
Abstract
The success of personalized medicine depends on the discovery of biomarkers that allow oncologists to identify patients that will benefit from a particular targeted drug. Molecular tests are mostly performed using tumor samples, which may not be representative of the tumor’s temporal and spatial heterogeneity. Liquid biopsies, and particularly the analysis of circulating tumor DNA, are emerging as an interesting means for diagnosis, prognosis, and predictive biomarker discovery. In this study, the amplification refractory mutation system (ARMS) coupled with high-resolution melting analysis (HRMA) was developed for detecting two of the most relevant KRAS mutations in codon 12. After optimization with commercial cancer cell lines, KRAS mutation screening was validated in tumor and plasma samples collected from patients with pancreatic ductal adenocarcinoma (PDAC), and the results were compared to those obtained by Sanger sequencing (SS) and droplet digital polymerase chain reaction (ddPCR). The developed ARMS-HRMA methodology stands out for its simplicity and reduced time to result when compared to both SS and ddPCR but showing high sensitivity and specificity for the detection of mutations in tumor and plasma samples. In fact, ARMS-HRMA scored 3 more mutations compared to SS (tumor samples T6, T7, and T12) and one more compared to ddPCR (tumor sample T7) in DNA extracted from tumors. For ctDNA from plasma samples, insufficient genetic material prevented the screening of all samples. Still, ARMS-HRMA allowed for scoring more mutations in comparison to SS and 1 more mutation in comparison to ddPCR (plasma sample P7). We propose that ARMS-HRMA might be used as a sensitive, specific, and simple method for the screening of low-level mutations in liquid biopsies, suitable for improving diagnosis and prognosis schemes.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Kirsten rat sarcoma viral oncogene homolog (KRAS) gene encodes for a small GTPase and is mutated in 25% of all cancers [1, 2]. The KRAS protein is cyclically in an active state, when bound to guanosine triphosphate (GTP), and inactive, when associated with guanosine diphosphate (GDP), thus functioning as a molecular switch responsible for activating signaling pathways involved in tumorigenesis [1, 2]. KRAS mutations are commonly missense mutations, the majority of which are in codon 12 of the gene [3,4,5,6], quite common in colorectal cancer (CRC) and non-small cell lung cancer (NSCLC), where they have been associated with poor prognosis [4, 5, 7,8,9]. Moreover, KRAS mutations are a negative predictive biomarker of epidermal growth factor receptor (EGFR)–targeted therapy response in CRC [10]. More importantly, KRAS is the most frequently mutated gene in pancreatic ductal adenocarcinoma (PDAC), showing mutations in more than 80% of patients [6]. In fact, pancreatic cancer stands out as that with the lowest survival rate in Europe, with PDAC accounting for 95% of cases [11]. The absence of screening tests and symptoms at the initial stages of the disease implies that, at the time of diagnosis, 80% of tumors are locally advanced or metastatic [11, 12]. The survival rate is further impacted by the recurrence levels, which occurs in 80% of patients after resection surgery [12].
Several techniques have been used for KRAS mutation detection, including single-strand conformation polymorphism (SSCP) [13], enriched-PCR and enzyme-linked mini-sequence assay (ELMA-PCR) [14], clamping peptide nucleic acids PCR (PNA-PCR) [15], restriction fragment length polymorphism (RFLP) assays [16], allele-specific locked nucleic acid PCR (AS-LNA PCR) [17], and Sanger sequencing (SS) [16]. Until recently, SS was considered the gold standard for KRAS mutation testing [18] but its limit of detection hampers its clinical application. Several recent studies have proved that methods with higher analytical sensitivities for KRAS mutation analysis may benefit treatment selection for metastatic cancer patients [18]. Between 2014 and 2017, the United States Food and Drug Administration (US FDA) approved three different strategies for KRAS mutation detection, the therascreen™ KRAS RGQ PCR Kit (Qiagen, Hilden, Germany), a combined approach of ARMS with bi-functional fluorescent primer/probe molecules, the real-time PCR cobas™ KRAS Mutation Test (Roche Molecular Systems, Inc., Switzerland), and the next-generation sequencing (NGS) Praxis™ Extended RAS Panel (Illumina, Inc., San Diego, CA, USA) with analytical sensitivities in the range of approximately 1–5% [19, 20].
