
Abstract
Late-onset Fuchs endothelial corneal dystrophy (FECD) is a disease affecting the corneal endothelium (CE), associated with a cytosine-thymine-guanine repeat expansion at the CTG18.1 locus in the transcription factor 4 (TCF4) gene. It is unknown whether CTG18.1 expansions affect global methylation including TCF4 gene in CE or whether global CE methylation changes at advanced age. Using genome-wide DNA methylation array, we investigated methylation in CE from FECD patients with CTG18.1 expansions and studied the methylation in healthy CE at different ages. The most revealing DNA methylation findings were analyzed by gene expression and protein analysis. 3488 CpGs had significantly altered methylation pattern in FECD though no substantial changes were found in TCF4. The most hypermethylated site was in a predicted promoter of aquaporin 1 (AQP1) gene, and the most hypomethylated site was in a predicted promoter of coagulation factor V (F5 for gene, FV for protein). In FECD, AQP1 mRNA expression was variable, while F5 gene expression showed a ~ 23-fold increase. FV protein was present in both healthy and affected CE. Further gene expression analysis of coagulation factors interacting with FV revealed a ~ 34-fold increase of thrombomodulin (THBD). THBD protein was detected only in CE from FECD patients. Additionally, we observed an age-dependent hypomethylation in elderly healthy CE.Thus, tissue-specific genome-wide and gene-specific methylation changes associated with altered gene expression were discovered in FECD. TCF4 pathological methylation in FECD because of CTG18.1 expansion was ruled out.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The corneal endothelium (CE) is a monolayer of cells on the posterior surface of the cornea that has a crucial role in maintaining the cornea in a semi-dry optical transparent state. Fuchs endothelial corneal dystrophy (FECD) is a bilateral disease caused by progressive loss of corneal endothelial cells, which at an advanced stage leads to corneal swelling and vision loss [1]. Hallmark of FECD is thickening of the Descemet’s membrane due to excessive accumulation of extracellular matrix and the presence of guttata; best described as penetrating droplets of extracellular matrix in the CE from the posterior collagenous layer of Descemet’s membrane [2, 3].
Two forms of FECD are known so far, one manifesting at a young age and therefore denoted early-onset FECD, and the second form starting later in life, termed late-onset FECD. Early-onset FECD is associated with mutations in COL8A2 and the disease is often advanced in the fourth decade of life [4,5,6]. Late-onset FECD with corneal guttata observed from the fourth decade in life and onward, was initially associated with an intronic SNP; rs613872 in transcription factor 4 gene (TCF4) [7]. Later, an even stronger association was found to a cytosine-thymine-guanine (CTG)n repeat expansion in an intron of TCF4, known as the CTG18.1 locus and located 43 kb from the original SNP [8]. Today, multiple studies on the TCF4 repeat expansion and FECD have been conducted in several populations, all showing convincing association to FECD with repeat lengths over 40–50 [9,10,11,12,13,14,15,16,17]. Expansion of the CTG18.1 locus in the TCF4 gene makes FECD one of the most common tri-nucleotide repeat disorders along with myotonic dystrophy (DM1 and DM2), Huntington disease (HD), Spinocerebellar ataxia (SCA), Friedreich ataxia (FA) and Fragile X syndrome (FRAXA) [18].
TCF4 is a member of the class I basic-helix-loop-helix (bHLH) family of transcription factors, that bind to the DNA motif called E-box (‘CANNTG’) and modulate transcription. These E-box regulatory sites found in promoters and enhancer elements of numerous genes regulate tissue-specific gene expression [19].
One of the proposed pathogenic mechanisms in FECD is the development of tissue-specific RNA nuclear foci formed from the expanded TCF4 repeat tract. The RNA foci present in CE cells were shown to sequester splicing factors such as Muscleblind Like Splicing Regulator 1 (MBNL1) and Muscleblind Like Splicing Regulator 2 (MBNL2) [20,21,22]. Another proposed mechanism is the repression of certain micro-RNAs that normally suppress the expression of extracellular matrix components in the posterior layer of the cornea, leading to excess of extracellular matrix [23,24,25].
In other trinucleotide repeat disorders, such as DM1, FA and FRAXA, an increase in DNA methylation upstream of the repeat expansion tracts was suggested as a pathogenic mechanism. These diseases are characterized by large expansions (> 100 repeats) in non-coding sequences, and their hypermethylation affects gene expression [26,27,28,29,30,31]. The CTG18.1 locus in TCF4 is also located in non-coding sequence within intron 2 (NM_001083962.2), and the median TCF4 repeat length is close to 100 repeats in the Swedish FECD cohort [17]. A well-known consequence of DNA methylation at gene promoter regions is gene silencing, whereas gene body methylation may be involved in the regulation of gene splicing [32, 33] and affect gene expression through i.a. enhancers [34, 35]. DNA methylation patterns in CE tissue from FECD patients with unknown TCF4 status have been investigated earlier, revealing altered methylation of promoter regions in FECD patients compared to controls [25, 36]. However, the effect of an expanded CTG18.1 locus on methylation in FECD remains unknown. Moreover, global hypomethylation is thought to contribute to age-related diseases such as degenerative joint diseases, cancer and progressive neurodegenerations [37,38,39], but the methylation pattern in the aged CE has never been investigated.
In this study, we performed a genome-wide DNA methylome analysis as well as targeted gene expression analysis in FECD patients and controls to gain deepened knowledge in FECD biology. Also, we specifically investigated whether an expanded CTG18.1 locus affects the TCF4 methylation in CE.
Materials and methods
Patients and controls
In this study approved by the Swedish Ethical Review Authority (2019–01,744), we used specimens consisting of corneal endothelial layer (CE) with Descemet’s membrane and white blood cells (WBCs) drawn from peripheral blood from FECD patients. All FECD CEs were taken during routine corneal transplantation using Descemet Stripping Automated Endothelial Keratoplasty (DSAEK) method [40, 41] and all patients had undergone cataract surgery with phacoemulsification and posterior chamber intraocular lens prior to transplantation. The patients received both oral and written information about the study before transplantation and written informed consent was obtained. CEs from non-FECD corneal donors were used as controls. These healthy human corneas were from deceased individuals who had chosen, when alive, to donate their corneas postmortem, through written consent and according to Swedish law. Three of the donors had undergone cataract surgery when alive. The donated corneas were kept in the Eye Bank at the Tissue Establishment in the University Hospital of Umeå, Sweden. If these healthy donated corneas were not used for transplantation after their collection, they were delivered to the laboratory for research purposes. All human specimens were handled under the guidelines based on the tenets of the Declaration of Helsinki developed by the World Medical Association (2013).
Biological samples
During the DSAEK of included FECD patients, the central 8 mm CE and Descemet’s membrane were removed with a Reversed Sinskey Hook after Trypan blue staining (descemetorhexis) and placed in RNAlater (Invitrogen, Waltham, USA).
The corneal tissues from non-FECD donors (controls) were stored in the Eye Bank in a nutritional solution for an average of 62.8 (± 34.7) days before DNA extraction. The nutritional solution was mixed in 500 ml volume, consisting of GIBCO’s Minimum Essential Medium (MEM) (Thermo Fisher Scientific, Waltham, USA) with 8% GIBCO’s Fetal Bovine Serum (Thermo Fisher Scientific), 100 mg Biklin (Vianex S.A., Athens, Greece) 1.25 mg Fungizone, (Cheplapharm, Greifswald, Germany), 12.5 mM GIBCO’s HEPES (Thermo Fisher Scientific) and 1 g/0.125 g Piperacillin/Tazobactam (Fresenius Kabi AB, Bad Homburg, Germany). The corneal endothelial layer together with its basement membrane was detached from the cornea and placed in RNAlater (Invitrogen, Waltham, USA). The remaining corneal tissue from non-FECD donors was placed into a separate RNAlater vial and used for TCF4 CTG18.1 genotyping. All corneas were washed twice with phosphate-buffered saline (PBS) and frozen at -80 °C on the same day of surgery or upon receiving corneal donor tissue. DNA extracted from CE of 16 FECD patients and eleven non-FECD donors (controls) were used for methylation array analysis (Fig. 1, Online Resource 1, Table S1). RNA extracted from 19 specimens from FECD patients, and seven samples from non-FECD controls were used for gene expression analyses. Both WBC, CE and stroma were used in the gene expression assays, and some samples were paired with the CE used in the methylation analysis (Fig. 1).

