
Abstract
Key message
The quantitative trait loci (QTLs) for grain morphological traits were identified via nested association mapping and validated in a natural wheat population via haplotype analysis.
Abstract
Grain weight, one of the three most important components of crop yield, is largely determined by grain morphological traits. Dissecting the genetic bases of grain morphology could facilitate the improvement of grain weight and yield production. In this study, four wheat recombinant inbred line populations constructed by crossing the modern variety Yanzhan 1 with three semi-wild wheat varieties (i.e., Chayazheda, Yutiandaomai, and Yunnanxiaomai from Xinjiang, Tibet, and Yunnan, respectively) and one exotic accession Hussar from Great Britain were investigated for grain weight and eight morphological traits in seven environments. Eighty-eight QTLs for all measured traits were totally identified through nested association mapping utilizing 14,643 high-quality polymorphic single nucleotide polymorphism (SNP) markers generated by 90 K SNP array. Among them, 64 (72.7%) QTLs have the most favorable alleles donated by semi-wild wheat varieties. For 14 QTL clusters affecting at least two grain morphological traits, nine QTL clusters were located in similar position with known genes/QTL, and the other five were novel. Three important novel QTLs (i.e., qTGW-1B.1, qTGW-1B.2, and qTGW-1A.1) were further validated in a natural wheat population via haplotype analysis. The favorable haplotypes for these three QTLs might be used in marker-assisted selection for the improvement of wheat yield by modifying morphological traits.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As one of the world’s major staple crops, wheat (Triticum aestivum L.) is critical for global food security (Li and Yang 2017). To feed the growing human population with decreasing area of land, wheat yield must be improved substantially (Simmonds et al. 2016). Grain weight, an important component of grain yield, is mainly determined by grain morphological traits, such as grain length, grain width, and grain area (Gegas et al. 2010). Therefore, dissecting the genetic basis of grain morphological traits is crucial for the improvement of grain weight and yield.
Grain morphology and weight are traits of quantitative inheritance, and many QTLs affecting wheat grain morphological traits and grain weight have been identified (Cheng et al. 2017; Cui et al. 2014; Heidari et al. 2011; Huang et al. 2003, 2006, 2004; Liu et al. 2014; Mccartney et al. 2005; Rasheed et al. 2014; Remington et al. 2001; Ren et al. 2018; Risch and Merikangas 1996; Stich et al. 2010; Wang et al. 2011; Wu et al. 2015; Yan et al. 2017; Yu et al. 2008; Zanke et al. 2015). Two complimentary tools that have been used to dissect the genetic architecture of complex quantitative traits are linkage mapping and genome-wide association studies (Risch and Merikangas 1996). Linkage mapping, possessing high power for QTL identification (Stich et al. 2010), has been extensively applied in the detection of QTL for wheat grain morphology and weight using biparental populations, such as recombinant inbred line (RIL) (Cheng et al. 2017; Jia et al. 2013; Liu et al. 2014; Wu et al. 2015), F2 (Yan et al. 2017), F2:3 (Wang et al. 2011), doubled haploid (DH) population (Heidari et al. 2011; Huang et al. 2006; Mccartney et al. 2005), and advanced backcross populations (Huang et al. 2003, 2004). Compared with linkage mapping, a genome-wide association study has a higher resolution for QTL detection (Remington et al. 2001). Recently, association studies have been applied in the analysis of grain morphology and weight in wheat (Rasheed et al. 2014; Zanke et al. 2015). To combine the advantages of two mapping methods, a nested association mapping (NAM) strategy was proposed for the genetic dissection of complex traits using several RIL populations derived from multiple-cross mating with a shared parent (Yu et al. 2008). However, only a few studies on QTL mapping through the NAM strategy in wheat have been conducted (Cui et al. 2014; Ren et al. 2018).
The increase in crop yield potential mainly benefited from artificial domestication (breeding). Grain morphology and weight have undergone a positive selection via open environments, which is one of the main components of domestication syndrome (Brown et al. 2009; Fuller 2007). Compared with ancestral wheat species, domesticated wheat species have a larger grain size (Fuller 2007). However, the crop domestication results in the improvement of grain yield but at a cost of reduced genetic variation which is referred to as the “domestication bottleneck” (Dempewolf et al. 2017). Luckily, genetic diversity is preserved in the wild relatives of wheat. Thus, exploiting and utilizing wild resources could break through the bottleneck in wheat breeding. Over the past few decades, the utilization of wild species in wheat breeding has achieved success. For instance, Tang et al. (2014) isolated a line from the cross between common wheat and Thinopyrum intermedium (Host) Barkworth and Dewey. This line carried the translocation of the T. intermedium chromosome replacing chromosome 6A of common wheat, which is highly resistant to powdery mildew. Similarly, a wheat Leymus mollis (Trin.) Pilg. 3D (3Ns) substitution line replacing chromosome 3D of common wheat was isolated by Pang et al. (2014). It was characterized by resistance to leaf rust and improved spike length and spike number. Zhang et al. (2014) developed some translocation lines carrying seed storage protein genes from chromosome 1 V of Dasypyrum villosum, which showed positive effects on qualities in wheat that are exploited for bread-making.
