
Abstract
Septic shock is associated with life-threatening vasodilation and hypotension. To cause vasodilation, vascular endothelium may release nitric oxide (NO), prostacyclin (PGI2), and the elusive endothelium-derived hyperpolarizing factor (EDHF). Although NO is critical in controlling vascular tone, inhibiting NO in septic shock does not improve outcome, on the contrary, precipitating the search for alternative therapeutic targets. Using a hyperacute tumor necrosis factor (TNF)-induced shock model in mice, we found that shock can develop independently of the known vasodilators NO, cGMP, PGI2, or epoxyeicosatrienoic acids. However, the antioxidant tempol efficiently prevented hypotension, bradycardia, hypothermia, and mortality, indicating the decisive involvement of reactive oxygen species (ROS) in these phenomena. Also, in classical TNF or lipopolysaccharide-induced shock models, tempol protected significantly. Experiments with (cell-permeable) superoxide dismutase or catalase, N-acetylcysteine and apocynin suggest that the ROS-dependent shock depends on intracellular \( ^\bullet {\hbox{OH}} \) radicals. Potassium channels activated by ATP (KATP) or calcium (KCa) are important mediators of vascular relaxation. While NO and PGI2-induced vasodilation involves KATP and large-conductance BKCa channels, small-conductance SKCa channels mediate vasodilation induced by EDHF. Interestingly, also SKCa inhibition completely prevented the ROS-dependent shock. Our data thus indicate that intracellular \( ^\bullet {\hbox{OH}} \) and SKCa channels represent interesting new therapeutic targets for inflammatory shock. Moreover, they may also explain why antioxidants other than tempol fail to provide survival benefit during shock.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Septic shock has become the main cause of death in intensive care units [1]. In the first week, refractory hypotension is the leading pathogenic feature underlying mortality; later deaths are mainly due to multiple organ failure resulting from hypoxia, cytotoxicity, mitochondrial dysfunction, and prolonged hypotension. Hypotension is caused by profound vasodilation due to the endothelial release of the vasodilators nitric oxide (NO), prostacyclin (PGI2), and/or endothelium-derived hyperpolarizing factor (EDHF), as well as decreased production of and refractoriness to vasoconstrictors and vasopressors [2, 3]. EDHF seems to be particularly important in the microcirculation, where systemic vascular resistance is regulated. Despite the on-going dispute over its identity, it is generally accepted that small-conductance Ca2+-activated K+ channels (SKCa) are critical for EDHF-dependent vasodilation [3–5]. NO can be produced by three different NO synthases (NOS); of which the inducible isoform iNOS (NOS2) is responsible for excessive and prolonged NO production following inflammation. Nevertheless, NO derived from the constitutive endothelial NOS isoform (eNOS and NOS3) may also mediate hypotension in both endotoxin-induced [6] and anaphylactic shock models [7, 8]. Besides modulating the contractile apparatus and intracellular calcium flux in vascular smooth muscle cells, NO also influences membrane potential and vascular tone via activating ATP-dependent (KATP) and large-conductance Ca2+-activated K+ channels (BKCa) [8, 9]. In septic shock, NO plays a critical but controversial role. Although inhibitors of NOS can improve hemodynamics, they also increase toxicity [10], and a phase III clinical trial had to be prematurely terminated because of increased mortality [11]. While part of this paradox might be explained by protective properties of NO such as its antioxidative effects [12], there is also growing evidence that the pathogenesis of inflammation-associated shock involves oxidative stress next to nitrosative stress.
The sequential donation of electrons to oxygen generates reactive oxygen species (ROS), which may cause oxidative damage at high concentrations, but whose signaling capacities and regulatory functions are becoming more and more evident as well. Donation of one electron to oxygen results in the formation of superoxide radicals \( \left( {{{\hbox{O}}_2}^{ \bullet - }} \right) \). Donation of a second electron generates peroxide, which then undergoes protonation to hydrogen peroxide (H2O2). Donation of a third electron, such as occurs in the Fenton reaction mediated by free Fe2+, results in the production of hydroxyl radicals \( \left( {^\bullet {\hbox{OH}}} \right) \). On the whole, \( {{\hbox{O}}_2}^{ \bullet - } \) is believed to contribute to cardiovascular pathologies such as hypertension and atherosclerosis because it reduces the bioavailability and the effects of NO [13, 14]. To the best of our knowledge, the contribution of various endogenous ROS \( \left( {{{\hbox{O}}_2}^{ \bullet - }{,}\;{{\hbox{H}}_2}{{\hbox{O}}_2}{,}{\;^\bullet }{\hbox{OH}}} \right) \) to inflammation-induced hypotension has not been extensively studied yet. The evaluation of the involvement of ROS in inflammatory hypotension in vivo is a complicated matter, given the vast amounts of NO produced following inflammation-induced iNOS transcription and the highly interactive chemistry of reactive oxygen and nitrogen species [15].