Schematic overview of the number of individuals used in the different assays with (a) display of non-FECD controls and (b) display of FECD patients. Overlapping circles represent numbers of paired samples between assays. CE—corneal endothelium, WBC—white blood cells, CTG18.1pos—individuals harboring TCF4 CTG18.1 > 40 repeats, CTG18.1neg—individuals harboring TCF4 CTG18.1 < 40 repeats, CTG18.1? —individuals with unknown TCF4 CTG18.1 repeat length
DNA extractions
Genomic DNA from human CE was extracted using NucleoSpin Tissue XS (Macherey–Nagel, Düren, Germany). The DNA was eluted in 20 µl BE buffer. Because of limited access to CE and the low yield of DNA extracted from CE cells, DNA concentration was only measured in one sample by spectrophotometry with DeNovix DS-11 FX (DeNovix, Wilmington, USA) to get an estimate of the extraction method yield. Genomic DNA from WBC was extracted using a modified standard salting-out-method [42, 43].
Methylation array analysis
The EZ DNA Methylation kit (Zymo Research, Irvine, USA) was applied for bisulfite conversion of genomic DNA from CE tissue according to the protocol, with the recommended temperature profile for Illumina Infinium Methylation assay: 16 cycles of 95 °C for 30 s and 50 °C for 60 min with a final step at 4 °C. The bisulfite-converted DNA was eluted in 12 µl M-elution buffer.
To confirm successful bisulfite conversion, a MethyLight analysis of the ALU gene with ALU-C4 primer probe set was used [44]. Modifications were made to the MethyLight qPCR reaction: 1X TaqMan Universal MasterMix II (no UNG) (Applied Biosystems, Waltham, USA), 0.6 µM ALU-C4 forward primer, 0.6 µM ALU-C4 reverse primer, 0.2 µM ALU probe (6-FAM, NFQ-MGB) and 1 µl of bisulfite converted DNA. The qPCR program was set at 40 cycles instead of 50 on QuantStudio 6 real-time PCR system (Applied Biosystems). DNA extracted from cell line CCRF-CEM (CCL-119™, American Type Culture Collection, Manassas, Virginia, USA) was also bisulfite converted and used as a standard curve in five tenfold serial dilutions ranging from 50 ng to 0.005 ng.
Genome-wide DNA methylation data were obtained by using Human Infinium MethylationEPIC array (Illumina, San Diego, USA). Briefly, 4 µl of speed-vac concentrated bisulfite-converted human CE DNA was used for array analysis according to the manufacturer’s protocol. The array was scanned with a HiScan array reader (Illumina), and the methylation level of each CpG site was determined by calculating the ratio (i.e., the β value) between the intensity of the methylated alleles and the total intensity using the minfi package version 1.32 [45]. The β value ranges from 0 (completely unmethylated) to 1 (fully methylated). Normalization was done with BMIQ version 1.3 [46], and annotation and pre-filtering were done in R version 3.6.2. The pre-filtering steps excluded CpGs with detection P-value > 0.05, CpG probes that aligned to multiple loci in the genome or were located less than 5 bp from a known single nucleotide polymorphism [47], and CpGs probes identified as meQTLs [48, 49]. CpGs with missing values in any of the samples were removed, and X and Y chromosomes were omitted from the analysis to avoid sex-related methylation biases. The genome build used was GRCh37/hg19. The previous dataset (GEO GSE94462) from [36] was also analyzed separately according to our method.
Genotyping and sanger sequencing
The assay to determine the TCF4 CTG18.1 repeat lengths for all samples included in this study (except two samples of CE with corneal stroma) was performed as described earlier [17]. Throughout this study, expanded TCF4 CTG18.1 alleles were defined as consisting of more than 40 (CTG)n repeats.
In addition, the FECD patients analyzed by methylation assay were genotyped with TaqMan® SNP Genotyping Assay (Thermo Fisher Scientific) for two common polymorphisms in coagulation factors. Commercially available assay C_11975250_10 (Thermo Fisher Scientific) was used for targeting rs6025 (chr1:169519049TG > A) known as factor V Leiden, (F5 for gene, FV for protein) and a custom design was used for targeting rs1799963 (chr11:46761055G > A) in coagulation factor 2/prothrombin), F2 (F: 5’- GTGTTTCTAAAACTATGGTTCCCAATAAAAGT-3’, R: 5’- CCATGAATAGCACTGGGAGCATT -3’, Probe VIC mutant: 5’- TCTCAGCGAGCCTC -3’, Probe FAM wildtype: 5’- CTCAGCAAGCCTC -3’).
Sanger sequencing was performed in all FECD patients and non-FECD controls analyzed with methylation array; 79 bps upstream and 150 bps downstream of F5 probe cg13122356 (F: 5’-GGGCAAAGGTCTGATTCACA-3’, R: 5’-GGGGCAGGACAAGTTAGAAC-3’) and 215 bps upstream and 199 bps downstream of AQP1 probe cg03310518 (F: 5’-CTCTCTTTCATGACCTGGGG-3’, R: 5’-TCCTACTTCTCAGAGCCCAG). In short, PCR amplified fragments were cleaned with Exonuclease I (Thermo Fisher Scientific) and sequencing reactions were set up with BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) according to the manufacturer’s protocol. Sequencing products were run on the ABI3500 Dx genetic analyzer (Applied Biosystems) and data were evaluated using Sequencher software version 4.9 (Gene Codes Corporation, Ann Arbor, USA).
RNA extractions
Peripheral blood was collected in whole blood vacutainer tubes with EDTA. WBCs were lysed in a buffer containing 130 mM NH3Cl, 2 mM NH3HCO3 and 0.02% diethylpyrocarbonate (DEPC). Total RNA from WBC was extracted using TRIzol Reagent™ (Invitrogen). For extraction of total RNA from the CE from FECD patients and non-FECD controls, miRNeasy Micro Kit (Qiagen, Hilden, Germany) was used.
cDNA synthesis and gene expression
The reverse transcriptase reaction was done according to the manufacturer’s instructions with SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme (Invitrogen). For each tissue type and each RNA extraction method, a “no reverse transcriptase control” (no RT) was included to verify the absence of genomic DNA contamination in subsequent analysis.
FAM- labeled TaqMan® gene expression assays (Thermo Fisher Scientific) were used to quantify coagulation factor V, F5 mRNA (Hs00914120_m1, targeting NM_000130.4), aquaporin, AQP1 mRNA (Hs01028916_m1, targeting NM_198098.2, NM_001185060.1, NM_001185061.1, NM_001185062.1 and NM_001329872.2), coagulation factor II, prothrombin, F2 mRNA (Hs01011988_m1, targeting NM_000506.4 and NM_001311257.1), coagulation factor II thrombin receptor, F2R mRNA (Hs00169258_m1, targeting NM_001992.4), coagulation factor III, tissue factor, F3 mRNA (Hs01076029_m1 targeting NM_001178096.1 and NM_001993.4), coagulation factor X, F10 mRNA (Hs00984442_m1 targeting NM_000504.3, NM_001312674.1 and NM_001312675.1), protein C, coagulation factor XIV, PROC mRNA (Hs00165584_m1 targeting NM_000312.3) and fibrinogen alpha chain, FGA mRNA (Hs00241027_m1, targeting NM_000508.4 and NM_021871.3) at exon–exon boundaries. Thrombomodulin, THBD mRNA was also assayed (Hs00264920_s1 targeting NM_000361.2); however, it consists of only one exon and therefore exon–exon boundary detection is unachievable.
VIC-labeled probe targeting exon–exon boundaries in RAS-associated protein, RAB7A mRNA (Hs01115139_m1, targeting NM_004637.5) was chosen as an internal reference gene due to its stable expression in 52 human tissues and cell lines [50].