The development of high-throughput technologies for molecular characterization of tumor cells has led to a better understanding of the genomic complexity of cancer [21, 22], which, as a result of new therapeutic agents aiming at specific targets, such as genes, proteins, and regulatory pathways [23, 24], allowed shifting from the traditional therapy of “one size fits all” to a more individualized approach based on each patient’s tumor-specific profiles [22, 25]. This new strategy of personalized medicine relies on the discovery of new biomarkers toward the molecular profiling of patients fitting to a specific targeted drug [23, 25]. Besides the impact for targeted therapy selection, molecular characterization of samples is also a powerful tool for early cancer detection and prognosis assessment [22, 26].
Molecular assays have been mostly performed on tumor biopsies [27, 28], which presents some limitations, such as the invasiveness and the use of single-site tissue biopsies collected from the primary tumor that may fall short of representing the tumor heterogeneity (including metastases) [27, 29,30,31]. Furthermore, samples collected at a single time point do not account for the dynamic of clonal evolution during treatment [32, 33]. Because of these limitations, current trends focus on the use of liquid biopsies, i.e., material recovered from circulating body fluids (e.g., blood and plasma), which can be easily retrieved through minimally invasive procedures, thus allowing for repeated sampling [22, 28]. Besides the longitudinal assessment of tumor-specific biomarkers, circulating tumor DNA (ctDNA) analysis provides a more comprehensive picture of intertumoral heterogeneity, since it derives from various cancer cells from both the primary tumor and metastases [27, 28, 34, 35]. In this regard, the analysis of ctDNA is gaining momentum in routine procedures of molecular profiling of cancer patients. Still, the biggest challenge in biomarker evaluation in ctDNA lies within the low percentage of ctDNA present in total cell-free DNA (cfDNA), and in the low frequency of mutant alleles found on a wide background of wild-type (wt) DNA [36, 37]. Considering the low amount of ctDNA found in body fluids, its isolation is a challenging procedure that severely impacts downstream molecular assessment [36, 38,39,40], namely by the well-known gold standard method SS [41]. Indeed, SS shows a modest limit of detection (allele frequency higher than 10%), is labor intensive, and is associated with high costs and a long run time [41,42,43]. In the clinical context, droplet digital PCR (ddPCR), a molecular detection method based on the partitioning of samples into thousands of nanoliter-sized droplets where individual PCRs take place, which, due to the low limit of detection and assessment of allele frequency, has gained momentum as a powerful tool for the detection of genetic alterations, such as single nucleotide and copy number variations, among others, in different biological fluids (e.g., peripheral blood (plasma and serum), urine, etc.) [44]. ddPCR application in the field of precision oncology has been crucial due to its extraordinarily accurate detection and allowing for low-abundance molecular target detection with high sensitivity, bypassing SS bottlenecks [44]. Nevertheless, ddPCR also has some disadvantages, such as slightly longer turnaround times compared to traditional PCR-based applications (due to droplet generation and reading) and its limited availability in all laboratories [45, 46]. There is plenty of room for new simple, fast, cost-effective and highly sensitive molecular methods for precision oncology.
High-resolution melting analysis (HRMA) is a fast and cost-effective approach for high-throughput mutation screening [41, 47]. HRMA is based on the dissociation behavior of DNA when subjected to increasing temperatures, in the presence of saturating fluorescent dyes with greater affinity for double-strand DNA than for single-strand DNA [41, 47, 48]. HRMA as a stand-alone technology has been shown to detect between 3 and 10% of mutant DNA in a background of wild-type (wt) DNA [41], which surpasses SS limitations. However, since any mutation in the analyzed region can result in a melting profile distinct from the wt, HRMA requires confirmatory testing of positive results [42, 43]. The amplification refractory mutation system (ARMS) stands out for its high sensitivity, run time, and specificity for genotyping [41, 43, 49], and is based on the use of primers whose 3′ nucleotides are allele-specific, thus resulting in amplification in samples containing the respective allele [41, 50]. The combination of ARMS and HRMA could circumvent some of the hurdles of current technologies.