This study was therefore conducted as a proof-of-principle, to evaluate whether ROS can cause inflammatory hypotension and shock in vivo. For this purpose, we used a rapid caspase-independent tumor necrosis factor (TNF)-induced murine shock model that starts to develop already before systemic NO production occurs, and that is accompanied by excessive oxidative stress [16]. Using this model, we found that ROS can indeed cause hypotension in inflammatory shock, and that Ca2+-dependent small-conductance SKCa channels are the only K+ channels that play a prominent role in this ROS-dependent shock.
Materials and methods
Mice
Female C57BL/6J were from Janvier (France); gp91phox−/−, 15-lipoxygenase−/−, iNOS−/− [17], and eNOS−/− [18] mice on a C57BL/6J background were from The Jackson Laboratory (Bar Harbor, ME, USA). To obtain double-deficient ixeNOS− −/− − animals, we crossed iNOS−/− with eNOS−/− mice and verified the knockout by polymerase chain reaction. Mice were housed in temperature-controlled, air-conditioned facilities with 14–10-h light/dark cycles and food and water ad libitum, and used at 8–12 weeks. All experiments were approved by the animal ethics committees of Ghent University, Belgium, and Maastricht University, The Netherlands.
Cytokines, reagents, and injections
Recombinant mouse TNF was produced in and purified from Escherichia coli; lipopolysaccharide (LPS) content was <0.02 ng/mg (chromogenic Limulus amoebocyte lysate assay, Kabivitrium, Sweden). TNF or ultrapure E. coli LPS (Invivogen, serotype 0111:B4) were injected i.v. in LPS-free phosphate buffered saline (PBS). The TNF LD100 was determined before each experiment and ranged from 8 to 18 μg depending on the TNF lot. zVAD-fmk (Bachem, Switzerland) was suspended in DMSO at 50 mg/ml, further diluted in PBS, and injected i.p. 15 min before (0.25 mg) and 1 h after TNF (0.1 mg). Mortality was scored up to 7 days. NG-nitro-l-arginine methyl ester (l-NAME, Novabiochem, 100 mg/kg) was given i.v. either 2 h or 30 min before, or together with TNF. For the hemodynamic studies, l-NAME or tempol were injected 45 min before TNF. Indomethacin (Sigma, 125 μg) was injected i.p. 1 h before challenge. Tempol (6 mg), SOD (7,500 U), PEG-SOD (400 U), catalase (7,500 U), and PEG-catalase (7,500 U), all from Sigma, were dissolved in PBS and injected i.p. 40 min before challenge. ABT (Sigma, 2.5 mg), SKF-525A (Biomol, 1.5 mg), and fluconazole (Pfizer, 1.2 mg) were injected i.p. 2 h before TNF. For K+ channel inhibition, apamin (ICN, 0.5 mg/kg i.v.), charybdotoxin (Sigma, 33 μg/kg i.v.), iberiotoxin (Sigma, 33 μg/kg i.v.), TEA (Sigma, 50 mg/kg i.p.), and glibenclamide (ICN, 25 mg/kg i.p.) were injected as previously described [19]. All treatment schedules and doses were based on other rodent studies in which they had a positive modulatory effect.
NO −x and cGMP measurement
For NO −x determination, serum was diluted 1:1 with 5.109 CFU/ml Pseudomonas oleovorans suspension (reducing nitrate to nitrite) and incubated at 37°C for 3–4 h. After centrifugation, the supernatant was diluted 1:2 with Griess reagent, and proteins were precipitated with 10% TCA. The absorbance of the supernatant was measured at 540 nm. Total NO −x was calculated from a nitrate standard curve. cGMP was determined by EIA (Amersham Pharmacia Biotech) after acetylation of the samples, according to the manufacturer's instructions. Plasma was collected by cardiac puncture using 7.5 mM EDTA and kept on ice until centrifugation. Dissected organs were immediately immersed in liquid nitrogen and stored at −20°C before homogenization and purification. Kidney cGMP levels were plotted as picomoles cGMP per milligram of TCA-precipitable protein solubilized with 1 N NaOH.