Droplet digital PCR (ddPCR) reactions were run in 20 µl reactions with 1 × ddPCR Supermix for Probes (no dUTP) (Bio-Rad, Hercules, USA), 1 × TaqMan probe (FAM), 1 × TaqMan probe (VIC) and 4 ng cDNA. For droplet generation, Droplet Generation Oil for probes (Bio-Rad) and QX200 Droplet Generator (Bio-Rad) were used. The PCR program was set as follows; initial denature step at 95 °C for 10 min, 40 cycles of; 94 °C for 30 s and 60 °C for 1 min (ramp rate -2 °C/sec), ending with 98 °C for 10 min and 40C infinity. QX200 Droplet Reader (Bio-Rad) was used for droplet detection and the Absolute Quantification (ABS) was used as the detection method. QuantaSoft software version 1.7.4.0917 was used for analysis. For all assays, absolute values of copies/µl were normalized against RAB7A, and the expression of the gene of interest (GoI) was presented as % of ratio GoI/RAB7A. The “no reverse transcriptase control” was run for each tissue type to rule out genomic DNA contamination. All biological samples were run in single technical replicates. To rule out false positives, more than one biological replicate had to have at least one positive droplet or a single positive sample had to have more than one positive droplet.
Fluorescent labeling and imaging of CE tissue
The endothelial layer attached to Descemet's membrane from one donor and two FECD patients was put in a nutrition medium described under the biological samples section.
The tissues were thereafter washed twice with PBS and fixed with 10% formalin neutral buffer for 60 min. Two washing steps with PBS were followed, and the tissues were permeabilized for 30 min at room temperature (RT) with penetration buffer (PBS, 0.2% Triton X-100, 0.3 M Glycine, 20% DMSO). Subsequently, the samples were washed for 40 min (4 × 10 min) with washing buffer (PBS, 0.2% Tween 20, 10 ug/ml heparin) and blocked for 60 min with blocking buffer (PBS, 0.2% Triton X-100, 6% Donkey serum, 10% DMSO). The recipes for penetration buffer, washing buffer and blocking buffer are adopted from Visikol (Visikol, Hampton, USA). Last, the tissues were divided into smaller pieces and were stained overnight with the primary antibodies: Thrombomodulin (1:100, MA5-11,454, Invitrogen) and factor V (1:100, PA5-81,998, Invitrogen). The next day the tissues were washed for 40 min (4 × 10 min) and incubated with secondary antibodies: Alexa fluor 488 (1:400, ab150113, Abcam, Cambridge, United Kingdom) and Alexa Fluor 594 (1:300, A32740, Invitrogen) for three hours at RT, then washed for 30 min (3 × 10 min), incubated 13 min with DAPI (1:3000, Thermo Fisher Scientific), and washed one more time for 10 min. Prior to imaging, the tissues were placed on microscope slides and a few drops of Prolong Gold Antifade Mountant (Invitrogen) were added onto the tissues before placing the coverslips, and the mounted samples were left overnight. Z-stack fluorescent images were acquired using a 40x, 1.4 numerical aperture oil-immersed lens on a Leica SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany). The Z-stacks of the images were combined in a single picture with open sourced Image J software version 2.3.0/1.53f51 [51], with Java version 1.8.0_172 (64 bit), using the maximum intensity projection method.
Statistical analysis and data visualization
Two-sided independent T-test, two-sided Mann–Whitney U and false discovery rate (FDR) with Benjamini-Hochberg [52] were calculated with Python-based open-source software Scipy version 1.5.4 (scipy.org) and Numpy version 1.16.4 (numpy.org) with Python version 3.7.2 in Jupyter Notebook version 6.0.0 with Pandas version 0.24.2 to detect significant differences between FECD patients and non-FECD controls. A CpG site was determined as differently methylated (DM-CpG) if the absolute value of the difference in beta value (Δβ) between the mean of FECD patients and the mean of non-FECD controls was ≥ 0.2 or ≤ -0.2 and if FDR q-value was < 0.05. Pearson’s Chi-squared test (df = 6) was calculated in R version 4.2.1 to compare the distribution of the number of CpG sites in the different target genomic regions (TSS1500, TSS200, 5’UTR, 1st Exon, Gene body, 3’UTR and intergenic) on the Human Infinium MethEPIC850K array in relation to the entire Human Infinium MethEPIC850K array, and to compare it to the distribution of CpGs located in each target genomic region on the array in relation to the DM-CpGs found in FECD.
Individual Fisher's exact tests (region vs non-region) and Bonferroni adjusted P-value were calculated in R version 4.2.1 for each genomic region of the DM-CpGs (TSS1500, TSS200, 5’UTR, 1st Exon, Gene body, 3’UTR and intergenic) and presented as odds ratio (OR, > 1 overrepresented, < 1 underrepresented).
Matplotlib version 3.1.0 (matplotlib.org) was used for plotting gene methylation levels and gene expression levels. Bioinfokit version 1.0.2 was used for producing volcano plots [53]. The open-sourced software CpGtools beta_PCA.py version 1.0.9 was used for principal component analysis [54]. Two-sided Mann–Whitney U was used to calculate the significance in gene expression results.
Results
TCF4 genotyping in FECD patients and non-FECD controls
All FECD patients and non-FECD controls included in this study were tested for CTG18.1 expansion in the TCF4, except for two anonymous non-FECD donor’s corneas used for gene expression analysis in the stroma. In the FECD group, successfully interrogated by methylation array 12 out of total 16 patients were females (75%) and the median age in this group was 72.5 years (IQR 16.5) (Online Resource 1, Table S1). All FECD patients had a TCF4 CTG18.1 expansion with more than 40 repeats and the median repeat length of the expanded allele was 94 repeats (IQR 15.5). In the non-FECD control group analyzed in the methylation assay (n = 11), all had less than 40 TCF4 (CTG)n repeats, and the median CTG18.1 repeat length was 16 (IQR 8.0) for the longest allele (Online Resource 1, Table S1). 55% of the non-FECD methylation controls were females and the median age of the elderly non-FECD methylation controls (≥ 57 years, n = 9) was 78 years (IQR 17.5) while the median age of the younger controls (< 30 years, n = 2) was 27 years (IQR 1), and for the whole group the median was 76 years (IQR 27) (Online Resource 1, Table S1).
Differential methylation analysis in FECD with TCF4 CTG18.1 expansion vs non-FECD controls
Initially, DNA from CE from 17 FECD patients with TCF4 CTG18.1 expansion (n > 40 repeats) and from 11 non-FECD controls (donors) without TCF4 CTG18.1 expansion (n < 40 repeats) were available for analysis with Human Infinium MethylationEPIC array, containing probes for ~ 850 000 CpG sites. However, one of the FECD patients (FECD case 15) was excluded from the downstream analysis due to the poor quality of the results on the methylation array, thus yielding methylation results from 16 FECD patients.
We observed an age-associated hypomethylation among the elderly non-FECD controls (≥ 57 years) compared to the younger non-FECD controls (< 30 years) (Online Resource 1, Fig. S1 and Fig. S2). Therefore, in our further analysis we used only age matched controls, and the two younger non-FECD controls were excluded from the comparison.
After filtering, subset of 686 247 CpG sites remained and was used for further analysis. A principal component analysis of FECD cases (n = 16) and non-FECD donors (n = 9) revealed two subtle clusters, one for FECD cases and one for non-FECD donors (Fig. 2). The mean β-value for each CpG site was calculated, and significant differences in methylation pattern between the FECD (n = 16) and non-FECD control group (n = 9) (Δβ = ± 0.2 with q-value < 0.05) were identified. In total, 3488 CpG sites were denoted as differentially methylated CpG sites (DM-CpGs) in the FECD group (P-value = < 2.2e-16, Chi-square), of which 1983 CpGs were hypomethylated and 1505 CpGs were hypermethylated (Fig. 3 and Online Resource 2) (NCBI GEO accession number GSE198917). The DM-CpG sites in FECD were overrepresented in gene bodies (OR 1.67, P-value = < 0.001), intergenic regions (OR 1.26, P-value = < 0.001) and 3’UTR regions, (OR 1.32, P-value = < 0.001) (Fig. 4), and underrepresented in TSS1500, TSS200 and 1st Exon, with OR of 0.34, 0.18 and 0.08, respectively (P-value = < 0.001). No difference in the distribution of DM-CpGs in 5’UTR was found (OR 0.95) (Fig. 4). No difference in methylation with the threshold of Δβ ± 0.2 was detected in the TCF4 gene (Online Resource 1, Fig. S3) nor 200 kb upstream or downstream of the gene.

