studies. In the case of Dicer1-deleted mice, rare sporadic occurrences of AML that exhibited cytogenetic alterations were reported [55]. Most remarkably, frequent recurrent chromosomal alterations and somatic mutations were detected in Ctnnb1 CAosb mice, including those in an ortholog region of human chromosome 7q, which is altered in AML and myelodysplastic syndromes. Notably, transformed HSCs from Ctnnb1 CAosb mice became autonomous and could transfer the AML to WT recipients [61].
In spite of these experimental models providing a proof of concept, we are still lacking direct demonstration that an initial lesion in the microenvironment may play a causative role in human leukemias. However, the situation is somewhat reminiscent of that traditionally observed in extraganglionar lymphomas associated with local infectious processes (such as Helicobacter pylori in MALT lymphomas) in which the pathogen may provide antigenic stimulation to malignant B-cells and the tumor remits after eradication of the infection [62]. In leukemias, available evidence is only indirect. Decreased expression of DICER, DROSHA and SBDS was detected in MSCs—but not in leukocytes—from MDS patients compared to healthy individuals [63]. SBDS is inactivated by mutations in the human Shwachman–Bodian–Diamond syndrome, featuring skeletal abnormalities, bone marrow failure and susceptibility to developing myelodysplastic syndrome and secondary AML [55]. CREBPP heterozygosity in humans causes the Rubinstein–Taybi syndrome, characterized by skeletal defects and cancer predisposition, including leukemias [64]. Activation of β-catenin in osteoblasts, accompanied by Notch activation in hematopoietic cells, was detected in over one-third of patients with myelodysplastic syndrome or AML [61].
Malignant cells create aberrant niches
Knowledge of how leukemic cells interact with their microenvironment has followed progress in the characterization of the normal HSC niche, and is affected by similar—if more notorious—uncertainties.
Xenografted primary human AML stem cells (CD34+ CD38−) were reported to exhibit preferential homing and engraftment to the endosteal, osteoblast-rich area of the BM [65], while acute lymphoblastic leukemia (ALL) cells tended to localize to vascular regions expressing E-selectin and Cxcl12, overlapping with perivascular HSPC niches [66, 67]; however, it was later recognized that these endothelial microdomains were juxtaposed to the endosteum [68], suggesting similar primary homing location of AML/ALL cells. Endosteal human leukemia stem cells (LSCs) causing AML were reported as characteristically quiescent and chemoresistant; cell cycle entry induced by G-CSF enhanced chemotherapy-induced apoptosis and elimination of LSCs [65, 69].
Xenotransplantation of ALL cell lines has been shown to disrupt the normal bone marrow microenvironment [66, 68]. Particularly, these cells can cause severe damage of the vasculature and endosteum-lining cells and lead to the formation of abnormal niches primarily formed by the mutated cells, which highly express stem cell factor but produce low levels of Cxcl12. In these aberrant niches, both the numbers and the traffic of normal HSPCs were reduced. This was proposed as a mechanism by which normal hematopoiesis could be impaired even in the presence of a low tumor burden. In the same xenotransplant model, chemotherapy was shown to induce the formation of transient niches consisting of small foci of surviving ALL cells and nestin+ LepR+ NG2+ stromal cells with properties of MSCs (multipotent sphere formation, in vitro differentiation). Formation of these structures required CCL3 and TGFb1 produced by ALL cells and, in spite of their short life, were proposed to protect ALL cells from chemotherapy-induced apoptosis. A recent study has shown that T-ALL cells require Cxcl12-producing endothelial cells, and CXCR4 antagonism suppressed T-ALL in primary xenografts [70].
It has been recently shown that MSCs exhibit an altered gene expression program in myelodysplastic syndromes, with increased expression of genes associated with osteoprogenitor cell fate, inflammation and fibrosis. These MSCs are capable of promoting the engraftment or propagation of myelodysplasia in orthotopic xenografts [71].