Body temperature, mean arterial pressure, and HR measurements
Rectal body temperature was recorded with an electronic thermometer (model 2001; Comark Electronics, Littlehampton, UK). Blood pressure and heart rate were measured continuously in conscious mice, either in tethetered permanently catheterized mice, as described [16], or via radiotelemetry. For the latter, PA-C10 telemetry probes (Data Sciences International) were surgically implanted. Mice were anesthetized with 2% isoflurane, and the cervical area skin was cleaned with povidone-iodine and alcohol. A 1-cm vertical incision was made in the left neck, and the left carotid artery was cannulated. The artery was punctured with a 26-gauge needle, the catheter tip of the transmitter was advanced to the aortic arch, and the catheter was sutured in place. A subcutaneous pocket was excavated over the right flank, the transmitter body was inserted into this, and the incision was closed with sutures. Ibuprofen was given in the drinking water from the day before surgery until 4 days after recovery. At day 10 after surgery, continuous, 24-h data collection began using the Dataquest ART Acquisition System (Data Sciences International, version 4.1).
Statistics
Statistics was performed using GraphPad Prism. NO −x or cGMP levels and body temperatures are presented as mean ± SEM. They were compared with a one-way analysis of variance test, with a Bonferroni post-test for comparison of all pairs. Survival curves and total percent mortality were compared with a log-rank (Mantel–Cox) test and a Chi-square test, respectively.
Results
NO or PGI2 do not mediate zVAD + TNF shock
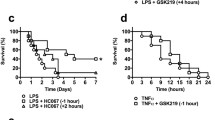
To evaluate the potential of ROS to cause hypotension, we used a hyperacute inflammatory shock model where excessive oxidative stress is a key feature [16]. In this model, TNF is combined with the caspase inhibitor zVAD, leading to abrupt, hyperacute hypotension and death within only a few hours after challenge. It is generally believed that inflammation-induced cardiovascular collapse is predominantly orchestrated by iNOS-derived NO [10]. However, the iNOS enzyme needs to be transcriptionally induced, and its action thus requires several hours [9]. In agreement with this, we find detectable systemic NO −x at the earliest 2–3 h after TNF [20]. Since the abrupt zVAD + TNF hypotension starts even before that [16], the involvement of iNOS-derived NO seems improbable. Nevertheless, we have identified eNOS-derived NO as the principal vasodilator in anaphylactic shock, causing hypotension within less than half an hour [7], making also this a potential candidate to mediate hyperacute shock. To evaluate the possible involvement of NO and its downstream mediator cGMP in the zVAD + TNF shock model, we first determined endogenously induced NO −x and cGMP, which were not increased at all but rather reduced to background levels (Fig. 1a–c). In addition, deficiency for iNOS, or iNOS and eNOS, could not protect mice from the abrupt zVAD + TNF toxicity (Fig. 1d), and the NOS inhibitor l-NAME even aggravated toxicity (Fig. 1e). To evaluate the effect of NOS inhibition on hypotension, we measured blood pressure in permanently catheterized conscious mice. After l-NAME treatment, zVAD + TNF still induced an acute and steep drop in blood pressure, which generally developed even more rapidly than without l-NAME (Fig. 1f). Together, these results imply that the hyperacute zVAD + TNF shock develops NO-independently.

NO and cGMP in zVAD + TNF shock. a NO −x (NO −2 + NO −3 ) in serum collected 3 h after PBS or TNF (n above the bars). b cGMP in plasma collected 3 h after PBS or TNF (n above the bars). c cGMP in homogenates from kidneys, collected 3 h after PBS or TNF (n above the bars). ***P < 0.001, **P < 0.01, compared with PBS. d WT, iNOS−/−, or double iNOS−/− and eNOS−/− mice (ixeNOS−−/−−) were injected with TNF (T, open symbols) or zVAD + TNF (zT, filled symbols; n in the legend). ***P < 0.001, *P < 0.05, compared with TNF alone. e WT mice were injected with TNF (open circles, n = 9) or TNF + zVAD (closed circles, n = 9), and the effect of l-NAME 2 h (triangles, n = 5) or 30 min (triangles, n = 5) before TNF or together with TNF (diamonds, n = 8) was tested. *P < 0.05, compared with zVAD + TNF. f Mean arterial pressure in conscious free-moving catheterized mice injected through a catheter with TNF, with or without zVAD i.p. To analyze the role of NO, a group of zVAD + TNF mice was treated with l-NAME 45 min before TNF. n = 5 for each group, plotted is the median response
Next to NO, endothelial cells may also produce vasorelaxing PGI2. However, inhibition of PGI2 producing cyclooxygenases with indomethacin could not prevent zVAD + TNF shock [16] and even exacerbated it (not shown).