Principal component analysis of normalized methylation β-values comprising 686 247 CpG sites in corneal endothelium of FECD patients (red) and non-FECD donors (green) with projection of samples onto the first two principal components (PC) of the methylation β-value measurements

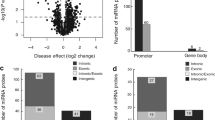
Volcano plot showing statistical significance and delta beta (Δβ) values of individual probes in the corneal endothelium (CE) from FECD patients (TCF4 CTG18.1 > 40 repeats) compared to CE from non-FECD controls TCF4 CTG18.1 < 40 repeats). Selected CpG extremes are displayed with gene names or genomic association. The red dots indicate CpG sites with Δβ values below -0.20 and green dots indicate CpG sites with Δβ values above 0.20 in FECD patients. The horizontal dotted line shows threshold significance of adjusted P-value 0.05, and the vertical dotted lines display thresholds of Δβ ± 0.2

Number of significant (FDR q < 0.05) differentially methylated (DM) CpG sites (Δβ ± 0.2) between FECD patients and age matched non-FECD controls, grouped by targeted genomic region. Grey stacks display hypomethylated DM-CpG sites and black stacks displays hypermethylated DM-CpG sites. Numbers on top of bars indicate odds ratio. *—P-value < 0.001. TSS1500 – 1500 bp from transcription start site, TSS200—200 bp from transcription start site
The CpG site (cg13122356) with the most pronounced decrease in methylation (Δβ = − 0.53) was in the coagulation factor V (F5) gene, (q-value = 0.00817) (Fig. 5a) on chromosome 1, at position g.169554071 within the predicted promoter/enhancer GH01J169584 (genecards.org) /ENSR00000376822 (ensembl.org). In contrast, the CpG site with the maximum increase in methylation (Δβ = + 0.495) matched the probe cg03310518 targeting the aquaporin gene, AQP1 (q-value = 0.00817) (Fig. 5b). This CpG site is located on chromosome 7, at position g.30954522 within the predicted promoter/enhancer GH07J030910 (genecards.org) /ENSR00000820824 (ensembl.org).

Mean methylation level (β) in the corneal endothelium from non-FECD controls (dotted line) and FECD patients (continuous line) in the (a) F5 gene and (b) AQP1 gene. Gene region annotation according to the MANE select and canonical transcripts NM_000130.5 (F5) and NM_198098.4 (AQP1). Vertical lines show standard deviation at each CpG site. Genomic positions are according to genome build GRCh37/hg19
Since all of our FECD cases had undergone cataract surgery prior to DSAEK, we wanted to rule out the DM-CpGs in F5 and AQP1 as a consequence of cataract surgery. Therefore, we compared elderly cataract operated non-FECD controls (n = 3) against elderly non-cataract non-FECD controls (n = 6) and found that Δβ was 0.03 for F5 and 0.04 for AQP1, implying that cataract surgery does not impact F5 and AQP1 methylation. Furthermore, we compared cataract operated non-FECD controls (n = 3) against the cataract operated FECD patients (n = 16) and here Δβ was -0.51 for F5 and + 0.47 for AQP1. These data conclude that cataract surgery is not the cause of the DM-CpGs in F5 and AQP1.
To rule out SNV at or in close proximity to DM probe positions cg13122356 (F5) and cg03310518 (AQP1), all FECD patients and non-FECD controls interrogated with methylation assay were analyzed by Sanger sequencing. No common SNV that could explain the differences in methylation between controls and FECD patients was found.
Forty-three probes targeting 30 different miRNA genes demonstrated a significant level of Δβ value ± 0.2. Nine of these miRNAs, miR-33B, miRNA-3681HG, miR-548A2, miR-548F1, miR-548H4, miR-661, miR-6787, miR-7853 and miR-942 had ≥ 2 CpG sites reaching above the threshold (Online Resource 3, pp 1–5).
Genes known to be associated with FECD (COL8A2, ZEB1, SLC4A11, AGBL1, LAMC1, LOXHD1, KANK4, ATP1B1 and DMPK) [5, 55,56,57,58,59,60] were analyzed for methylation differences between FECD patients and non-FECD controls. None of the CpG sites in these genes reached the significant threshold of Δβ ± 0.20 (Online Resource 3, pp 6–10).
We analyzed the GEO dataset GSE94462 [36] by running the raw data through our pipeline, and we found that probe cg03310518 in AQP1 had a Δβ of + 0.35 in the FECD group. The probe cg13122356 in F5 does not exist on the Illumina 450 K methylation array and could therefore not be analyzed. Also, in our dataset, we analyzed the methylation level in genes for which probes in our filtered data overlap with the top 20 probes found by Khuc et al. [36] (EML3 and MYADML) (Online Resource 3, p11) and Pan et al. [25] (miR199B, miR33B, miR1286, miR1306, miR130A, miR942, miR499, miR184, miR194-2, miR25, miR1471, miR199A1 and miR320D1) (Online Resource 3, pp 12–18), both studies based on the GSE94462 dataset. The probe in EML3 had a Δβ of + 0.22 and the probe in MYADML had Δβ of -0.05 in our dataset (Online Resource 3, p 11). From the top 20 probes of Pan et al. study, only the probe in miR33B reached our threshold of Δβ of ± 0.20 at + 0.25 (Online Resource 3, pp12-18). However, for miR942 in our dataset, two consecutive probes, downstream of the probe listed as top 20 [25], reached our significant threshold and were DM-CpGs in our study (Online Resource 3, p13).
Gene expression of F5 and AQP1
Considering the significant difference in methylation in the predicted promoter/enhancer regions of F5 and AQP1 genes in this study, we examined their mRNA expression. Absolute quantification of targeted genes was performed by ddPCR on RNA extracted from CE from FECD patients (n = 5) and from non-FECD controls (n = 4) (Fig. 6, Table 1). In addition, gene expression was analyzed using RNA extracted from WBC from ten FECD patients with TCF4 CTG18.1 expansion, four FECD patients without TCF4 CTG18.1 expansion and one non-FECD control (Table 1). Thus, FECD diagnosis and TCF4 CTG18.1 genotype were taken into account when gene expression was analyzed in CE and WBC.

mRNA gene expression levels normalized against gene coding for RAS-associated protein (RAB7A) in the corneal endothelium (CE) from FECD patients and non-FECD controls for (a) factor V (F5) gene and (b) aquaporin 1 (AQP1) gene. mRNA gene expression levels normalized against RAB7A in CE from FECD patients, non-FECD controls and corneal grafts containing corneal stroma and CE from non-FECD controls for (c) tissue factor (F3) gene and (d) thrombomodulin (THBD) gene. P-values displayed are a comparison between FECD patients and CE from non-FECD controls only (not stroma). Filled horizontal lines within boxes display means, and dotted lines within boxes display medians
In our experiments, the mRNA expression of F5 gene was ~ 23-fold higher in CE from FECD patients compared to non-FECD controls (P-value = 0.02) (Fig. 6a, Table 1). Moreover, F5 expression in WBC from FECD patients was substantially lower than in FECD CE (Table 1). No significant differences were observed for F5 expression in WBC from FECD patients with and without TCF4 CTG18.1 expansion (Table 1). These data indicate tissue-specific increase of F5 expression in CE from FECD patients.
The differentially methylated AQP1 gene (hypermethylated in CE tissue from FECD patients) has previously been associated with disorders involving imbalance in ocular fluid movement [61], which is also a characteristic feature in FECD. We observed a noteworthy variation although not statistically significant (P-value = 0.71) in AQP1 expression levels in CE from FECD patients compared to CE from non-FECD controls (Fig. 6b, Table 1). AQP1 was barely expressed in WBC from FECD patients with and without TCF4 CTG18.1 expansion and in the non-FECD control without TCF4 repeat expansion (Table 1).