Yunnan wheat (Triticum aestivum ssp. yunnanense King), Tibetan wheat (Triticum aestivum ssp. tibetanum Shao), and Xinjiang wheat (T. petropavloski Udatsz. et Migusch.) are three unique wheat subspecies that were discovered in western China (Wang et al. 2007). Yunnan wheat, characterized by tough and hard glumes, is a semi-wild cultivar that was discovered in Yunnan (Dong et al. 1981). It has the advantages of good quality, preharvest sprouting resistance, high tolerance to poor soil fertility, drought, cold, and frost and is considered one of the most precious resources for wheat breeding (Chen et al. 2007; Dong et al. 1981). Tibetan wheat is primitive semi-wild hexaploid wheat from Tibet (Shao et al. 1980), which has a spike morphology similar to that of common wheat but is resistant to abiotic stresses and has a strong seed dormancy and strong nutrition deficiency tolerance (Liu et al. 2014; Sun et al. 1998). As one of the Chinese endemic wheat landraces, Xinjiang wheat, known as “Daosuimai,” or rice-head wheat, is characterized by a long glume (Chen et al. 2013). It is a hexaploid wheat species that was discovered in the Talimu basin of Xinjiang, and it has a similar morphology as the tetraploid T. turgidum ssp. polonicum. These semi-wild wheat varieties possess many advanced traits; however, only a few studies have been conducted on mining the favorable alleles of semi-wild wheat cultivars to control grain morphology and weight (Liu et al. 2014; Luo et al. 2016).
Therefore, to better exploit desirable QTL/genes from wild relatives and exotic germplasms for grain morphology and weight, four RIL populations were constructed, with a modern wheat variety Yanzhan 1 (YZ) as the common female parent and four male parents including (three unique semi-wild subspecies from China and one dwarf cultivar Hussar from Great Britain). Nested association mapping was performed to identify the favorable alleles for wheat grain weight and grain morphological traits. Some important QTLs were further validated in a wheat natural population. Our study could help to provide a better understanding of the genetic base that controls grain morphological traits; therefore, it would be helpful for the genetic improvement of wheat grain weight and yield potential.
Materials and methods
Plant materials
The study comprised four RIL populations derived from crosses between YZ (the common female parent) and four semi-wild or exotic germplasms. YZ, a high-yielding and good-quality winter wheat variety released in Henan province of the Huanghuai region in China, possessed small seed size. The four male parents included Hussar (HR, a dwarf winter wheat cultivar) from Great Britain and three semi-wild wheat varieties including Yunnanxiaomai (YN, T. aestivum ssp. yunnanense King, semi-winterness variety) from Yunnan, Chayazheda (CY, T. aestivum ssp. tibetanum Shao, winter variety) from Tibet, and Yutiandaomai (YT, T. aestivum. petropavloski Udats et Migusch, winter variety) from Xinjiang. Single seed descent method was applied to develop the RIL populations by eight times of self-pollination from F2 generation. The population sizes for YN, CY, YT, and HR populations were 97, 82, 96, and 94, respectively.
Field trials
The four RIL populations, along with their corresponding parents, were planted in seven environments in the Shandong Province of China in two locations Heze (115.51°E, 35.58°N) and Dezhou (116.39°E, 37.38°N) in 2015 and 2016 and one location Taian (117.17°E, 36.17°N) from 2015 to 2017. The environment names were coded by year of evaluation and abbreviation of location name, e.g., 15H refers to environment in 2015 at Heze. Each line was planted in a two-row plot with 50 seeds per row with a row length of 2.0 m and a row spacing of 0.25 m. Two replicates were performed under each environment. The local farmers’ standard management practices were followed for the field of each environment.
Trait measurements
At maturity, 10 uniform plants in the middle of each plot were bulk harvested. The seeds were fully cleaned and dried before trait measurement. The thousand grain weight (TGW, in g), grain length (GL, in mm), grain width (GW, in mm), grain area (GA, in mm2), grain perimeter (GP, in mm), grain diameter (GD, in mm), grain shape (GS), and grain roundness (GR) were measured using a grain seed measurement machine (SC-E, Wanshen Technology Company, Hangzhou, China). Due to some uncontrollable factors, the traits for the CY-RIL population in 15 T, 15H, and 15D were not collected.
Genotyping
The genomic DNA of the four interconnected RIL populations and five parents was extracted from fresh seedling leaves using the CTAB method (Doyle 1987). The qualified DNA was genotyped using the wheat 90 K iSelect array (Wang et al. 2014). The physical positions for all the SNP markers were determined through a BLAST alignment to the IWGSC Reference Sequence v1.0 (Alaux et al. 2016) using the short sequences that harbored SNP loci, and only the uniquely matched SNP loci were retained. The heterozygous alleles were regarded as missing, and the SNP loci with a missing rate over 20% and minor allele frequency (MAF) less than 0.05 were removed. Finally, a total of 14,643 high-quality SNP markers were used in the following analysis.
Population structure and kinship
The population structure and kinship of the four interconnected RIL populations sharing one common parent (nested association population) were analyzed using the 14,643 high-quality SNP markers. A model-based Bayesian clustering analysis method implemented in STRUCTURE software version 2.3.4 (Pritchard et al. 2000) was utilized for structure analysis. The parameters were set as follows: k, the number of groups in the panel varying from 1 to 5; 5 runs each k value; 10,000 burn-in iterations followed by 10,000 MCMC (Markov chain Monte Carlo) iterations. For kinship calculation, the Centered_IBS method implemented in TASSEL5.2.23 was used (Bradbury et al. 2007). The kinship heatmap was constructed using the R package “d3heatmap.”
Phenotypic analysis
The best linear unbiased prediction (BLUP) across the seven evaluated environments was calculated using the “lmer” function implemented in R package lme4, with both genotypes and environments considered as random factors. Phenotypic correlations were computed using the BLUP value of each line via the “rcorr” function implemented in the R package Hmisc (Harrell and Dupont 2013).