EDHF candidates do not mediate zVAD + TNF shock
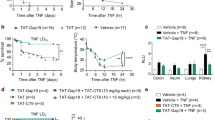
NO- and PGI2-independent vasodilation is especially significant in resistance vessels, which are particularly important for blood pressure regulation, and is mediated via a mechanism collectively labeled EDHF [3]. The (patho)physiological involvement of EDHF in vivo is hard to investigate, as there is still no consensus about its nature and exact mode of action. H2O2 and epoxyeicosatrienoic acids (EETs) have been proposed as EDHF candidates [3]. To investigate the contribution of H2O2, we treated mice with catalase or cell-permeable PEG-catalase. However, they could not protect and rather aggravated the acute toxicity (Fig. 2a). In addition, also the CYP inhibitors 1-aminobenzotriazole (ABT), fluconazole, or SKF-525A could not provide significant protection, excluding a role for EETs in zVAD + TNF shock (Fig. 2b). Also a 15-lipoxygenase derivative has been proposed as a vasorelaxing arachidonic acid metabolite [3], but 15-lipoxygenase−/− mice were as sensitive to zVAD + TNF shock as wild-type animals (not shown).

ROS and EETs in zVAD + TNF toxicity. a Effect of catalase, PEG-catalase, or tempol on mortality induced by zVAD ± TNF. Plotted is the percent survival of all mice used in three independent experiments; the total number is indicated between brackets in the legend. Sensitization by zVAD corresponds to mortality within 6 h. Mice injected with TNF alone are also plotted for comparison. ***P < 0.0001 compared with zVAD + TNF (black bar). b Effect of the CYP inhibitors ABT, fluconazole, and SKF-525A. Plotted is the percent survival of all mice used in up to four independent experiments; the total number is indicated between brackets in the legend. Differences between zVAD + TNF and SKF-525A + zVAD + TNF are not significant (P > 0.07). c Blood pressure and (d) heart rate were monitored in conscious radiotelemetred mice injected with zVAD ± TNF. Two mice were treated with tempol 45 min before TNF. e, f Effect of tempol, SOD, or PEG-SOD on hypothermia (e) and mortality (f) induced by zVAD + TNF (n = 5), ***P < 0.001 compared with zVAD + TNF. g Tempol protects against TNF shock. Mice were injected i.v. with a lethal dose of TNF alone (n = 6), **P = 0.0012. h Tempol protects against LPS shock. Mice were injected i.v. with a lethal dose of LPS (n = 6), ***P = 0.0006
Antioxidant protection against shock induced by zVAD + TNF, TNF, or LPS
ROS are thought to contribute to morbidity in sepsis because of their direct cytotoxic and organ-damaging capacities. To investigate whether they also might be involved in causing the hypotension, we measured blood pressure in conscious radiotelemetred mice pretreated with tempol, a cell-permeable SOD mimetic, radical scavenger and inhibitor of the Fenton reaction [21, 22]. Surprisingly, while NOS inhibition did not prevent the abrupt zVAD + TNF hypotension at all (Fig. 1f), tempol very efficiently did (Fig. 2c). Moreover, while the blood pressure of mice treated with TNF or zVAD + TNF dropped substantially following TNF challenge, blood pressure did not decrease at all for at least 2 h if the mice had been pretreated with tempol (Fig. 2c). In addition, pretreatment with tempol completely prevented the severe bradycardia that normally develops shortly after zVAD ± TNF challenge (Fig. 2d). The effect of tempol on blood pressure and heart rate was mirrored by its effect on peripheral body temperature (Fig. 2e).
To investigate the involvement of ROS in other models, we used tempol in shock induced by a high dose of TNF or LPS. Also in these models, tempol completely prevented mortality (Fig. 2g, h).
To better understand the exact mechanism of tempol's protective action, we compared its effect on ROS-dependent zVAD + TNF shock with SOD and cell-permeable PEG-SOD. Surprisingly, only tempol fully protected against acute hypothermia (Fig. 2e) and prevented both early and late mortality (Fig. 2a, f). As tempol is also an efficient scavenger of \( ^\bullet {\hbox{OH}} \) radicals and reduces the formation of \( ^\bullet {\hbox{OH}} \) [21, 22], these results suggest that not \( {{\hbox{O}}_2}^{ \bullet - } \) but \( ^\bullet {\hbox{OH}} \) radicals are crucial. Apocynin, an NADPH oxidase (NOX) inhibitor and \( ^\bullet {\hbox{OH}} \) scavenger [23], also prevented hyperacute zVAD + TNF shock, but could not offer long-term protection (Fig. 3a, b). In combination with tempol, apocynin also conferred some additional protection against both hypothermia (Fig. 3c) and mortality (Fig. 3d).