Gene expression of clotting factors and F5, F2 genotyping
Considering that coagulation factor V (FV) is a central player in the coagulation process, involved in numerous interactions with other clotting factors, we investigated mRNA expression of its direct and indirect interacting targets. Expression of the genes coding for coagulation factor II, thrombin (F2), coagulation factor II, thrombin receptor (F2R), coagulation factor III, tissue factor (F3), coagulation factor X (F10), coagulation factor XIV, protein C (PROC), thrombomodulin (THBD) and fibrinogen alpha chain (FGA) was analyzed in the same tissues as for F5 (Table 1). Barely detectable or no expression of F2 was found in CE and WBC from FECD patients and non-FECD controls (Table 1, Online Resource 4). No difference in F2R gene expression was detected between CE from FECD patients and CE from non-FECD controls (Table 1). F2R gene expression in WBC was similar between the three groups investigated (Table 1). Variable levels of F3 expression were detected in CE from FECD patients with a mean value of 61% compared to 3.6% in CE from non-FECD controls (P-value = 0.08) (Fig. 6c, Table 1). No expression of PROC was seen in CE from FECD patients nor in CE from non-FECD controls; however, scarcely detectable levels were seen in WBC in all groups tested (Table 1). High levels of THBD gene expression were seen in CE from FECD patients (mean = 1092%) compared to non-FECD controls (mean = 32%) (P-value = 0.08) (Fig. 6d, Table 1), deducing a ~ 34-fold increase of THBD gene expression in FECD CE. This increase was not seen in WBC since all three groups tested had similar THBD expression levels (mean = ~ 6–9%) (Table 1). No expression of F10 or FGA was detected in CE from FECD patients nor in CE from controls (Table 1).
Since blood clotting occurs extracellularly, we investigated if factor V interactors were expressed in adjacent tissues of the CE. The corneal stroma contains nuclear cells and is the closest tissue of the central CE, with aqueous humor residing posteriorly of the CE. For mRNA expression of F2, F3, F10, PROC, THBD and FGA gene in the corneal stroma, we analyzed corneal grafts from two non-FECD controls containing CE, Descemet’s membrane and about 100 microns of stroma. In these specimens, F3, F10, and THBD was expressed and no expression of F2, PROC nor FGA was found in these samples (Table 1).
“No reverse transcriptase” controls for CE, stroma and WBC were blank when F5, AQP1, F2, F2R, F3, F10, PROC and FGA were tested. However, since THBD is an intronless gene, very low levels of genomic THBD amplification were seen in the CE and stroma. These levels were more than eightfold lower than the lowest THBD expression level from a CE and stroma cDNA sample, and not considered to have a big impact on the gene expression results (Online Resource 4).
In addition, to exclude the presence of the two most common polymorphisms in F5 and F2 genes known to increase blood clotting and possible fibrin accumulation, all FECD patients examined for methylation pattern were genotyped for F5 rs6025 and F2 rs1799963. Only one FECD patient was found to be heterozygous carrier of the F2 rs1799963G > A variant, and it was the same individual (FECD case 15) that was excluded from the analysis due to poor quality of the methylation data.
Protein expression of F5 and THBD
As both F5 and THBD gene expression levels were significantly increased in the CE from FECD patients compared to non-FECD controls, we sought to investigate whether the protein expression also could be altered. For this, we used confocal microscopy with primary antibody against FV and THBD proteins in CE from one non-FECD control and two FECD patients. The longest CTG18.1 of the donor was 18 repeats and both FECD patients had CTG18.1 alleles > 80 repeats. Due to poorly preserved CE morphology of FECD CE, the images of FV and THBD protein analysis were taken from two separate FECD patients (Fig. 7). FV protein was seen in both the FECD patient and the non-FECD control (Fig. 7). For THBD, we found very scarce expression in the non-FECD control, while distinct protein expression of THBD was revealed in the CE from the FECD patient (Fig. 7).

Fluorescent labeling and imaging in human corneal endothelium at 40 × magnification for (a) FV in non-FECD control and (b) FV in FECD patient and c) THBD in non-FECD control and (d) THBD in FECD patient. Nucleus are stained with DAPI (blue) and FV and THBD are shown in red. Due to highly degenerated FECD tissue, the imaging of FV and THBD presented are from two separate FECD patients
Discussion
According to the latest research, Fuchs endothelial corneal dystrophy (FECD), strongly associated with expansion of the CTG18.1 locus in the transcription factor 4 gene, represents one of the most common trinucleotide repeat disorders [8, 18, 62, 63]. In this study we tested the hypothesis of aberrant TCF4 methylation as a pathogenic mechanism, previously shown for trinucleotide repeat disorders such as DM1, FRDA and FRAXA [64]. The inclusion of only FECD patients with TCF4 CTG18.1 expansion (n > 40 repeats) in our study allowed to investigate the relation between the TCF4 CTG18.1 expansion and TCF4 methylation. As a result, no DM-CpG sites were found in the TCF4 gene among FECD patients, nor upstream or downstream of the gene. This ruled out a pathogenic role of TCF4 methylation as a consequence of expanded CTG18.1 locus in FECD.
We also investigated global methylation in the corneal endothelium from FECD patients and non-FECD controls. Multiple DM-CpG sites were found among FECD patients, especially sites in the gene body and intergenic regions. In general, an age-dependent global hypomethylation was seen among the elderly non-FECD controls (Online Resource 1, Fig. S1). This is in line with other studies demonstrating that aging strongly correlates with decreased global methylation and, supposedly contributes to age-related diseases such as neurodegeneration, cancer, and degenerative joint diseases [37,38,39] though methylation changes in CE at advanced age has never been studied. One drawback of our analysis is that only two individuals were considered young (< 30 years), and no reliable statistics could be performed on the data. The global hypomethylation pattern seen in CE from non-FECD elderly donors could however reflect on the pathogenesis of FECD as a late-onset disease.
Results of the global methylation analysis revealed nine miRNAs that had more than 2 DM-CpG sites reaching the significant threshold of Δβ ± 0.2. These miRNAs have been implicated in many different diseases, such as cancer, heart defects, attention deficit, hyperactive disorder, viral infections, and oral ulcers [65,66,67,68,69,70,71,72] without indications of involvement in the FECD pathogenesis.
Based on our global methylation results, we focused on two DM-genes, the most hypermethylated (AQP1) and the most hypomethylated (F5), by conducting gene expression analysis on these two genes. Due to the limited number of cells in CE from FECD patients and non-FECD controls, CE from additional FECD patients and non-FECD controls were collected for these experiments. The AQP1 gene was hypermethylated in the predicted promoter/enhancer region, however, the AQP1 mRNA expression in CE from FECD patients was fluctuating, with the mean value higher in this group compared to controls. Thus, our results did not support earlier reported decreased protein levels of AQP1 in CE from FECD patients [73, 74]. AQP1 is a water channel protein that facilitates water movement across the corneal endothelium to and from the corneal stroma [61], and the shifting AQP1 expression seen in our study may be a compensatory mechanism of the cells, to keep water movement stable when the surrounding cells have been lost.
The most striking finding in the global methylation analysis was hypomethylation of F5 gene, which prompted us to examine the F5 mRNA expression. We demonstrated a ~ 23-fold increase of F5 gene expression in CE from FECD patients compared to non-FECD controls, which was in line with the loss of methylation in the predicted enhancer/promoter at position chr1:169,554,071 (GRCh37/hg19) in the FECD group. We also found that protein FV was present in both a non-FECD control and a FECD patient, though quantitative analysis was impossible to conduct. If FV levels are sustained while the mRNA is increased in FECD, there may be some sort of translational buffering that regulates FV quantity in the cells [75]. If FV levels are increased, it could contribute to FECD pathogenesis and, possibly, to the thickening of Descemet's membrane since FV is a secreted extracellular protein.
F5 gene encodes for coagulation factor V (F5 for gene, FV for protein), a protein mainly produced in the liver. FV is a participant of the blood clotting process characterized by both anticoagulant and procoagulant activities (Online Resource 1, Fig. S4) [76]. In addition to being part of the blood clotting cascade, prothrombin, the direct downstream target of FV can activate the prothrombin receptor; protease activated receptor 1 (PAR-1, F2R) [77]. PAR-1 signaling leads to increased Ca2+ levels and the phosphorylation of myosin light chain II (MLC) with subsequent contraction of the actin cytoskeleton, which results in gaps and breakdown of barrier integrity in vascular endothelial cells (Online Resource 1, Fig. S4) [78, 79].
FV’s involvement in blood clotting and possibly in PAR-1 signaling, impelled us to investigate the mRNA expression of six coagulation factors (F2, F3, F10, FGA, PROC and THBD) and F2R in the CE and stroma (not F2R in stroma), that are downstream or upstream targets in FV signaling [76, 77, 80].