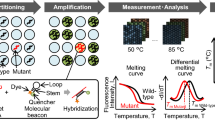
Herein, we developed an ARMS-HRMA assay for the detection of the KRAS mutations suitable for application in solid and liquid (plasma) biopsies. We targeted G12D and G12V, two of the most relevant KRAS mutations in cancer, and the two mutations most detected in PDAC [6]. After assay optimization using immortalized cell lines, the methodology was validated in tumor and plasma samples from patients with PDAC, and the performance compared to the gold standard (SS) and droplet digital PCR (ddPCR)—see Fig. 1.

Schematic representation of the proposed method for KRAS mutation detection based on combinatory ARMS-HRMA assay. 1. First, tumor and plasma samples were collected from patients with PDAC. 2. Next, genomic DNA was extracted from the samples and further purified and quantified. 3a. ARMS reactions were performed aiming at the detection of wild-type, G12V, and G12D phenotypes. 3b. The obtained ARMS products were subjected to HRMA for unequivocal phenotype discrimination. The results previously obtained by ARMS-HRMA were compared to those attained by 4a. SS and 4b. ddPCR. 5. The results obtained by the three methods were compared for sample genotyping
Materials and methods
The tumor samples used in this study were obtained from 30 patients with PDAC from Hospital Beatriz Ângelo and Hospital da Luz, Lisbon, Portugal. Plasma samples were isolated by centrifugation immediately after whole blood (6 mL) collection from each patient and were immediately stored at – 80 °C until DNA extraction (IRB HBA 00,184). Written informed consent was obtained from all patients.
DNA extraction
DNA extraction from PDAC patients’ samples was performed using the High Pure PCR Template Preparation Kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions for extraction from solid tissue and whole blood/plasma. A small modification of the protocol referred to the elution volume: 50 μL for tumor samples and 30 μL for plasma. DNA yields were quantified using a Nanodrop spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Control DNA was extracted from colorectal cancer cell lines SW48 – wildtype genotype (American Type Culture Collection [ATCC]® reference no. CCL-231), SW480 – G12V mutation (ATCC® reference no. CCL-228), and LS174T – G12D mutation (ATCC® reference no. CL-188) as previously described [51].
PCR amplification for SS
To assess KRAS exon 2 mutational status, 50 ng of template DNA was used for standard PCR amplification using 0.12 μM of each primer (forward: 5′-GGTGGAGTATTTGATAGTGTA-3′; reverse: 5′-TGGACCCTGACATACTCCCAAG-3′), 2 mM of MgCl2 (NzyTech, Lisbon, Portugal), 0.8 mM of dNTP Mix (NzyTech, Lisbon, Portugal), and 0.15 units of NZYTaq II DNA Polymerase (NzyTech, Lisbon, Portugal) in a final reaction volume of 20 μL. Reactions were performed on a DNA Engine® thermocycler (Bio-Rad, Hercules, CA, USA) as follows: 95 °C for 5 min, followed by 30 cycles of 95 °C for 30 s, 61 °C for 30 s for tumor samples and 53 °C for 30 s for plasma samples, and 72 °C for 20 s. For plasma samples, four independent amplification reactions were performed for each sample, which were later concentrated using the SpeedVac Concentrator (Thermo Fisher Scientific, Waltham, MA, USA) and resuspended in 15 μL of DEPC-treated water.
PCR products were then direct sequenced at STAB VIDA (Setubal, Portugal), and the chromatograms analyzed using FinchTV software (Geospiza, Inc) for sequence characterization and identification of possible mutations in codon 12.