Apocynin protects against acute zVAD + TNF shock. a Effect of apocynin on hypothermia induced by zVAD + TNF, *P < 0.05, **P < 0.01, ***P < 0.001 compared with zVAD ± TNF. b Effect of apocynin on zVAD ± TNF mortality, **P = 0.0013 compared with zVAD ± TNF. c, d Effect of apocynin on protection by tempol. *P < 0.05, **P < 0.01, ***P < 0.001 compared with zVAD ± TNF
SKCa channels are essential for ROS-mediated zVAD + TNF shock
NO and PGI2 activate large-conductance BKCa and KATP channels [2, 8, 9]. Vascular smooth muscle BKCa channels may also be activated by EETs or H2O2, which have been suggested as EDHFs in certain systems [3]. In contrast, EDHF-dependent hyperpolarization specifically depends on endothelial SKCa channels, and not on BKCa or KATP channels [3, 4]. To study K+ channels, we used apamin (SKCa inhibitor), iberiotoxin (BKCa inhibitor), charybdotoxin (inhibits BKCa, IKCa, and certain voltage-gated Kv), glibenclamide (KATP inhibitor), and TEA (inhibits BKCa, KATP, and some Kv). Only apamin completely prevented hyperacute (within 6 h) zVAD + TNF-induced hypothermia and mortality (Fig. 4a–c). Furthermore, apamin also significantly protected against long-term TNF-induced mortality (Fig. 4a, c). To evaluate the cardiovascular effects of SKCa inhibition, blood pressure and heart rate were measured in radiotelemetred mice. In two of the three animals pretreated with apamin, hypotension and bradycardia were efficiently prevented (one representative mouse is shown in Fig. 4d, e), resulting in survival. In one mouse, apamin could not prevent but delayed hypotension, bradycardia, and mortality (Fig. 4d, e, dotted line)

The role of K+ channels and H2O2 in ROS-dependent zVAD + TNF shock. a Effect of different K+ channel inhibitors. Plotted is the percent survival of all mice used in up to five independent experiments; total numbers are indicated between brackets in the legend. ***P < 0.0001 compared with zVAD + TNF (black bar). b, c Effect of apamin on hypothermia (b) and mortality (c) induced by zVAD ± TNF in a representative experiment (n in the legend), *P < 0.05, **P = 0.0049, ***P < 0.001 compared with zVAD ± TNF. d, e Mean arterial pressure and HR were monitored in conscious radiotelemetred mice injected with zVAD ± TNF. Three mice were treated with apamin 2 h before TNF, plotted are the non-survivor and one of the two survivors. f Effect of catalase and PEG-catalase on protection by tempol, ***P < 0.001 compared with tempol + zVAD + TNF (diamonds); data shown are from one individual representative experiment
H2O2 antagonizes TNF-induced shock
As shown in Fig. 2a, catalase could not improve but rather worsened zVAD + TNF shock. When catalase was combined with a protective tempol treatment, it even reversed the long-term protection provided by tempol (Fig. 4f). PEG-catalase, accumulating intracellularly, did not revert tempol protection (Fig. 4f), suggesting that extracellularly produced H2O2 may counteract shock.