In CE, we found no significant difference in the expression of F2, F2R, F10, PROC or FGA between FECD patients and non-FECD controls. In fact, we found no expression of F10, PROC nor FGA in CE in either group. F3 showed an increase in expression in CE from FECD patients, although the expression was very shifting among FECD patients (4.2 to 144%, n = 5) compared to controls (3.5 to 3.7%, n = 2). The negative drawback of this data is that only two controls were used in the experiment and the numbers could therefore mirror mere coincidence and not a true difference. Moreover, no DM-CpG site was found in the F3 gene that could explain the difference in gene expression (Online Resource 1, Fig. S5a). A more interesting finding was the distinctive ~ 34-fold increase in THBD expression in CE from FECD cases compared to non-FECD controls. The elevated level of THBD expression could not be explained by hypomethylation, since no DM-CpG sites were found in the THBD gene (Online Resource 1, Fig. S5b). This increase may reflect an unknown (to our knowledge) positive regulation of THBD expression, as a consequence of elevated F5 mRNA expression, possibly to maintain FV inactivated in the CE. However, since PROC was not expressed in the CE, the direct link between THBD and FV gene regulation remains to be studied. THBD protein was highly expressed in CE from a FECD patient and scarcely present in the non-FECD control. Again, this significant difference may contribute to FECD pathogenesis and possibly to the thickening of Descemet’s membrane since THBD is a membrane-bound protein that is shed from the cell’s surface during wound healing [81]. However, these conclusions drawn from an experiment on a single individual should be interpreted cautiously and require further investigation.
In samples containing both corneal stroma and CE from non-FECD controls, we observed a consistent higher expression of F3 (13%) and F10 (1%) than in CE-only from non-FECD controls (4 and 0%, respectively). However, given the small number of analyzed samples (n = 2 versus n = 4), this observation should also be interpreted with some uncertainty. THBD had variable expression in both non-FECD groups, ranging from 2 to 16% for CE with corneal stroma and 14–49% for CE only. In WBC, no difference in gene expression was seen for any gene expression assay in any group, implying that the gene expression pattern for AQP1, F5 and THBD in CE is tissue specific.
Both factor V mRNA and prothrombin mRNA and protein have previously been shown to be expressed in human CE [82]. Moreover, increased F5 gene expression, placing as one of the top 20 most upregulated genes, was shown in CE with TCF4 CTG18.1 expansion, regardless of FECD status [83]. Furthermore, RNA sequencing performed on CE from individuals with and without FECD, all carriers of TCF4 CTG18.1 expansion did not reveal any differences in F5 gene expression between the two groups [84]. To answer the question of whether CTG18.1 expansion might influence F5 methylation resulting in mRNA overexpression, the inclusion of FECD patients without TCF4 CTG18.1 expansion is certainly warranted for future DNA methylation studies.
It is known that prothrombin and fibrinogen are constituents of the aqueous humor in the eye, the liquid formed from filtered blood in the ciliary body [85, 86]. We have, however, not investigated the presence of factor V interacting proteins extracellularly of the CE. In regard to our findings, the aqueous humor and corneal stroma could be valuable components to study blood clotting proteins in FECD patients.
Lack of the TCF4 genotype and different filtering methods in previous FECD methylation studies could be one of the reasons why there is not much agreement in our results compared to Khuc et al. [36] and Pan et al. [25] who used Illumina 450 K methylation array on samples with unknown TCF4 CTG18.1 genotype. We applied a different method to calculate significant changes between FECD and controls; β-values versus M-values and an absolute difference versus linear model, which could also bring the dissimilarities. Moreover, 90% (18 out of 20) of Khuc et al. [36] top 20 CpG sites and 35% (7 out of 20) of Pan et al. [25] top 20 CpG sites were filtered out in our pre-analysis as either meQTL, SNP, aligned to multiple loci or background noise. We did however analyzed the previous dataset (GEO GSE94462) according to our method, and found that the same probe in AQP1 corroborated with our results. The probe cg13122356 in F5 does not exist on the Illumina 450 K methylation array and could therefore not be analyzed.
We also acknowledge that it is challenging to filter out all possible CpG sites that may be affected by meQTLs. However, in order to study non-genetic epigenetic changes, we have applied best practice to filter out regions denoted as meQTL with current knowledge.
Conclusions
In conclusion, significant global methylation changes in corneal endothelium from FECD patients with TCF4 CTG18.1 repeat expansion are an indisputable fact. Hypomethylation of coagulation factor V and the significant increase of its and thrombomodulin’s mRNA expression might reveal novel mechanisms in the appearance and/or progress of FECD. At the same time, currently unknown functions of factor V, besides its important role in the coagulation cascade, can be discovered. Notably, that in FECD no methylation changes of CpGs located in the key gene TCF4 were detected, which rules out hypermethylation as a pathogenic disease mechanism.
Data availability
The datasets generated and/or analyzed during the current study are available in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) repository at https://www.ncbi.nlm.nih.gov/geo under the accession number: GSE198917.
References
Tuft SJ, Coster DJ (1990) The corneal endothelium. Eye 4(Pt 3):389–424. https://doi.org/10.1038/eye.1990.53
Xia D, Zhang S, Nielsen E et al (2016) The ultrastructures and mechanical properties of the descement’s membrane in fuchs endothelial corneal dystrophy. Sci Rep 6:23096. https://doi.org/10.1038/srep23096
Klintworth GK (2009) Corneal dystrophies. Orphanet J Rare Dis 4:7. https://doi.org/10.1186/1750-1172-4-7
Mok JW, Kim HS, Joo CK (2009) Q455V mutation in COL8A2 is associated with Fuchs’ corneal dystrophy in Korean patients. Eye 23:895–903. https://doi.org/10.1038/eye.2008.116
Gottsch JD, Sundin OH, Liu SH et al (2005) Inheritance of a novel COL8A2 mutation defines a distinct early-onset subtype of fuchs corneal dystrophy. Invest Ophthalmol Vis Sci 46:1934–1939. https://doi.org/10.1167/iovs.04-0937
Biswas S, Munier FL, Yardley J et al (2001) Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet 10:2415–2423. https://doi.org/10.1093/hmg/10.21.2415
Baratz KH, Tosakulwong N, Ryu E et al (2010) E2–2 protein and Fuchs’s corneal dystrophy. N Engl J Med 363:1016–1024. https://doi.org/10.1056/NEJMoa1007064
Wiben ED, Aleff RA, Tosakulwong N et al (2012) A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy. PLoS One. https://doi.org/10.1371/journal.pone.0049083
Foja S, Luther M, Hoffmann K et al (2017) CTG18.1 repeat expansion may reduce TCF4 gene expression in corneal endothelial cells of German patients with Fuchs’ dystrophy. Graefes Arch Clin Exp Ophthalmol 255:1621–1631. https://doi.org/10.1007/s00417-017-3697-7
Rao BS, Tharigopala A, Rachapalli SR et al (2017) Association of polymorphisms in the intron of TCF4 gene to late-onset fuchs endothelial corneal dystrophy: an Indian cohort study. Indian J Ophthalmol 65:931–935. https://doi.org/10.4103/ijo.IJO_191_17
Nanda GG, Padhy B, Samal S et al (2014) Genetic association of TCF4 intronic polymorphisms, CTG18.1 and rs17089887, with Fuchs’ endothelial corneal dystrophy in an Indian population. Invest Ophthalmol Vis Sci 55:7674–7680. https://doi.org/10.1167/iovs.14-15297
Okumura N, Hayashi R, Nakano M et al (2019) Effect of trinucleotide repeat expansion on the expression of TCF4 mRNA in Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci 60:779–786. https://doi.org/10.1167/iovs.18-25760
Kuot A, Hewitt AW, Snibson GR et al (2017) TGC repeat expansion in the TCF4 gene increases the risk of Fuchs’ endothelial corneal dystrophy in Australian cases. PLoS One. https://doi.org/10.1371/journal.pone.0183719