Nested association mapping
NAM was performed both on the BLUP for each trait from the combined analysis of seven environments and on the mean values for each trait in each individual environment from individual environment analyses using the NAM package (Xavier et al. 2015) in R. In the “NAM” package, an association analysis was carried out through the efficient massive mapping algorithm (EMMA). The P3D strategy was utilized to avoid updating the polygenic term for every marker. Using the empirical Bayes approach, each molecular marker was treated as a random effect, and the model was refitted using Eigen decomposition and evaluated with the likelihood ratio test (Xavier et al. 2015). A QTL was claimed when the test statistics reached P < 5 × 10−3. The QTLs affecting different traits with overlapped chromosome interval were designated as QTL clusters. In this study, two kinds of QTL clusters were defined. The clusters affecting both grain weight (TGW) and size (GL, GW, GA, GD, or GP), were given the prefix “QW,” and the clusters affecting grain shape (GS or GR) and size (GL, GW, GA, GD, or GP) were given the prefix “QS.” The annotated genes inside the QTL intervals were extracted from the IWGSC RefSeq Annotations database v1.0 (https://wheat-urgi.versailles.inra.fr/Seq-Repository/Annotations).
Validation of important QTLs by haplotype analysis
A validation was carried out for important QTLs through haplotype analysis using wheat natural population comprising 574 wheat cultivars or advanced breeding lines originated from 43 regions worldwide. QTLs that met the following criteria were regarded as important: (1) affecting at least four grain-size-related traits simultaneously; (2) being consistently identified in at least three individual environments. First, several KASP (Kompetitive Allele Specific PCR) markers inside the interval of important QTLs were developed from SNP markers linked to the target QTL or SNP markers with the unique position on wheat reference genome selected from Wheat 660 K SNP array designed by the Chinese Academy of Agricultural Sciences and synthesized by Affymetrix. (https://wheat.pw.usda.gov/ggpages/topics/Wheat660_SNP_array_developed_by_CAAS.pdf). Then, all the available KASP markers inside the QTL region were used to genotype 574 wheat cultivars or lines. Finally, haplotype analysis was performed by comparing the significant differences among major haplotypes (containing more than 8 samples) for each important QTL through analysis of variance (ANOVA).
Results
Traits variations and correlations
ANOVA results showed that differences among genotypes and environments were highly significant for all measured traits (Table S1). Genotype (G) explained an average of 51.8 ± 6.9% of the phenotypic variance, ranging from 39.4% for GW to 62.6% for GL. Environment (E) accounted for an average of 12.9 ± 5.1% of the phenotypic variation, ranging from 6.0% for GL to 21.8% for GW. G × E interaction was also significant for all measured traits and the phenotypic variance accounted for was 27.9% ± 2.0%, ranging from 25.3% for GP to 30.8% for GR.
The BLUP values for each line were used to draw the boxplots and conduct the phenotypic correlation analysis. The five parents showed great differences in grain morphology (Fig. 1). The common parent YZ exhibited a moderate grain size compared with the parent CY, which was characterized by the largest GW, GD, GA, and GP, and the parent YT had the largest GL compared with the other three parents. Thus, the TGW of CY (52.70 g) and YT (49.05 g) was higher, followed by YZ (38.24 g), YN (31.85 g), and HR (29.58 g) (Fig. 1). Wide variations were observed for all measured traits in the four RIL populations. Moreover, most of the traits in each RIL population appeared to be normally distributed, and strong transgressive segregations toward both directions were observed. The trend of differences in mean values among the populations was consistent with the differences among their male parents (Fig. 1). The phenotype pairwise correlations between the eight measured traits are illustrated in Fig. 2. Significant correlations between the traits were observed at the level of p = 0.01. The GA, GP, GL, GW, GS, and GD were positively correlated with one another (except for GS with GW), and the correlation coefficients varied from 0.26 of GS with GD to 1.00 of GA with GD. The GR was negatively correlated with all other grain morphological traits except for GW, with correlation coefficients ranging from − 0.29 with GD to − 0.99 with GS. TGW was positively correlated with all the grain morphological traits (except for GR), especially with GA and GD, with correlation coefficients being 0.95 in both cases. To investigate the environmental stability of the measured traits, the correlations between seven environments for each trait were calculated (Fig. S1). For all the traits, the positive correlations between different environments were significant at the level of p = 0.01, with most of the correlation coefficients being larger than 0.50. The GL showed the best environmental stability, with correlation coefficients ranging from 0.70 to 0.92, while the GW showed unstable correlation coefficients ranging from 0.25 to 0.79.

a Grain morphology of five parents in Taian of 2017. The scale bar represents 3 mm. b Boxplots for eight wheat grain morphology traits and grain weight. CY, HR, YN, and YT represented the RIL populations coming from the cross of the common parent YZ and the corresponding male parent CY, HR, YN, and YT, respectively. TGW, thousand grain weight; GA, grain area; GP, grain perimeter; GS, grain shape; GL, grain length; GW, grain width; GD, grain diameter; GR, grain roundness. The black dotted line represented the common female parent YZ. The dashed line of red, green, blue, and yellow represented the male parent CY, HR, YN, and YT, respectively

Phenotypic correlations between eight evaluated traits using the BLUP value for each line. In the lower triangular, the values were correlation coefficients (r) multiplied by 100. In the upper triangular, the areas and colors of ellipses showed the absolute value of corresponding r. Right and left oblique ellipses indicated positive and negative correlations, respectively. The values without glyphs indicated insignificant at 0.01
Basic statistics of markers
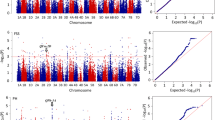
For the 14,643 high-quality SNP markers, there were 5881, 6483, and 2279 markers on sub-genomes A, B, and D, respectively. The number of markers per chromosome ranged from 112 on chromosome 4D to 1292 on chromosome 2B (Table S2 and Fig. 3). The coverage rate of the 14,643 SNP markers was 82.70% on average, ranging from 66.28% for chromosome 6D to 90.48% for chromosome 3A. The average marker spacing was 1.28 Mb, with spacing ranging from 0.60 Mb for chromosome 1A and 4.54 Mb for chromosome 4D. On average, the marker spacing for sub-genomes A, B, and D was 0.87 Mb, 0.85 Mb, and 2.14 Mb, respectively (Table S2).