Discussion
Our results indicate a key role for ROS and SKCa channels in inflammatory hypotension and shock. To prove the involvement of ROS in inflammation-induced shock is not easy, as ROS and their production systems are efficiently antagonized by NO, which is massively produced as soon as iNOS is transcribed. Therefore, we used this specific TNF-induced caspase-independent hyperacute shock model as a proof-of-principle, because it develops even before NO metabolites can be detected in circulation [16]. In this shock model, we found that NOS inhibition or deficiency did not prevent hypotension and shock, while the antioxidants N-acetylcysteine (NAC), butylated hydroxyanisole (BHA), or tempol completely precluded the fast hyperacute mortality induced by zVAD ± TNF [16]. In general, antioxidants are thought to protect against morbidity and mortality in sepsis because they prevent the cytotoxic and tissue-damaging effects of ROS. Our cardiovascular studies now reveal that tempol not just protects against oxidative toxicity and injury but also prevents the dramatic drop in mean arterial pressure and HR that normally develops abruptly after zVAD ± TNF. The possible involvement of ROS in hypotension creates an intriguing paradox, since vascular ROS (particularly superoxide) have traditionally been associated with hypertension. A possible answer to this paradox could be that different ROS have different specific properties [24]. Therefore, we tried to pinpoint which ROS exactly could be the hypotensive culprit in the ROS-dependent zVAD + TNF shock. Tempol, which was clearly very proficient to prevent hypotension and mortality, is generally referred to as a “cell-permeable SOD mimetic.” However, since cell-permeable SOD could not protect, the ROS-dependent shock is most probably not directly triggered by \( {{{\hbox{O}}_2}^{ \bullet - }} \). Tempol is also an efficient radical scavenger, as well as an inhibitor of Fenton-type reactions that occur between peroxide and transition metals and result in the formation of \( ^\bullet {\hbox{OH}} \) radicals, and it has already been suggested that many of tempol's effects are probably due to its ability to scavenge \( ^\bullet {\hbox{OH}} \) radicals [21, 22]. Also, the NOX inhibitor apocynin significantly delayed the hyperacute shock, in contrast to NOX2-deficiency [16]. This may indicate the involvement of NOX1 or NOX4, but the protective effect of apocynin may also be due to its \( ^\bullet {\hbox{OH}} \) scavenging potential [23]. Interestingly, endothelial NOX was recently identified as a functionally relevant mediator of vasodilation in human coronary microcirculation [25]. We have previously shown that the hyperacute shock induced by zVAD + TNF may also be prevented by NAC [16], a scavenger of \( ^\bullet {\hbox{OH}} \) radicals and precursor for cytoplasmic glutathione essential to prevent intracellular \( ^\bullet {\hbox{OH}} \) production [26, 27]. Together, the protective effects of tempol, apocynin, and NAC, compared with the failure of (cell-permeable) SOD or catalase to protect, thus suggest the possibility that intracellular \( ^\bullet {\hbox{OH}} \) radicals are the shock-causing ROS. The fact that NOS inhibition significantly exacerbated hypotension and toxicity may further corroborate this hypothesis, since NO can not only scavenge \( ^\bullet {\hbox{OH}} \) radicals at near diffusion control but also prevent \( ^\bullet {\hbox{OH}} \) formation from H2O2 by scavenging superoxide radicals at diffusion control (thus inhibiting Fe3+ reduction in ferritin and Fe-S clusters) and by reacting directly with free Fe2+ to form dinitrosyliron complexes [15, 28]. The vasorelaxing and maybe even hypotensive potential of \( ^\bullet {\hbox{OH}} \) radicals had already been suggested in the past [29, 30] but has not been given much attention since.
Surprisingly, catalase, but not PEG-catalase, aggravated zVAD + TNF toxicity and even reverted tempol's protection, demonstrating the detrimental effect of extracellular H2O2 removal. Tempol is known to increase H2O2 formation, vasoconstriction, and hypertension in vivo, effects that are abolished by catalase coinfusion [31]. Together, this suggests that extracellular H2O2 may exert a protective role in shock, and that part of the protective capacity of tempol may reside in its ability to allow H2O2 accumulation while efficiently interfering with intracellular \( ^\bullet {\hbox{OH}} \) formation and accumulation at the same time. In this way, our results may explain why antioxidants other than tempol fail to provide survival benefit during shock [32], because they inhibit the accumulation of not only the detrimental \( ^\bullet {\hbox{OH}} \) but also of the protective H2O2. Despite the fact that H2O2 has been put forward as a potential EDHF [3, 33], due to the capability of catalase to diminish NO- and PGI2-independent vasorelaxation in isolated arteries, H2O2 has also been demonstrated to antagonize vascular relaxation through various mechanisms [33–35] and was even identified as an important endogenous vasoconstrictor contributing to blood pressure regulation in vivo [36].
In addition to the effect of tempol on blood pressure, whose decline was prevented the first hours after TNF injection, there was also a remarkable effect on HR. Following injection with zVAD ± TNF, mice respond with a transient compensatory tachycardia and a severe loss of HR variability, followed shortly afterwards by severe and abrupt bradycardia. Although the loss of HR variability was not affected by tempol, the initial tachycardia was not followed by bradycardia, indicating that ROS are involved in cardiac dysfunction during shock as well.