Okumura N, Puangsricharern V, Jindasak R et al (2020) Trinucleotide repeat expansion in the transcription factor 4 (TCF4) gene in Thai patients with fuchs endothelial corneal dystrophy. Eye 34:880–885. https://doi.org/10.1038/s41433-019-0595-8
Skorodumova LO, Belodedova AV, Antonova OP et al (2018) CTG18.1 expansion is the best classifier of late-onset Fuchs’ Corneal dystrophy among 10 biomarkers in a cohort from the European part of Russia. Invest Ophthalmol Vis Sci 59:4748–4754. https://doi.org/10.1167/iovs.18-24590
Xing C, Gong X, Hussain I et al (2014) Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs’ corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci 55:7073–7078. https://doi.org/10.1167/iovs.14-15390
Viberg A, Westin IM, Golovleva I, Bystrom B (2021) TCF4 trinucleotide repeat expansion in Swedish cases with Fuchs’ endothelial corneal dystrophy. Acta Ophthalmol. https://doi.org/10.1111/aos.15032
Ramakrishnan S, Gupta V (2022) Trinucleotide Repeat Disorders. In StatPearls Publishing, Treasure Island
Massari ME, Murre C (2000) Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 20:429–440. https://doi.org/10.1128/MCB.20.2.429-440.2000
Mootha VV, Hussain I, Cunnusamy K et al (2015) TCF4 triplet repeat expansion and nuclear RNA foci in Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci 56:2003–2011. https://doi.org/10.1167/iovs.14-16222
Hu J, Rong Z, Gong X et al (2018) Oligonucleotides targeting TCF4 triplet repeat expansion inhibit RNA foci and mis-splicing in Fuchs’ dystrophy. Hum Mol Genet 27:1015–1026. https://doi.org/10.1093/hmg/ddy018
Zarouchlioti C, Sanchez-Pintado B, Hafford Tear NJ et al (2018) Antisense therapy for a common corneal dystrophy ameliorates TCF4 repeat expansion-mediated toxicity. Am J Hum Genet 102:528–539. https://doi.org/10.1016/j.ajhg.2018.02.010
Matthaei M, Hu J, Kallay L et al (2014) Endothelial cell microRNA expression in human late-onset Fuchs’ dystrophy. Invest Ophthalmol Vis Sci 55:216–225. https://doi.org/10.1167/iovs.13-12689
Toyono T, Usui T, Villarreal G Jr et al (2016) MicroRNA-29b overexpression decreases extracellular matrix mRNA and protein production in human corneal endothelial cells. Cornea 35:1466–1470. https://doi.org/10.1097/ICO.0000000000000954
Pan P, Weisenberger DJ, Zheng S et al (2019) Aberrant DNA methylation of miRNAs in Fuchs endothelial corneal dystrophy. Sci Rep 9:16385. https://doi.org/10.1038/s41598-019-52727-z
Sutcliffe JS, Nelson DL, Zhang F et al (1992) DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1:397–400. https://doi.org/10.1093/hmg/1.6.397
Verkerk AJ, Pieretti M, Sutcliffe JS et al (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65:905–914. https://doi.org/10.1016/0092-8674(91)90397-h
Brook JD, McCurrach ME, Harley HG et al (1992) Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 68:799–808. https://doi.org/10.1016/0092-8674(92)90154-5
Klesert TR, Otten AD, Bird TD, Tapscott SJ (1997) Trinucleotide repeat expansion at the myotonic dystrophy locus reduces expression of DMAHP. Nat Genet 16:402–406. https://doi.org/10.1038/ng0897-402
Alwazzan M, Newman E, Hamshere MG, Brook JD (1999) Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum Mol Genet 8:1491–1497. https://doi.org/10.1093/hmg/8.8.1491
Eriksson M, Hedberg B, Carey N, Ansved T (2001) Decreased DMPK transcript levels in myotonic dystrophy 1 type IIA muscle fibers. Biochem Biophys Res Commun 286:1177–1182. https://doi.org/10.1006/bbrc.2001.5516
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13:484–492. https://doi.org/10.1038/nrg3230
Bird AP, Wolffe AP (1999) Methylation-induced repression–belts, braces, and chromatin. Cell 99:451–454. https://doi.org/10.1016/s0092-8674(00)81532-9
Weigel C, Chaisaingmongkol J, Assenov Y et al (2019) DNA methylation at an enhancer of the three prime repair exonuclease 2 gene (TREX2) is linked to gene expression and survival in laryngeal cancer. Clin Epigenetics 11:67. https://doi.org/10.1186/s13148-019-0666-5
Zhou S, Shen Y, Zheng M et al (2017) DNA methylation of METTL7A gene body regulates its transcriptional level in thyroid cancer. Oncotarget. https://doi.org/10.18632/oncotarget.16147
Khuc E, Bainer R, Wolf M et al (2017) Comprehensive characterization of DNA methylation changes in Fuchs endothelial corneal dystrophy. PLoS ONE 12:e0175112
Johnson AA, Akman K, Calimport SRG et al (2012) The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res 15:483–494. https://doi.org/10.1089/rej.2012.1324
Noroozi R, Ghafouri-Fard S, Pisarek A et al (2021) DNA methylation-based age clocks: from age prediction to age reversion. Ageing Res Rev. https://doi.org/10.1016/j.arr.2021.101314
Xiao F-H, Kong Q-P, Perry B, He Y-H (2016) Progress on the role of DNA methylation in aging and longevity. Brief Funct Genomics 15:454–459. https://doi.org/10.1093/bfgp/elw009
Boynton GE, Woodward MA (2015) Evolving techniques in corneal transplantation. Curr Surg Rep. https://doi.org/10.1007/s40137-014-0079-5
Fuest M, Ang M, Htoon HM et al (2017) Long-term visual outcomes comparing descemet stripping automated endothelial keratoplasty and penetrating keratoplasty. Am J Ophthalmol 182:62–71. https://doi.org/10.1016/j.ajo.2017.07.014
Maniatis T, Sambrook FE (1982) JK (1983) Molecular cloning. a laboratory manual by T Maniatis, E F Fritsch and J Sambrook. pp 545. Cold Spring Harbor Laboratory, New York. 1982. $48 ISBN 0-87969-136-0. Biochem Educ 11:82–82. https://doi.org/10.1016/0307-4412(83)90068-7
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215–1215
Weisenberger DJ, Campan M, Long TI et al (2005) Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res 33:6823–6836. https://doi.org/10.1093/nar/gki987
Aryee MJ, Jaffe AE, Corrada-Bravo H et al (2014) Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369. https://doi.org/10.1093/bioinformatics/btu049
Teschendorff AE, Marabita F, Lechner M et al (2013) A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29:189–196. https://doi.org/10.1093/bioinformatics/bts680
Zhou W, Laird PW, Shen H (2017) Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw967
Gaunt TR, Shihab HA, Hemani G, et al (2016) Systematic identification of genetic influences on methylation across the human life course. Genome Biology 17
McClay JL, Shabalin AA, Dozmorov MG et al (2015) High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol 16:291. https://doi.org/10.1186/s13059-015-0842-7
Hounkpe BW, Chenou F, de Lima F, De Paula EV (2021) HRT Atlas v1.0 database: redefining human and mouse housekeeping genes and candidate reference transcripts by mining massive RNA-seq datasets. Nucleic Acids Res 49:D947–D955
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. https://doi.org/10.1038/nmeth.2089
Martins FP (2017) FDR.py. GitHub Accessed 22 Oct 2021:https://github.com/CoBiG2/cobig_misc_scripts/blob/master/FDR.py
Bedre R (2020) reneshbedre/bioinfokit: Bioinformatics data analysis and visualization toolkit. Zenodo March 5: https://doi.org/10.5281/zenodo.3698145
Wei T, Nie J, Larson NB et al (2021) CpGtools: a python package for DNA methylation analysis. Bioinformatics 37:1598–1599. https://doi.org/10.1093/bioinformatics/btz916
Vithana EN, Morgan PE, Ramprasad V et al (2008) SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet 17:656–666. https://doi.org/10.1093/hmg/ddm337
Riazuddin SA, Vasanth S, Katsanis N, Gottsch JD (2013) Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am J Hum Genet 93:758–764. https://doi.org/10.1016/j.ajhg.2013.08.010
Riazuddin SA, Zaghloul NA, Al-Saif A et al (2010) Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet 86:45–53. https://doi.org/10.1016/j.ajhg.2009.12.001
Riazuddin SA, Parker DS, McGlumphy EJ et al (2012) Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am J Hum Genet 90:533–539. https://doi.org/10.1016/j.ajhg.2012.01.013
Mootha VV, Hansen B, Rong Z et al (2017) Fuchs’ endothelial corneal dystrophy and RNA foci in patients with myotonic dystrophy. Invest Ophthalmol Vis Sci 58:4579–4585. https://doi.org/10.1167/iovs.17-22350
Afshari NA, Igo RP Jr, Morris NJ et al (2017) Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat Commun 8:14898. https://doi.org/10.1038/ncomms14898