QTL clusters for eight traits evaluated. The QTLs in green were mapped to a similar location with related cloned genes or mapped QTL reported previously
Population genetic structure
The entire nested association population comprised four RIL populations. According to values of LnP(D) generated from STRUCTURE, with its modal value used to detect the true k of three groups, k = 3 was recommended where the ascent changed gradually according to the method of Evanno et al. (2005). A kinship analysis also suggested three distinct subgroups (Fig. S2). Group I consisted of 75 lines from the CY-RIL population and 90 lines from the YT-RIL population. Group II consisted of 116 accessions, with most of the lines being from the HR-RIL population. Group III comprised 88 lines, and all of them were from the YN-RIL population. In the entire nested association population, 80.2% (296/369) of the lines did not show any admixture, and 6.0% (22/369) showed less than 20% admixture (Fig. S2B).
QTL mapping
A total of 88 QTLs were identified for eight grain morphological traits through combined analysis of seven environments, which were located on all 21 wheat chromosomes except for 2A, 3D, 4A, 4D, and 7D (Table S3). These QTLs were also detected via individual environment analysis in between one to five environments (Tables 1 and S4). Among the 88 QTLs, 64 (72.7%) had the most favorable alleles (i.e., the largest absolute additive effects in increasing grain weight or decreasing grain shape) donated from the semi-wild cultivars (Tables S3 and S5). CY, YN, and YT contributed the most favorable alleles for 43, 0, and 21 QTLs, respectively. Moreover, the exotic germplasm HR donated the most favorable alleles, amounting to 19 QTLs (21.6%). For the remaining five QTLs (5.7%), the most favorable alleles were from the common parent YZ.
For TGW, 14 QTLs were detected on chromosomes 1A, 1B, 2B, 3A, 3B, 6A, 6B, 7A, and 7B. For seven QTLs, including qTGW-1A.1, qTGW-1A.2, qTGW-2B, qTGW-3A, qTGW-3B, qTGW-7A, and qTGW-7B.2, the most favorable alleles were donated by the CY. The favorable alleles of qTGW-1B.1, qTGW-1B.2, and qTGW-7B.1 were contributed by YT. For qTGW-1B.3, qTGW-6A, and qTGW-6B, the most favorable alleles were contributed by HR. The most favorable allele of qTGW-7B.3 was from the common parent YZ.
For GA, 13 QTLs were detected on chromosomes 1A, 1B, 2B, 3A, 5A, 7A, and 7B. The most favorable alleles of seven QTLs (qGA-1A.1, qGA-1A.2, qGA-1A.3, qGA-2B, qGA-3A, qGA-5A, and qGA-7A), three QTLs (qGA-1B.2, qGA-1B.1, and qGA-7B.1), two QTLs (qGA-1A.4 and qGA-1B.3), and one QTL (qGA-7B.2) were contributed by CY, YT, HR, and YZ, respectively.
For GP, 14 QTLs were identified on chromosomes 1A, 1B, 1D, 2B, 5A, 7A, and 7B. The most favorable alleles of seven QTLs (qGP-1A.1, qGP-1A.2, qGP-2B, qGP-5A, qGP-7A.1, qGP-7A.2, and qGP-7B.2), four QTLs (qGP-1B.1, qGP-1B.2, qGP-1D, and qGP-7B.1), two QTLs (qGP-1A.3 and qGP-1B.3), and one QTL (qGP-7B.3) were donated by CY, YT, HR, and YZ, respectively.
Eight QTLs for GS were identified on chromosomes 2D, 4B, 5A, 5B, 5D, and 7A. The alleles of CY, YT, and HR decreased the GS for two QTLs (qGS-5B and qGS-5D), three QTLs (qGS-4B, qGS-7A.1, and QS-7A.2), and three QTLs (qGS-2D, qGS-5A.1, and qGS-5A.2), respectively.
Nine QTLs for GL were identified on chromosomes 1A, 1B, 1D, 2B, 5A, 7A, and 7B. The most favorable alleles of six QTLs (qGL-1A.1, qGL-2B, qGL-5A, qGL-7A.1, qGL-7A.2, and qGL-7B), two QTLs (qGL-1B and qGL-1D) and one QTL (qGL-1A.2) were contributed by CY, YT, and HR, respectively.
Nine QTLs for GW were detected on chromosomes 1A, 3B, 5A, 6A, 6B, 6D, and 7B. The most favorable alleles of four QTLs (qGW-1A.1, qGW-1A.2, qGW-3B, and qGW-7B), four QTLs (qGW-6A.1, qGW-6A.2, qGW-6B, and qGW-6D), and one QTL (qGW-5A) were donated by CY, HR, and YZ, respectively.
In total, 14 QTLs for GD were detected on chromosomes 1A, 1B, 2B, 3A, 3B, 5A, 7A, and 7B. The most favorable alleles of eight QTLs (qGD-1A.1, qGD-1A.2, qGD-1A.3, qGD-2B, qGD-3A, qGD-3B, qGD-5A, and qGD-7A), three QTLs (qGD-1B.1, qGD-1B.2, and qGD-7B.1) and two QTLs (qGD-1A.4 and qGD-1B.3), and one QTL (qGD-7B.2) were contributed by CY, YT, HR, and YZ, respectively.
For GR, seven QTLs were detected on chromosomes 2D, 4B, 5A, 5B, and 7A. The alleles of CY, HR, and YT increased the GR for two QTLs (qGR-5B and qGR-5D), two QTLs (qGR-2D and qGR-5A) and three QTLs (qGR-4B, qGR-7A.1, and qGR-7A.2), respectively.