Severe osteoblastic defects were found in a model of myeloid blast-crisis chronic myeloid leukemia (CML, driven by BCR-ABL;Nup98/HoxA9), with decreased osteoprogenitors, endosteal-lining osteoblasts and bone mass, associated with increased CCL3 expression [72]. Using a mouse model of chronic-phase, BCR-ABL-induced CML (SCL-tTA;TRE-BCR/ABL mice), two studies have provided evidence for the concept that leukemic cells can remodel the bone marrow environment into a self-supporting niche that impairs normal hematopoiesis. Leukemic cells were shown to stimulate excessive production of osteoblastic cells. These cells had reduced expression of HSC maintenance factors and impaired ability to support normal HSCs, while having limited impact on LSCs. Abnormal osteoblastic cells overexpressed inflammation- and myelofibrosis-related genes, contributed to bone marrow fibrosis, and their expansion was driven by TPO, CCL3 and direct contact with leukemic cells [73]. In another study, G-CSF overproduction by CML cells reduced Cxcl12 expression by bone marrow stromal cells and increased several pro-inflammatory cytokines, leading to selective impairment of normal HSCs that also favored the growth of CML stem cells [74]. Pro-inflammatory cytokines produced by mature leukemic cells had also direct effects on the leukemic HSPC; thus, BCR-ABL-dependent secretion of IL-6 by CML cells drives myeloid differentiation of leukemic multipotent MPP, establishing a feed-forward loop [75].
Recent work from our laboratory has demonstrated that the manifestation of JAK2 V617F-induced myeloproliferative neoplasms (MPN) requires the loss of sympathetic regulation in the HSC niche [76]. Sympathetic fibers and associated Schwann cells are consistently decreased in the bone marrow of JAK2 V617F-positive MPN patients and mice due to apoptosis induced by MPN cell-derived IL-1β. In turn, neuroglial damage compromises the survival of nestin+ cells, which are also reduced in number over the course of the disease. This loss of MSCs plays a driving role in MPN development, since selective depletion of nestin+ cells, or Cxcl12 produced by them, is sufficient to accelerate disease progression. Conversely, pharmacological compensation of neural damage blocks MPN progression (see below).
The leukemic niche as a therapeutic target
Growing evidence of the active participation of the HSC niche in hematological malignancies, either by providing an initiating lesion or by responding to leukemia-induced remodeling, suggests that its therapeutic targeting may have therapeutic benefit. This could be potentially achieved through inhibition of essential interactions between LSCs and supporting cells, or by reversion of niche-related changes that drive the course of the disease. We have provided direct evidence of the latter in the context of JAK2 V617F-induced MPN, in which loss of nestin+ MSCs caused by neuroglial damage promotes disease progression [76]. Rescue of the defective sympathetic stimulation by chronic administration of β3-adrenergic agonists prevented the loss of nestin+ MSC and inhibited disease progression at both early and late stages, preventing neutrophilia, thrombocytosis, IL-1β production, bone marrow fibrosis and expansion of LSCs, while having negligible effects on normal HSCs. Similarly, treatment of MPN-affected mice with the neuroprotective molecule 4-methylcatecol, which protects sympathetic fibers and Schwann cells, abolished MPN-related neutrophilia. These results demonstrate that specific reversion of malignancy-induced changes in the bone marrow microenvironment may be sufficient to block MPN. They also prove that niche transformation represents a major driving force and a requirement for disease progression, and provide a novel, potential safe therapeutic approach where hematopoietic cell-directed therapies have previously shown limited efficacy.
However, this is very unlikely the case of more aggressive leukemias, such as AML, despite some similarities shown in a recent study suggesting that sympathetic neuropathy caused by acute myeloid leukemic cells promotes malignancy in an altered hematopoietic stem cell niche [77]. Unlike in MPN [76], adrenergic drugs did not significantly impact AML progression in this study. However, emerging evidence suggests that the microenvironment might represent a feasible therapeutic target for AML. For instance, inhibition of Cxcr4/Cxcl12 signaling may show some therapeutic advantage in AML. Cxcr4 antagonists (AML3100/Plerixafor and the novel LY2510924) induce leukemic cell mobilization and enhance the antileukemic action of chemotherapy in xenotransplant models [78, 79]. Furthermore, addition of AMD3100 to cytotoxic chemotherapy increased remission rates in patients with relapsed or refractory AML in a phase 1/2 clinical study [80].