Schey KL, Wang Z, Wenke L (2014) Aquaporins in the eye: expression, function, and roles in ocular disease. Biochim Biophys Acta 1840:1513–1523. https://doi.org/10.1016/j.bbagen.2013.10.037
Krachmer JH, Purcell JJ Jr, Young CW, Bucher KD (1978) Corneal endothelial dystrophy. a study of 64 families. Arch Ophthalmol 96:2036–2039. https://doi.org/10.1001/archopht.1978.03910060424004
Zoega GM, Fujisawa A, Sasaki H et al (2006) Prevalence and risk factors for cornea guttata in the reykjavik eye study. Ophthalmology 113:565–569
Pook MA (2012) DNA methylation and trinucleotide repeat expansion diseases. DNA Methylation—From Genomics to Technology
Gong C, Sun S, Liu B et al (2017) Identification of potential therapeutic target genes, key miRNAs and mechanisms in oral lichen planus by bioinformatics analysis. Arch Oral Biol 78:122–128. https://doi.org/10.1016/j.archoralbio.2017.02.013
Kang S-H, Choi J-S (2019) MicroRNA-661 upregulation in myelodysplastic syndromes induces apoptosis through p53 activation and associates with decreased overall survival. Leuk Lymphoma 60:2779–2786. https://doi.org/10.1080/10428194.2019.1608528
Yang X, Zhao C, Bamunuarachchi G et al (2019) miR-193b represses influenza a virus infection by inhibiting Wnt/β-catenin signalling. Cell Microbiol. https://doi.org/10.1111/cmi.13001
Luo Y, Wang Q, Teng L et al (2020) LncRNA DANCR promotes proliferation and metastasis in pancreatic cancer by regulating miRNA-33b. FEBS Open Bio 10:18–27. https://doi.org/10.1002/2211-5463.12732
Radhakrishna U, Albayrak S, Zafra R et al (2019) Placental epigenetics for evaluation of fetal congenital heart defects: ventricular septal defect (VSD). PLoS One. https://doi.org/10.1371/journal.pone.0200229
Coskun S, Karadag M, Gokcen C, Oztuzcu S (2021) miR-132 and miR-942 expression levels in children with attention deficit and hyperactivity disorder: a controlled study. Clin Psychopharmacol Neurosci 19:262–268. https://doi.org/10.9758/cpn.2021.19.2.262
Niu R, Xiao X, Liu B et al (2019) Retraction Note: Inhibition of airway inflammation in a cockroach allergen model of asthma by agonists of miRNA-33b. Sci Rep 9:16790. https://doi.org/10.1038/s41598-019-53536-0
Wilkins OM, Titus AJ, Gui J et al (2017) Genome-scale identification of microRNA-related SNPs associated with risk of head and neck squamous cell carcinoma. Carcinogenesis 38:986–993
Macnamara E, Sams GW, Smith K et al (2004) Aquaporin-1 expression is decreased in human and mouse corneal endothelial dysfunction. Mol Vis 10:51–56
Kenney MC, Atilano SR, Zorapapel N et al (2004) Altered expression of aquaporins in bullous keratopathy and Fuchs’ dystrophy corneas. J Histochem Cytochem 52:1341–1350. https://doi.org/10.1177/002215540405201010
Kusnadi EP, Timpone C, Topisirovic I et al (2022) Regulation of gene expression via translational buffering. Biochim Biophys Acta Mol Cell Res. https://doi.org/10.1016/j.bbamcr.2021.119140
Lam W, Moosavi L (2021) Physiology, Factor V. In: StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC Treasure Island (FL)
Couhglin S (1999) How the protease thrombin talks to cells. PNAS 96(20):11023–11027. https://doi.org/10.1073/pnas.96.20.11023
Garcia JG, Davis HW, Patterson CE (1995) Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol 163:510–522. https://doi.org/10.1002/jcp.1041630311
Vouret-Craviari V, Boquet P, Pouysségur J, Van Obberghen-Schilling E (1998) Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell 9:2639–2653. https://doi.org/10.1091/mbc.9.9.2639
Conway EM (2012) Thrombomodulin and its role in inflammation. Seminars in Immunopathology 34:107–125
Cheng T-L, Wu Y-T, Lin H-Y et al (2011) Functions of rhomboid family protease RHBDL2 and thrombomodulin in wound healing. J Invest Dermatol 131:2486–2494. https://doi.org/10.1038/jid.2011.230
Ayala A, Warejcka DJ, Olague-Marchan M, Twining SS (2007) Corneal activation of prothrombin to form thrombin, independent of vascular injury. Invest Ophthalmol Vis Sci 48:134–143. https://doi.org/10.1167/iovs.06-0339
Chu Y, Hu J, Liang H et al (2020) Analyzing pre-symptomatic tissue to gain insights into the molecular and mechanistic origins of late-onset degenerative trinucleotide repeat disease. Nucleic Acids Res 48:6740–6758. https://doi.org/10.1093/nar/gkaa422
Wieben ED, Baratz KH, Aleff RA et al (2019) Gene expression and missplicing in the corneal endothelium of patients with a TCF4 trinucleotide repeat expansion without Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci 60:3636–3643. https://doi.org/10.1167/iovs.19-27689
Kodeboyina SK, Lee TJ, Churchwell L et al (2020) The constitutive proteome of human aqueous humor and race specific alterations. Proteomes. https://doi.org/10.3390/proteomes8040034
Kliuchnikova AA, Samokhina NI, Ilina IY et al (2016) Human aqueous humor proteome in cataract, glaucoma, and pseudoexfoliation syndrome. Proteomics 16:1938–1946. https://doi.org/10.1002/pmic.201500423
Acknowledgements
We acknowledge Mona M Lindström, Biomedical Scientist, Eye Bank and Tissue Establishment, University Hospital of Umeå, Sweden, for help with corneal donor tissue and Ludvig Backman, Associate Professor at the Department of Integrative Medical Biology, Umea University, for advices regarding immunofluorescence assay. We would also like to acknowledge Nina Norgren at the National Bioinformatics Infrastructure Sweden at SciLifeLab for bioinformatics advice. We are grateful for the opportunity to use digital droplet PCR resources at Translational Research Center (TRC) at Umeå University and thankful for all help in microscopy at the Biochemical Imaging Center at Umeå University and the National Microscopy Infrastructure, NMI (VR-RFI 2016-00968).
Funding
Open access funding provided by Umea University. This research was funded by regional agreement between Umeå University and Västerbotten County Council on cooperation in the field of Medicine, Odontology and Health (ALF) (BB, IG), by Ögonfonden (Gothenburg, Sweden) (IG) and by Kronprinsessan Margaretas Arbetsnämnd för Synskadade (Valdemarsvik, Sweden) (BB, IG). The study was also supported by the Medical Faculty of Umeå University (IG, SD) and the Kempe Foundation (SD, IMW).
Author information
Authors and Affiliations
Contributions
AV and BB recruited all participants to the study and performed all clinical examinations and contributed to the manuscript. PO performed the wet lab for the methylation array experiments, including bisulfite conversion and participated in writing the manuscript. ML performed the quality analysis, most of the filtering of the methylation data, statistics and participated in writing the manuscript. IMW did filtering, statistical analysis on the methylation data, performed all wet labs regarding nucleic extraction, genotyping and gene expression experiments and created all the figures in the manuscript and was a major contributor to the conceptualization of the project, interpreting all the data and in writing the manuscript. AG performed the wet lab for the immunofluorescence experiments and confocal imaging and participated in writing the manuscript. SD contributed to the methodology and conceptualization of the methylation experiments and writing of the manuscript. IG was a major contributor on deciding the methodologies used, conceptualization, project administration, supervision and writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests. The authors have no relevant financial or non-financial interests to disclose.
Ethical approval and consent to participate
The study was approved by the Swedish Ethical Review Authority (2019–01744) and all human tissues were handled under the guidelines based on the tenets of the Declaration of Helsinki developed by the World Medical Association (2013). The participating patients received both oral and written information about the study, and written informed consent was obtained.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Westin, I.M., Landfors, M., Giannopoulos, A. et al. DNA methylation changes and increased mRNA expression of coagulation proteins, factor V and thrombomodulin in Fuchs endothelial corneal dystrophy. Cell. Mol. Life Sci. 80, 62 (2023). https://doi.org/10.1007/s00018-023-04714-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-023-04714-x