QTL clusters
The 14 QTLs governing TGW were always co-located with the QTL for grain size traits, such as GA, GP, GD, GW, and GL, which merged into 14 QTLs clusters located on chromosomes 1A, 1B, 2B, 3A, 5A, 6A, 6B, 7A, and 7B (Table 1). The semi-wild relatives, CY and YT, contributed the most favorable alleles of the seven and three QTL clusters, respectively, and the exotic line HR donated the most favorable alleles of the three clusters. The other one was from the common parent YZ. QW-1A.1 consisted of five QTLs for GA, GD, GP, GW, and TGW, and the most favorable alleles were from CY. The cluster QW-1A.2 for GA, GD, GW, and TGW were detected, and the most favorable alleles were contributed by CY. Four QTL clusters, QW-1B.3, QW-1B.2, QW-7B.1, and QW-7B.3, were identified to affect GA, GD, GP, and TGW, and the most favorable alleles were donated by HR, YT, YT, and YZ, respectively. QW-1B.1 and QW-2B, which consisted of QTL for GA, GD, GP, GL, and TGW, were detected, and the most favorable alleles came from YT and CY, respectively. The QTL cluster QW-3B, which harbored QTL for GD, GW, and TGW, was identified, with the most favorable alleles contributed by CY. Three QTL clusters for GW and TGW, including QW-6A, QW-6B, and QW-7B.2, were identified, and the most favorable alleles were from HR, HR, and CY, respectively. Two QTL clusters for GA, GD, and TGW, QW-7A and QW-3A.1, were detected, and the most favorable alleles were contributed by CY.
The QTLs affecting the GS were co-located with QTL for GR or GW, which merged into eight QTL clusters on chromosome 2D, 4B, 5A, 5B, 5D, and 7A (Table 1). The alleles of HR decreased GS for QS-2D, QS-5A.1, and QS-5A.2. For QS-5B and QS-5D, the alleles decreasing GS were contributed by CY. The alleles of YT decreased GS for QS-4B, QS-7A.1 and QS-7A.2.
Validation and haplotype analysis for important QTLs
Haplotype analysis was conducted using a natural wheat population containing 574 cultivars or lines to validate three important QTLs: qTGW-1B.1, qTGW-1B.2, and qTGW-1A.1 (Table S7). qTGW-1B.1, which affected GA, GD, GP, GL, and TGW, was identified in the region of 49,926,288–53,251,268 ( ~ 3.3 Mb) on chromosome 1B in individual environments harboring 30 annotated genes (Tables 1 and S6). This QTL was consistently detected in four individual environments and showed excellent environment stability (Table S4). Subsequently, seven KASP markers in the target region of qTGW-1B.1 were developed for haplotype analysis through a natural wheat population (Tables S7 and S8). Significant differences were detected for the eight haplotypes at a significant level of p = 0.001 (Fig. 4). Haplotypes H7 and H8 showed significantly higher TGW than the other haplotypes. qTGW-1B.2, affecting GA, GD, GP, and TGW, was detected in the region of 368,543,950–376,616,816 ( ~ 8.07 Mb) on chromosome 1B in three individual environments harboring 38 annotated genes (Tables 1 and S6). Four KASP markers in the target region of qTGW-1B.2 were developed for haplotype analysis, and significant differences were detected for the four haplotypes at a significant level of p = 0.001 (Tables S7 and S8, Fig. 4). The TGW for haplotypes H3 and H4 was significantly higher than that of haplotypes H1 and H2. For, qTGW-1A.1, affecting GA, GD, GP, GW, and TGW, one KASP marker was developed in the target region of 1,339,530–3,556,253 bp ( ~ 2.2 Mb) on chromosome 1A harboring 39 annotated genes (Tables 1 and S6). Haplotype analysis revealed that the two haplotypes showed significant differences at a significant level of p = 0.01 (Tables S7 and S8, Fig. 4). Haplotype H2 had a significantly higher TGW than Haplotype H1 did.

Haplotype analysis for three important QTL qTGW-1B.1, qTGW-1B.2, and qTGW-1A.1 using a natural wheat population containing 574 cultivars or lines. The ** and *** suggested significance of ANOVA at p < 0.01 and p < 0.001, respectively. The letter on histogram (a, b, and c) indicated multiple comparisons result at the significant level 0.01. The red and blue bars indicated the TGW in Taian of 2017 and 2018, respectively
Discussion
Trait correlations
Grain weight is largely determined by grain morphology. We observed that TGW was highly correlated with the measured grain morphological traits, including GA (0.95), GD (0.95), GP (0.88), GW (0.85), and GL (0.79), which could be partially explained by the co-located QTLs that were identified (Table 1, Fig. 2). The 14 detected QTLs affecting TGW were always co-located with the QTL for the above five morphological traits with the same direction of additive effects. The results suggested that modifying grain morphology is a promising way to increase grain weight. However, the correlations of TGW with GS were very weak, and no QTL affecting both TGW and GS was detected, suggesting that grain weight was not affected by grain shape. Moreover, TGW was more correlated with GW (r = 0.85) than with GL (r = 0.79), and five and two QTL clusters affecting GW/TGW and GL/TGW were detected, respectively, indicating that GW had stronger effects on TGW than GL did at the QTL level.
The trait correlation results were consistent with previous reports by Cui et al. (2014) and Cheng et al. (2017). However, it was surprising that the correlation between GL and GW was positive (r = 0.44), but no QTL affecting both of them was detected; this suggested that the genetic mechanisms of GL and GW may be different and independent. For modern wheat varieties, the accomplishment of high yields benefited from the trade-off among the three yield component traits (spike number per unit, grain number per spike, and grain weight). Thus, the positive phenotypical correlation between GL and GW may have been due to the results of co-directional artificial selection for increasing grain size.