Considerations for the future
As outlined at the beginning of this review, elucidation of the cellular and molecular interactions in the HSC niche has two major practical motivations. One is the identification of key molecules that would allow the production, maintenance and expansion of HSCs. Identification of the specific factors that contribute to maintaining stemness in vivo would facilitate the development of chemically defined culture media, avoiding the use of animal-derived culture supplements and possibly the need for time-consuming coculture protocols. The identification of specific microenvironments and factors required for the maintenance of different types of hematopoietic progenitors could make possible the selective ex vivo generation of different lineages for specific applications.
Another aspect with translational potential is the identification of alterations in the bone marrow niche directly associated with, and required for, the development of each type of leukemia. It is expected that different malignancies will induce a specific set of abnormalities and will also differ in their dependence on the niche. Thus, it would not be surprising to find that aggressive, acute leukemias with accumulated mutations may become relatively cell autonomous and less sensitive to microenvironmental regulation. Most relevant will be to identify cellular processes or pathways selectively required for the maintenance of malignant progenitors—as opposed to normal HSCs—and susceptible to pharmacological targeting. The success of niche-directed therapies will likely depend on how accurately we can validate preclinical findings from mouse models in the human system, for which it will be critical to develop parallel sets of molecular markers for human HSCs and niche cells, as well as more faithful xenotransplant models.
From the more theoretical point of view, research in the immediate future is expected to refine our knowledge of the composition of the HSC niche, dissecting between direct interactions with bona fide “niche cells” and indirect signals from more distant components. It would also be advantageous to clarify some of the confusion in the field resulting from the use of incompletely characterized genetic drivers and partially overlapping markers (reviewed in [19]). Again, it is also expected that improved xenograft models and human cell markers will allow a more direct investigation of the biology of the human HSC niche.
Abbreviations
- HSC:
-
Hematopoietic stem cell
- HSPC:
-
Hematopoietic stem cells and progenitors
- MPP:
-
Multipotent progenitors
- LSC:
-
Leukemia stem cell
- MSC:
-
Mesenchymal stem cell
- AML:
-
Acute myeloid leukemia
- ALL:
-
Acute lymphoblastic leukemia
- MPN:
-
Myeloproliferative neoplasm
- CML:
-
Chronic myeloid leukemia
References
Acar M, Kocherlakota KS, Murphy MM, Peyer JG, Oguro H, Inra CN, Jaiyeola C, Zhao Z, Luby-Phelps K, Morrison SJ (2015) Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526(7571):126–130. doi:10.1038/nature15250
Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A (2008) Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135(6):1118–1129
Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ (2006) Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest 116(10):2808–2816. doi:10.1172/JCI28310
McKenzie JL, Gan OI, Doedens M, Wang JC, Dick JE (2006) Individual stem cells with highly variable proliferation and self-renewal properties comprise the human hematopoietic stem cell compartment. Nat Immunol 7(11):1225–1233. doi:10.1038/ni1393
Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ (2005) SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121(7):1109–1121. doi:10.1016/j.cell.2005.05.026
Matsuzaki Y, Kinjo K, Mulligan RC, Okano H (2004) Unexpectedly efficient homing capacity of purified murine hematopoietic stem cells. Immunity 20(1):87–93
Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, Sanchez Castillo M, Oedekoven CA, Diamanti E, Schulte R, Ponting CP, Voet T, Caldas C, Stingl J, Green AR, Theis FJ, Gottgens B (2015) Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell 16(6):712–724. doi:10.1016/j.stem.2015.04.004
Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE (2011) Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 333(6039):218–221. doi:10.1126/science.1201219
Sieburg HB, Cho RH, Dykstra B, Uchida N, Eaves CJ, Muller-Sieburg CE (2006) The hematopoietic stem compartment consists of a limited number of discrete stem cell subsets. Blood 107(6):2311–2316. doi:10.1182/blood-2005-07-2970
Dykstra B, Kent D, Bowie M, McCaffrey L, Hamilton M, Lyons K, Lee SJ, Brinkman R, Eaves C (2007) Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 1(2):218–229. doi:10.1016/j.stem.2007.05.015
Lu R, Neff NF, Quake SR, Weissman IL (2011) Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat Biotechnol 29(10):928–933. doi:10.1038/nbt.1977
Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S (2008) Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 453(7194):544–547. doi:10.1038/nature06965
Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Hofer T, Rodewald HR (2015) Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518(7540):542–546. doi:10.1038/nature14242
Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD (2014) Clonal dynamics of native haematopoiesis. Nature 514(7522):322–327. doi:10.1038/nature13824
Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, David E, Cohen N, Lauridsen FK, Haas S, Schlitzer A, Mildner A, Ginhoux F, Jung S, Trumpp A, Porse BT, Tanay A, Amit I (2015) Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 163(7):1663–1677. doi:10.1016/j.cell.2015.11.013
Ding L, Morrison SJ (2013) Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495(7440):231–235. doi:10.1038/nature11885
Ding L, Saunders TL, Enikolopov G, Morrison SJ (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481(7382):457–462. doi:10.1038/nature10783
Morrison SJ, Scadden DT (2014) The bone marrow niche for haematopoietic stem cells. Nature 505(7483):327–334. doi:10.1038/nature12984
Mendez-Ferrer S, Scadden DT, Sanchez-Aguilera A (2015) Bone marrow stem cells: current and emerging concepts. Ann N Y Acad Sci 1335:32–44. doi:10.1111/nyas.12641
Fulzele K, Krause DS, Panaroni C, Saini V, Barry KJ, Liu X, Lotinun S, Baron R, Bonewald L, Feng JQ, Chen M, Weinstein LS, Wu JY, Kronenberg HM, Scadden DT, Divieti Pajevic P (2013) Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood 121(6):930–939. doi:10.1182/blood-2012-06-437160
Asada N, Katayama Y, Sato M, Minagawa K, Wakahashi K, Kawano H, Kawano Y, Sada A, Ikeda K, Matsui T, Tanimoto M (2013) Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell 12(6):737–747. doi:10.1016/j.stem.2013.05.001
Kamioka H, Honjo T, Takano-Yamamoto T (2001) A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone 28(2):145–149
Taylor AF, Saunders MM, Shingle DL, Cimbala JM, Zhou Z, Donahue HJ (2007) Mechanically stimulated osteocytes regulate osteoblastic activity via gap junctions. Am J Physiol Cell Physiol 292(1):C545–C552. doi:10.1152/ajpcell.00611.2005
Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, Taketo MM, Karlsson S, Iwama A, Nakauchi H (2011) Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 147(5):1146–1158. doi:10.1016/j.cell.2011.09.053
Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, Mar JC, Bergman A, Frenette PS (2013) Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502(7473):637–643. doi:10.1038/nature12612
Wang L, Benedito R, Bixel MG, Zeuschner D, Stehling M, Savendahl L, Haigh JJ, Snippert H, Clevers H, Breier G, Kiefer F, Adams RH (2013) Identification of a clonally expanding haematopoietic compartment in bone marrow. EMBO J 32(2):219–230. doi:10.1038/emboj.2012.308
Tamplin OJ, Durand EM, Carr LA, Childs SJ, Hagedorn EJ, Li P, Yzaguirre AD, Speck NA, Zon LI (2015) Hematopoietic stem cell arrival triggers dynamic remodeling of the perivascular niche. Cell 160(1–2):241–252. doi:10.1016/j.cell.2014.12.032
Rashidi NM, Scott MK, Scherf N, Krinner A, Kalchschmidt JS, Gounaris K, Selkirk ME, Roeder I, Lo Celso C (2014) In vivo time-lapse imaging shows diverse niche engagement by quiescent and naturally activated hematopoietic stem cells. Blood 124(1):79–83. doi:10.1182/blood-2013-10-534859
Tokoyoda K, Egawa T, Sugiyama T, Choi BI, Nagasawa T (2004) Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 20(6):707–718. doi:10.1016/j.immuni.2004.05.001
Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, Scadden DT, Kronenberg HM (2008) Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci USA 105(44):16976–16981. doi:10.1073/pnas.0802898105
Yu VW, Saez B, Cook C, Lotinun S, Pardo-Saganta A, Wang YH, Lymperi S, Ferraro F, Raaijmakers MH, Wu JY, Zhou L, Rajagopal J, Kronenberg HM, Baron R, Scadden DT (2015) Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J Exp Med 212(5):759–774. doi:10.1084/jem.20141843
Ludin A, Itkin T, Gur-Cohen S, Mildner A, Shezen E, Golan K, Kollet O, Kalinkovich A, Porat Z, D’Uva G, Schajnovitz A, Voronov E, Brenner DA, Apte RN, Jung S, Lapidot T (2012) Monocytes-macrophages that express alpha-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat Immunol 13(11):1072–1082. doi:10.1038/ni.2408
Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC (2011) Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med 208(2):251–260. doi:10.1084/jem.20101700
Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, Tanaka M, Merad M, Frenette PS (2011) Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 208(2):261–271. doi:10.1084/jem.20101688
Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, Pinho S, Leboeuf M, Noizat C, van Rooijen N, Tanaka M, Zhao ZJ, Bergman A, Merad M, Frenette PS (2013) CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med 19(4):429–436. doi:10.1038/nm.3057
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, Levesque JP (2010) Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116(23):4815–4828. doi:10.1182/blood-2009-11-253534
Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SE, Wakkach A, Blin-Wakkach C (2012) Osteoclasts promote the formation of hematopoietic stem cell niches in the bone marrow. J Exp Med 209(3):537–549. doi:10.1084/jem.20110994
Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T (2006) Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med 12(6):657–664. doi:10.1038/nm1417
Miyamoto K, Yoshida S, Kawasumi M, Hashimoto K, Kimura T, Sato Y, Kobayashi T, Miyauchi Y, Hoshi H, Iwasaki R, Miyamoto H, Hao W, Morioka H, Chiba K, Yasuda H, Penninger JM, Toyama Y, Suda T, Miyamoto T (2011) Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J Exp Med 208(11):2175–2181. doi:10.1084/jem.20101890
Flores C, Moscatelli I, Thudium CS, Gudmann NS, Thomsen JS, Bruel A, Karsdal MA, Henriksen K, Richter J (2013) Osteoclasts are not crucial for hematopoietic stem cell maintenance in adult mice. Haematologica 98(12):1848–1855. doi:10.3324/haematol.2013.089466
Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, Frenette PS (2014) Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. doi:10.1038/nm.3707
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, Ahamed J, Li L (2014) Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. doi:10.1038/nm.3706
Nakamura-Ishizu A, Takubo K, Kobayashi H, Suzuki-Inoue K, Suda T (2015) CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J Exp Med 212(12):2133–2146. doi:10.1084/jem.20150057
Nakamura-Ishizu A, Takubo K, Fujioka M, Suda T (2014) Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem Biophys Res Commun 454(2):353–357. doi:10.1016/j.bbrc.2014.10.095
Olson TS, Caselli A, Otsuru S, Hofmann TJ, Williams R, Paolucci P, Dominici M, Horwitz EM (2013) Megakaryocytes promote murine osteoblastic HSC niche expansion and stem cell engraftment after radioablative conditioning. Blood 121(26):5238–5249. doi:10.1182/blood-2012-10-463414
Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chevre R, Noelia AG, Kunisaki Y, Zhang D, van Rooijen N, Silberstein LE, Weber C, Nagasawa T, Frenette PS, Castrillo A, Hidalgo A (2013) Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 153(5):1025–1035. doi:10.1016/j.cell.2013.04.040
Mendez-Ferrer S, Lucas D, Battista M, Frenette PS (2008) Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452(7186):442–447. doi:10.1038/nature06685
Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo Celso C, Tsuyuzaki H, Sato T, Cote D, Sykes M, Strom TB, Scadden DT, Lin CP (2011) In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 474(7350):216–219. doi:10.1038/nature10160
Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 129(6):1097–1110. doi:10.1016/j.cell.2007.05.014
Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH (2007) Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 129(6):1081–1095. doi:10.1016/j.cell.2007.03.055
Rupec RA, Jundt F, Rebholz B, Eckelt B, Weindl G, Herzinger T, Flaig MJ, Moosmann S, Plewig G, Dorken B, Forster I, Huss R, Pfeffer K (2005) Stroma-mediated dysregulation of myelopoiesis in mice lacking I kappa B alpha. Immunity 22(4):479–491. doi:10.1016/j.immuni.2005.02.009
Kim YW, Koo BK, Jeong HW, Yoon MJ, Song R, Shin J, Jeong DC, Kim SH, Kong YY (2008) Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood 112(12):4628–4638. doi:10.1182/blood-2008-03-148999
Zimmer SN, Zhou Q, Zhou T, Cheng Z, Abboud-Werner SL, Horn D, Lecocke M, White R, Krivtsov AV, Armstrong SA, Kung AL, Livingston DM, Rebel VI (2011) Crebbp haploinsufficiency in mice alters the bone marrow microenvironment, leading to loss of stem cells and excessive myelopoiesis. Blood 118(1):69–79. doi:10.1182/blood-2010-09-307942
Raaijmakers MH (2012) Myelodysplastic syndromes: revisiting the role of the bone marrow microenvironment in disease pathogenesis. Int J Hematol 95(1):17–25. doi:10.1007/s12185-011-1001-x
Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, Aung Z, Matza M, Merkenschlager M, Lin C, Rommens JM, Scadden DT (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464(7290):852–857. doi:10.1038/nature08851
Rodda SJ, McMahon AP (2006) Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133(16):3231–3244. doi:10.1242/dev.02480
Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13(1):133–140. doi:10.1038/nn.2467
Liu Y, Strecker S, Wang L, Kronenberg MS, Wang W, Rowe DW, Maye P (2013) Osterix-cre labeled progenitor cells contribute to the formation and maintenance of the bone marrow stroma. PLoS One 8(8):e71318. doi:10.1371/journal.pone.0071318
Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM (2010) Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 19(2):329–344. doi:10.1016/j.devcel.2010.07.010
Maes C, Goossens S, Bartunkova S, Drogat B, Coenegrachts L, Stockmans I, Moermans K, Nyabi O, Haigh K, Naessens M, Haenebalcke L, Tuckermann JP, Tjwa M, Carmeliet P, Mandic V, David JP, Behrens A, Nagy A, Carmeliet G, Haigh JJ (2010) Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. EMBO J 29(2):424–441. doi:10.1038/emboj.2009.361
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, Brum A, Park D, Galili N, Mukherjee S, Teruya-Feldstein J, Raza A, Rabadan R, Berman E, Kousteni S (2014) Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature 506(7487):240–244. doi:10.1038/nature12883
Herreros B, Sanchez-Aguilera A, Piris MA (2008) Lymphoma microenvironment: culprit or innocent? Leukemia 22(1):49–58. doi:10.1038/sj.leu.2404970
Santamaria C, Muntion S, Roson B, Blanco B, Lopez-Villar O, Carrancio S, Sanchez-Guijo FM, Diez-Campelo M, Alvarez-Fernandez S, Sarasquete ME, de las Rivas J, Gonzalez M, San Miguel JF, Del Canizo MC (2012) Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica 97(8):1218–1224. doi:10.3324/haematol.2011.054437
Roelfsema JH, Peters DJ (2007) Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med 9(23):1–16. doi:10.1017/S1462399407000415
Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, Fukata M, Miyamoto T, Lyons B, Ohshima K, Uchida N, Taniguchi S, Ohara O, Akashi K, Harada M, Shultz LD (2007) Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 25(11):1315–1321. doi:10.1038/nbt1350
Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA (2008) Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 322(5909):1861–1865. doi:10.1126/science.1164390
Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, Luster AD, Scadden DT, Lin CP (2005) In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435(7044):969–973. doi:10.1038/nature03703
Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, Zhou XC, Cai YJ, Li ZQ, Zhang F, Yin MZ, Tao Y, Mi JQ, Li LH, Enver T, Chen GQ, Hong DL (2014) Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 25(6):778–793. doi:10.1016/j.ccr.2014.04.015
Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A, Taniguchi S, Shultz LD, Ishikawa F (2010) Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol 28(3):275–280. doi:10.1038/nbt.1607
Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, Morrison SJ, Fooksman DR, Aifantis I, Schwab SR (2015) CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell 27(6):755–768. doi:10.1016/j.ccell.2015.05.002
Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, Lier A, Eisen C, Nowak V, Zens B, Mudder K, Klein C, Oblander J, Fey S, Vogler J, Fabarius A, Riedl E, Roehl H, Kohlmann A, Staller M, Haferlach C, Muller N, John T, Platzbecker U, Metzgeroth G, Hofmann WK, Trumpp A, Nowak D (2014) Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 14(6):824–837. doi:10.1016/j.stem.2014.02.014
Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM (2012) Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood 119(2):540–550. doi:10.1182/blood-2011-04-348151
Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, Passegue E (2013) Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 13(3):285–299. doi:10.1016/j.stem.2013.06.009
Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, Hair A, Holyoake TL, Huettner C, Bhatia R (2012) Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 21(4):577–592. doi:10.1016/j.ccr.2012.02.018
Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, Sala-Torra O, Radich JP, Passegue E (2011) IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 20(5):661–673. doi:10.1016/j.ccr.2011.10.012
Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, Lundberg P, Muntion S, Tzeng YS, Lai DM, Schwaller J, Skoda RC, Mendez-Ferrer S (2014) Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. doi:10.1038/nature13383
Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Duhrsen U, Frenette PS (2014) Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. doi:10.1016/j.stem.2014.06.020
Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, Prior JL, Piwnica-Worms D, Bridger G, Ley TJ, DiPersio JF (2009) Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 113(24):6206–6214. doi:10.1182/blood-2008-06-162123
Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, Levis M, Rubin JB, Negrin RR, Estey EH, Konoplev S, Andreeff M, Konopleva M (2009) Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 113(24):6215–6224. doi:10.1182/blood-2008-05-158311
Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, Kulkarni S, Abboud CN, Cashen AF, Stockerl-Goldstein KE, Vij R, Westervelt P, DiPersio JF (2012) A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 119(17):3917–3924. doi:10.1182/blood-2011-10-383406
Sugiyama T, Kohara H, Noda M, Nagasawa T (2006) Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25(6):977–988. doi:10.1016/j.immuni.2006.10.016
Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS (2010) Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466(7308):829–834. doi:10.1038/nature09262
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ (2009) Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460(7252):259–263. doi:10.1038/nature08099
Acknowledgments
We thank members of the SM-F group for helpful discussions. This work was supported by core support grants from the Wellcome Trust and MRC to the Cambridge Stem Cell Institute, the Spanish Ministry of Economy and Competitiveness (SAF-2011-30308), Pro-CNIC Foundation, Severo Ochoa Center of Excellence award SEV-2015-0505 to CNIC, TerCel (Spanish Cell Therapy Network), Ramón y Cajal Program grants RYC-2011-09726 to AS-A and RYC-2009-04703 to SM-F), Marie Curie Career Integration Program grants (FP7-PEOPLE-2011-RG-294262/294096) to AS-A and SM-F; and a ConSEPOC-Comunidad de Madrid grant (S2010/BMD-2542) and Horizon2020 (ERC-2014-CoG-64765 grant to SM-F. This research was partly funded by a European Hematology Association Research Fellowship awarded to AS-A and an International Early Career Scientist Grant from the Howard Hughes Medical Institute to SM-F.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sánchez-Aguilera, A., Méndez-Ferrer, S. The hematopoietic stem-cell niche in health and leukemia. Cell. Mol. Life Sci. 74, 579–590 (2017). https://doi.org/10.1007/s00018-016-2306-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2306-y