The opening of potassium (K+) channels is the main determinant of cell membrane potential, and thus the activation of vascular K+ channels is critical for the regulation of vascular tone. The vasodilators NO, PGI2, and EDHF may all activate various K+ channels in either endothelial or smooth muscle membranes, allowing K+ efflux out of the cell, causing hyperpolarization and vasodilation. More specifically, NO and PGI2 are known to hyperpolarize vascular smooth muscle cells by activating Ca2+-dependent large-conductance BKCa channels or ATP-sensitive KATP channels [2]. Although the exact molecular identity and mechanism of EDHF is still a matter of debate, it is generally agreed that EDHF-dependent hyperpolarization depends on endothelial SKCa, and not on BKCa or KATP channels [3, 4]. Using different inhibitors, we could clearly demonstrate the decisive contribution of SKCa channels to ROS-dependent shock, as apamin efficiently prevented hypotension, bradycardia, hypothermia, and mortality. Interestingly, both SK2 and SK3 channel conductances have been demonstrated to be increased by intracellular oxidative stress [37].
In conclusion, our data suggest that in an early phase of inflammation, important ROS-dependent cardiovascular effects are taking place. Interfering with these ROS-dependent effects efficiently antagonizes the progressive hypotension and bradycardia that normally ensues, resulting in fast recovery and survival. In general models of (septic) shock, the ROS-dependent effects may be easily overlooked, because of concomitant iNOS induction and massive NO production, which scavenges ROS and prevents further ROS production [15]. By acutely exacerbating oxidative stress (for example, by inhibiting caspases via the application of zVAD-fmk) and/or inhibiting NO production, the ROS-induced hypotension is amplified and results in lethal shock that is entirely ROS-dependent and efficiently prevented with the antioxidant tempol. In addition, ROS-dependent shock specifically relies on SKCa channels only. Importantly, ROS and SKCa-dependent events are also critical in the progression of shock in the absence of zVAD-fmk, as tempol (shown in this report) and apamin [19] also significantly improve survival in normal TNF- or LPS-induced shock models. Thus, specific inhibitors of vascular/endothelial SKCa channels and/or scavengers of intracellular \( ^\bullet {\hbox{OH}} \) radicals and inhibitors of their formation (which may include vascular NOX1 or NOX4 enzymes) could represent interesting new therapeutics for the treatment of hypotension, cardiac dysfunction, and shock associated with inflammation.
References
Nguyen HB, Rivers EP, Abrahamian FM, Moran GJ, Abraham E, Trzeciak S, Huang DT, Osborn T, Stevens D, Talan DA (2006) Severe sepsis and septic shock: review of the literature and emergency department management guidelines. Ann Emerg Med 48:28–54
Landry DW, Oliver JA (2001) The pathogenesis of vasodilatory shock. N Engl J Med 345:588–595
Feletou M, Vanhoutte PM (2006) Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol 26:1215–1225
Crane GJ, Gallagher N, Dora KA, Garland CJ (2003) Small- and intermediate-conductance calcium-activated K + channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J Physiol 553:183–189
Grgic I, Kaistha BP, Hoyer J, Kohler R (2009) Endothelial Ca + -activated K + channels in normal and impaired EDHF-dilator responses—relevance to cardiovascular pathologies and drug discovery. Br J Pharmacol 157:509–526
Connelly L, Madhani M, Hobbs AJ (2005) Resistance to endotoxic shock in endothelial nitric-oxide synthase (eNOS) knock-out mice: a pro-inflammatory role for eNOS-derived no in vivo. J Biol Chem 280:10040–10046
Cauwels A, Janssen B, Buys E, Sips P, Brouckaert P (2006) Anaphylactic shock depends on PI3K and eNOS-derived NO. J Clin Invest 116:2244–2251
Cauwels A (2007) Nitric oxide in shock. Kidney Int 72(5):557–565
Fernandes D, Assreuy J (2008) Nitric oxide and vascular reactivity in sepsis. Shock 30(Suppl 1):10–13
Hauser B, Bracht H, Matejovic M, Radermacher P, Venkatesh B (2005) Nitric oxide synthase inhibition in sepsis? Lessons learned from large-animal studies. Anesth Analg 101:488–498
Lopez A, Lorente JA, Steingrub J, Bakker J, McLuckie A, Willatts S, Brockway M, Anzueto A, Holzapfel L, Breen D et al (2004) Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: effect on survival in patients with septic shock. Crit Care Med 32:21–30
Cauwels A, Bultinck J, Brouckaert P (2005) Dual role of endogenous nitric oxide in tumor necrosis factor shock: induced NO tempers oxidative stress. Cell Mol Life Sci 62:1632–1640