Comparison with previous studies
In total, 88 QTLs affecting all measured traits were identified, including 14 QTL clusters affecting grain size and weight. Of these, nine clusters were mapped to a similar location, with related cloned genes or mapped QTL reported previously. At the region of QTL cluster QW-1A.1 detected in the present study, Heidari et al. (2011) detected the QTL region for grain number (GN), fertile spikes/m2, and grain weight per spike (GWP) and Zanke et al. (2015) identified the QTL for TGW. The QTL cluster QW-3A.1, associated with GA, GD, and TGW in our study, harbored the Cytokinin Oxidase gene Tackx4, which has been found to be associated with flag leaf chlorophyll content and TGW (Chang et al. 2015). Moreover, Liu et al. (2014) detected QTL for grain weight per spike and TGW and Jia et al. (2013) identified QTL for grain weight in the same region. In the region of QW-7B.2, affecting GW and TGW, wheat sucrose synthase gene TaSus1 was found; it participated in the conversion of sucrose to starch and was associated with TGW (Hou et al. 2014).
The QTL cluster QW-1B.3 and QW-2B, associated with GA, GD, GP, GL, and TGW, corresponded to the mapped QTL for TGW on chromosome 1B and 2B, respectively, as detected by Sukumaran et al. (2018). QW-3B for GD, GW, and TGW corresponded to the QTL region affecting GA, GW, and TGW detected by Gegas et al. (2010). Moreover, QW-6A for GW and TGW in the present study were located in the same chromosome region for GW, TGW and factor form density detected by Gegas et al. (2010) and for grain GN, GW, and TGW identified by Huang et al. (2004). At the region of QW-6B for GW and TGW in our study, Wu et al. (2015) and Mccartney et al. (2005) identified QTL for GL, GW, GT, and TGW, and Luo et al. (2016) detected QTL for tiller number. QW-7A, governing GA, GD, and TGW, was in a similar position as QTL for TGW detected by Cui et al. (2014) and Huang et al. (2004). The other five QTL clusters were located on chromosome regions where no QTL for measured traits has been reported; these can be regarded as novel. The results indicated that a joint multiple related population analysis by NAM was an effective way to detect QTL for interested traits.
Among the eight QTL clusters for grain shape, QS-5A, comprising qGR-5A.2 and qGS-5A.3, harbored the major wheat domestication gene Q, a member of the APETALA2-like transcription factor family (Simons et al. 2006). The transformation from allele q to Q on chromosome 5A was found to simultaneously increase GR and decrease GS (Xie et al. 2018). Another two QTL clusters, QS-5B and QS-5D, covered the homologous genes of Q (Simons et al. 2006). Zhang et al. (2011) reported that the chromosome 5A alleles played a key role in domestication-related traits, whereas the chromosome 5B alleles indirectly suppressed the speltoid phenotype, and chromosome 5D alleles were sub-functionalized. The contrasting functions of the chromosome 5A and chromosome 5B alleles for the Q gene could explain why the additive effects for QS-5A.1 and QS-5B were opposite in each population. In this study, the alleles from the semi-wild cultivars CY and YT increased the GS and decreased the GR in QS-5A.1, which indicated that the domestication from semi-wild cultivar to domesticated cultivar accompanied the transition from slender grains to rounder grains (Charmet 2011; Gegas et al. 2010). Our results support the notion that the domestication of grain size accompanied the formation of free-threshing wheat (Kerber and Rowland 1974; Yan et al. 2017).
Application potential of semi-wild cultivars in wheat high-yield breeding
Utilizing semi-wild relatives or importing exotic accessions can be an effective way to broaden the genetic diversity of local breeding materials for breeders (Liu et al. 2014; Luo et al. 2016). In this study, we constructed four interconnected RIL populations by crossing local high-yielding wheat variety, YZ, with three semi-wild relatives and one exotic elite variety. These RILs, carrying exotic chromosome fragments, can serve as precious materials for further breeding and genetic studies.
Through NAM for grain morphological traits and grain weight, we could not only identify QTL but also estimate allelic effects from different parents, which gave us the ability to choose the most favorable alleles for wheat high-yield breeding. For the 14 QTL clusters affecting weight detected in the present study, CY, YT, and HR contributed the most favorable alleles for seven, three, and three QTL clusters, respectively, which indicated that these parents carried precious genes for increasing grain weight. Therefore, we could pyramid the most favorable alleles from different parents for high-yield breeding after converting the peak and flanking SNP of identified QTL into other high-throughput genetic markers, such as KASP, to carry out marker-assisted breeding. In this study, three important QTL clusters (qTGW-1B.1, qTGW-1B.2, and qTGW-1A.1) were validated through a natural wheat population containing 574 cultivars or lines via haplotype analysis. The haplotypes of H7/H8, H3/H4, and H2 for qTGW-1B.1, qTGW-1B.2, and qTGW-1A.1 showed larger TGW than the other haplotypes did. The KASP markers developed for these three important QTLs could be used for marker-assisted high-yield breeding through the improvement of TGW.
Author contribution statement
LK and HW designed the experiments and revise the manuscript. XW analyzed the data and wrote the manuscript. LD, JH, and XM participated in field experiments and the data collection. XW and LD contributed to the genotyping of a natural wheat population. YP assisted in the data analysis and paper revision. XK and JJ provided the RIL populations.
References
Alaux M, Letellier T, Alfama-Depauw F, Jamilloux V, Rogers J, Choulet F, Guerche C, Loaec M, Flores R-G, Michotey C (2016) IWGSC Sequence Repository: Moving towards tools to facilitate data integration for the reference sequence of wheat. In: PAG XXIV-Plant and Animal Genome Conference, San Diego, USA, 2016-01-09-2016-01-13
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brown TA, Jones MK, Powell W, Allaby RG (2009) The complex origins of domesticated crops in the fertile crescent. Trends Ecol Evol 24:103–109
Chang C, Lu J, Zhang HP, Ma CX, Sun G (2015) Copy number variation of cytokinin oxidase gene Tackx4 associated with grain weight and chlorophyll content of flag leaf in common wheat. PLoS ONE 10:e0145970
Charmet G (2011) Wheat domestication: lessons for the future. CR Biol 334:212
Chen F, Yu Y, Xia X, He Z (2007) Prevalence of a novel puroindoline b allele in Yunnan endemic wheats (Triticum aestivum ssp. yunnanense King). Euphytica 156:39–46
Chen Q, Kang HY, Fan X, Wang Y, Sha LN, Zhang HQ, Zhong MY, Xu LL, Zeng J, Yang RW (2013) Evolutionary history of Triticum petropavlovskyi Udacz. et Migusch. Inferred from the sequences of the 3-phosphoglycerate kinase gene. PLoS ONE 8:e71139
Cheng R, Kong Z, Zhang L, Xie Q, Jia H, Yu D, Huang Y, Ma Z (2017) Mapping QTLs controlling kernel dimensions in a wheat inter-varietal RIL mapping population. Theoret Appl Genet 130:1405–1414
Cui F, Zhao C, Ding A, Li J, Wang L, Li X, Bao Y, Li J, Wang H (2014) Construction of an integrative linkage map and QTL mapping of grain yield-related traits using three related wheat RIL populations. Theoret Appl Genet 127:659
Dempewolf H, Baute G, Anderson J, Kilian B, Smith C, Guarino L (2017) Past and future use of wild relatives in crop breeding. Crop Sci 57(3):1070–1082
Dong Y, Zeng X, Chen X (1981) Expedition and investigation of yunnan wheat (Triticum aestivum ssp. yunnanense king). Acta Agronomica Sinica 7(3):145–146
Doyle J (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull Bot Soc Am 19:11–15
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Fuller DQ (2007) Contrasting patterns in crop domestication and domestication rates: recent archaeobotanical insights from the Old World. Ann Bot 100:903–924
Gegas VC, Nazari A, Griffiths S, Simmonds J, Fish L, Orford S, Sayers L, Doonan JH, Snape JW (2010) A genetic framework for grain size and shape variation in wheat. Plant Cell 22:1046–1056
Harrell FE, Dupont C (2013) Hmisc: Harrell Miscellaneous (R Package Version 3)
Heidari B, Sayed-Tabatabaei BE, Saeidi G, Kearsey M, Suenaga K (2011) Mapping QTL for grain yield, yield components, and spike features in a doubled haploid population of bread wheat. Genome 54:517–527
Hou J, Jiang Q, Hao C, Wang Y, Zhang H, Zhang X (2014) Global selection on sucrose synthase haplotypes during a century of wheat breeding. Plant Physiol 164:1918
Huang XQ, Cöster H, Ganal MW, Röder MS (2003) Advanced backcross QTL analysis for the identification of quantitative trait loci alleles from wild relatives of wheat (Triticum aestivum L.). Theoret Appl Genet 106:1379–1389
Huang XQ, Kempf H, Ganal MW, Röder MS (2004) Advanced backcross QTL analysis in progenies derived from a cross between a German elite winter wheat variety and a synthetic wheat (Triticum aestivum L.). Theoret Appl Genet 109:933–943
Huang XQ, Cloutier S, Lycar L, Radovanovic N, Humphreys DG, Noll JS, Somers DJ, Brown PD (2006) Molecular detection of QTLs for agronomic and quality traits in a doubled haploid population derived from two Canadian wheats (Triticum aestivum L.). Theoret Appl Genet 113:753–766
Jia H, Wan H, Yang S, Zhang Z, Kong Z, Xue S, Zhang L, Ma Z (2013) Genetic dissection of yield-related traits in a recombinant inbred line population created using a key breeding parent in China’s wheat breeding. Theoret Appl Genet 126:2123–2139
Kerber ER, Rowland GG (1974) Origin of the free-threshing character in hexaploid wheat. Can J Genet Cytol 16:145–154
Li W, Yang B (2017) Translational genomics of grain size regulation in wheat. Theoret Appl Genet 130:1765–1771
Liu G, Jia L, Lu L, Qin D, Zhang J, Guan P, Ni Z, Yao Y, Sun Q, Peng H (2014) Mapping QTLs of yield-related traits using RIL population derived from common wheat and Tibetan semi-wild wheat. Theoret Appl Genet 127:2415–2432
Luo W, Ma J, Zhou XH, Sun M, Kong XC, Wei YM, Jiang YF, Qi PF, Jiang QT, Liu YX (2016) Identification of quantitative trait loci controlling agronomic traits indicates breeding potential of Tibetan semiwild wheat (ssp.). Crop Sci 56(5):2410–2420
Mccartney CA, Somers DJ, Humphreys DG, Lukow O, Ames N, Noll J, Cloutier S, Mccallum BD (2005) Mapping quantitative trait loci controlling agronomic traits in the spring wheat cross RL4452x'AC Domain'. Genome 48:870
Pang Y, Chen X, Zhao J, Du W, Cheng X, Wu J, Li Y, Wang L, Wang J, Yang Q (2014) Molecular cytogenetic characterization of a wheat-leymus mollis 3D (3Ns) substitution line with resistance to leaf rust. J Genet Genom 41:205–214
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rasheed A, Xia X, Ogbonnaya F, Mahmood T, Zhang Z, Mujeebkazi A, He Z (2014) Genome-wide association for grain morphology in synthetic hexaploid wheats using digital imaging analysis. BMC Plant Biol 14:128