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Schmidt HH, Schmidt PM, Stasch JP (2009) NO- and haem-independent soluble guanylate cyclase activators. Handb Exp Pharmacol 191:309–339
Wink DA, Miranda KM, Espey MG, Pluta RM, Hewett SJ, Colton C, Vitek M, Feelisch M, Grisham MB (2001) Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal 3:203–213
Cauwels A, Janssen B, Waeytens A, Cuvelier C, Brouckaert P (2003) Caspase inhibition causes hyperacute tumor necrosis factor-induced shock via oxidative stress and phospholipase A2. Nat Immunol 4:387–393
Laubach VE, Shesely EG, Smithies O, Sherman PA (1995) Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci USA 92:10688–10692
Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O (1996) Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93:13176–13181
Cauwels A, Brouckaert P (2008) Critical role for small and large conductance calcium-dependent potassium channels in endotoxemia and TNF toxicity. Shock 29(5):577–582
Cauwels A, Van Molle W, Janssen B, Everaerdt B, Huang P, Fiers W, Brouckaert P (2000) Protection against TNF-induced lethal shock by soluble guanylate cyclase inhibition requires functional inducible nitric oxide synthase. Immunity 13:223–231
Thiemermann C (2003) Membrane-permeable radical scavengers (tempol) for shock, ischemia-reperfusion injury, and inflammation. Crit Care Med 31:S76–S84
Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y (2005) Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 45:860–866
Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP (2008) Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51:211–217
Hartzell HC (2007) The stress of relaxation. Science 317:1331–1332
Larsen BT, Bubolz AH, Mendoza SA, Pritchard KA Jr, Gutterman DD (2009) Bradykinin-induced dilation of human coronary arterioles requires NADPH oxidase-derived reactive oxygen species. Arterioscler Thromb Vasc Biol 29:739–745
Gillissen A, Nowak D (1998) Characterization of N-acetylcysteine and ambroxol in anti-oxidant therapy. Resp Med 92:609–623
Jiang M, Wei Q, Pabla N, Dong G, Wang CY, Yang T, Smith SB, Dong Z (2007) Effects of hydroxyl radical scavenging on cisplatin-induced p53 activation, tubular cell apoptosis and nephrotoxicity. Biochem Pharmacol 73:1499–1510
Toledo JC Jr, Bosworth CA, Hennon SW, Mahtani HA, Bergonia HA, Lancaster JR Jr (2008) Nitric oxide-induced conversion of cellular chelatable iron into macromolecule-bound paramagnetic dinitrosyliron complexes. J Biol Chem 283:28926–28933
Rubanyi GM, Vanhoutte PM (1986) Oxygen-derived free radicals, endothelium, and responsiveness of vascular smooth muscle. Am J Physiol 250:H815–H821
Prasad K, Bharadwaj LA (1996) Hydroxyl radical—a mediator of acetylcholine-induced vascular relaxation. J Mol Cell Cardiol 28:2033–2041
Makino A, Skelton MM, Zou AP, Cowley AW Jr (2003) Increased renal medullary H2O2 leads to hypertension. Hypertension 42:25–30
Taylor DE, Kantrow SP, deBoisblanc B (2001) Tempering the temptation to treat with tempol. Crit Care Med 29:212–214
Ellis A, Triggle CR (2003) Endothelium-derived reactive oxygen species: their relationship to endothelium-dependent hyperpolarization and vascular tone. Can J Physiol Pharmacol 81:1013–1028
Tang XD, Santarelli LC, Heinemann SH, Hoshi T (2004) Metabolic regulation of potassium channels. Ann Rev Physiol 66:131–159
Ardanaz N, Pagano PJ (2006) Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp Biol Med 231:237–251
Suvorava T, Lauer N, Kumpf S, Jacob R, Meyer W, Kojda G (2005) Endogenous vascular hydrogen peroxide regulates arteriolar tension in vivo. Circulation 112:2487–2495
Keating DJ, Rychkov GY, Giacomin P, Roberts ML (2005) Oxygen-sensing pathway for SK channels in the ovine adrenal medulla. Clin Exp Pharmacol Physiol 32:882–887
Acknowledgements
We thank our animal caretakers, as well as J. Debets and A. Brouns for preparation of mice for the hemodynamic studies in Maastricht. Research was supported by the Fonds voor Wetenschappelijk Onderzoek (FWO)-Vlaanderen and the Universiteit Gent-Geconcerteerde Onderzoeks Acties (GOA). A.C. was a postdoctoral fellow of the FWO-Vlaanderen.
Conflicts of interest
The authors declare no conflict of interests related to this study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cauwels, A., Rogge, E., Janssen, B. et al. Reactive oxygen species and small-conductance calcium-dependent potassium channels are key mediators of inflammation-induced hypotension and shock. J Mol Med 88, 921–930 (2010). https://doi.org/10.1007/s00109-010-0633-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-010-0633-2