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Iv ESB (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Ren D, Fang X, Jiang P, Zhang G, Hu J, Wang X, Meng Q, Cui W, Lan S, Ma X, Wang H, Kong L (2018) Genetic architecture of nitrogen-deficiency tolerance in wheat seedlings based on a nested association mapping (NAM) population. Front Plant Sci 9:845
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Epidemiology 273:350–354
Shao Q, Li C, Besang C (1980) Semi-wild wheat from Xizang (Tibet). Acta Genet Sin 7:149–156
Simmonds J, Scott P, Brinton J, Mestre TC, Bush M, Blanco AD, Dubcovsky J, Uauy C (2016) A splice acceptor site mutation in TaGW2-A1 increases thousand grain weight in tetraploid and hexaploid wheat through wider and longer grains. Theoret Appl Genet 129:1099–1112
Simons KJ, Fellers JP, Trick HN, Zhang Z, Tai YS, Gill BS, Faris JD (2006) Molecular characterization of the major wheat domestication gene Q. Genetics 172:547–555
Stich B, Utz HF, Piepho HP, Maurer HP, Melchinger AE (2010) Optimum allocation of resources for QTL detection using a nested association mapping strategy in maize. Theoret Appl Genet 120:553–561
Sukumaran S, Lopes M, Dreisigacker S, Reynolds M (2018) Genetic analysis of multi-environmental spring wheat trials identifies genomic regions for locus-specific trade-offs for grain weight and grain number. Theoret Appl Genet 131(4):985–998
Sun Q, Ni Z, Liu Z, Gao J, Huang T (1998) Genetic relationships and diversity among Tibetan wheat, common wheat and European spelt wheat revealed by RAPD markers. Euphytica 99:205–211
Tang X, Dong S, Xu J, Li Y, Li W, Ren Z, Fu T (2014) Molecular cytogenetic characteristics of a translocation line between common wheat and Thinopyrum intermedium with resistance to powdery mildew. Euphytica 197:201–210
Wang H, Wang X, Chen P, Liu D (2007) Assessment of genetic diversity of Yunnan, Tibetan, and Xinjiang wheat using SSR markers. J Genet Genom 34:623–633
Wang J, Liu W, Hui W, Li L, Wu J, Yang X, Li X, Gao A (2011) QTL mapping of yield-related traits in the wheat germplasm 3228. Euphytica 177:277–292
Wang S, Wong D, Forrest K, Allen A, Chao S, Huang BE, Maccaferri M, Salvi S, Milner SG, Cattivelli L (2014) Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796
Wu QH, Chen YX, Zhou SH, Fu L, Chen JJ, Xiao Y, Zhang D, Ouyang SH, Zhao XJ, Cui Y (2015) High-density genetic linkage map construction and QTL mapping of grain shape and size in the wheat population Yanda 1817 × Beinong6. PLoS ONE 10:e0118144
Xavier A, Xu S, Muir W, Rainey K (2015) NAM: association studies in multiple populations. Bioinformatics 31:3862–3864
Xie Q, Li N, Yang Y, Lv Y, Yao H, Wei R, Sparkes DL, Ma Z (2018) Pleiotropic effects of the wheat domestication gene Q on yield and grain morphology. Planta 247:1089–1098
Yan L, Liang F, Xu H, Zhang X, Zhai H, Sun Q, Ni Z (2017) Identification of QTL for grain size and shape on the D genome of natural and synthetic allohexaploid wheats with near-identical AABB genomes. Front Plant Sci 8:1705
Yu J, Holland JB, Mcmullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551
Zanke CD, Ling J, Plieske J, Kollers S, Ebmeyer E, Korzun V, Argillier O, Stiewe G, Hinze M, Neumann F (2015) Analysis of main effect QTL for thousand grain weight in European winter wheat (Triticum aestivum L.) by genome-wide association mapping. Front Plant Sci 6:644
Zhang Z, Belcram H, Gornicki P, Charles M, Just J, Huneau C, Magdelenat G, Couloux A, Samain S, Gill BS, Rasmussen JB, Barbe V, Faris JD, Chalhoub B (2011) Duplication and partitioning in evolution and function of homoeologous Q loci governing domestication characters in polyploid wheat. Proc Natl Acad Sci USA 108:18737–19187
Zhang R, Zhang M, Wang X, Chen P (2014) Introduction of chromosome segment carrying the seed storage protein genes from chromosome 1V of Dasypyrum villosum showed positive effect on bread-making quality of common wheat. Theoret Appl Genet 127:523–533
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFD0100602), the Transgenic Special Item of China (2016ZX08002003-002), National Natural Science Foundation of China (31520103911), and Shandong Modern Agricultural Technology &Industry System (SDAIT-01-01).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical standards
I declare on behalf of my co-authors that the work described is original, previously unpublished research and not under consideration for publication elsewhere. The experiments in this study comply with the current laws of China.
Additional information
Communicated by Steven S. Xu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2019_3410_MOESM2_ESM.tif
Fig. S1 Phenotypic correlations between seven environments for eight traits. Blue shades indicate significant at P < 0.01. TGW, thousand grain weight; GA, Grain area; GP, Grain perimeter; GS, Grain shape; GL, Grain length; GW, Grain width; GD, Grain diameter; GR, Grain roundness.(TIFF 1399 kb)
122_2019_3410_MOESM3_ESM.tif
Fig. S2 (a) Values of LnP(D) generated from STRUCTURE, with its modal value used to detect the true k of three groups (k = 3). (b) Heat map of kinship from TASSEL with the tree shown on the top and left. (c) Bayesian clustering for NAM populations using STRUCTURE program. (TIFF 526 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, X., Dong, L., Hu, J. et al. Dissecting genetic loci affecting grain morphological traits to improve grain weight via nested association mapping. Theor Appl Genet 132, 3115–3128 (2019). https://doi.org/10.1007/s00122-019-03410-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03410-4