Droplet digital PCR
For droplet digital PCR (ddPCR) analysis, genomic DNA and cfDNA concentrations were measured by Qubit Fluorometer using Qubit dsDNA High Sensitivity Assay Kit (Life Technologies), following the manufacturer’s instructions. ddPCR was performed using the ddPCR™ KRAS G12/G13 Screening Kit (Biorad, CA, USA) according to the manufacturer’s instructions for the droplet digital amplification of KRAS G12/13 mutations using 0.5–10 ng of DNA per reaction, depending on the quantity of DNA available. The droplet generation, reaction amplification, and droplet readout were conducted on a QX200 Droplet Digital PCR System (Biorad, CA, USA). The final score of the positive/negative droplets and the concentration in copies/microliter were determined using QuantaSoft™ software (Bio-Rad, CA, USA). Additionally, for SW48 (wt), LS174T (G12D), and SW480 (G12V), the following controls were used for the indicated mutation profile: Caco-2 for wild-type status, HD701 (Horizon) for G12D and G13D, and genomic DNA from FFPE cancer samples positive by NGS analysis for KRAS G12A, G12C, G12R, or G12S. For cfDNA analysis, the control HD917 (Horizon) was also used in the wild type and 5% allele frequency version. Non-template controls (only with water) were used to discard any contamination.
ARMS-HRMA for the detection of KRAS G12V and KRAS G12D mutations
Sample DNA was analyzed for the G12D and G12V KRAS point mutations through ARMS coupled with HRMA (amplicon size 96 bp). The reaction mixture consisted of 0.3 μM of forward and reverse primers (primer forward G12V 5′-CTTGTGGTAGTTGGAGCTTT-3′; primer forward G12D 5′-CTTGTGGTAGTTGGAGCTTA-3′; primer reverse 5′-CTCTATTGTTGGATCATATTCG-3′), 2% (v/v) of DMSO (Sigma-Aldrich, St. Louis, MO, USA), 1 × of NZYTaq II 2 × Green Master Mix (NzyTech, Lisbon, Portugal) or NZYSpeedy qPCR Green Master Mix (2 ×) (NzyTech, Lisbon, Portugal), and 4 ng of template DNA in a final reaction volume of 10 μL.
ARMS-HRMA reactions were performed on a Corbett Rotor-Gene 6000 (Qiagen, Hilden, Germany) thermocycler, and conditions included an initial denaturation at 95 °C for 3 min; 10 cycles of 95 °C for 30 s, 52 °C for 15 s for G12V mutation and 54 °C for 15 s for G12D mutation, and 72 °C for 10 s; followed by 25 cycles of 95 °C for 30 s, 60 °C for 45 s, and 72 °C for 10 s. Afterward, ARMS products were assessed by HRMA through temperature increase from 45 to 95 °C, rising at 0.2 °C per step/wait 5 s each step. The resulting derivative plot was generated using Rotor-Gene 6000 Series Software 1.7 (Qiagen, Hilden, Germany). For the difference plot, the melting curve of each sample was subtracted from that corresponding to the SW48 cell line (wt) [52].
Controls in each ARMS-HRMA reaction included a non-template-control, a “wt” control (gDNA from SW48 cell line), and a “mutant” control (gDNA from LS174T and SW480 cell lines for G12D mutation and G12V mutation, respectively). The final mutation scoring was attained by the normalization of the fluorescence value at 79.5 °C to the correspondent positive and negative cell-line controls of each reaction. The average normalized result was then scored using a threshold of 0.5 (based on the average results of the mutant and non-mutated cell lines). Accordingly, samples with a final fluorescence above the threshold (0.5) were scored mutated and the ones with fluorescence below the threshold as non-mutated.
Results and discussion
Herein, we present a simple yet robust approach for the rapid molecular profiling of KRAS mutations in PDAC patients, which combines the amplification refractory mutation system followed by high-resolution melting analyses (ARMS-HRMA), for the detection of two of the most relevant KRAS mutations (G12V and G12D) in these patients. Because these two alterations are single-point missense mutations, we used an allele-specific amplification (ARMS) where the forward primers were designed to favor amplification of the target mutated allelic sequence to the detriment of the wt allele. Even though ARMS-primers provide for the allele specificity of amplification, mismatched priming at 3′ still allows DNA polymerase to yield residual amplification at a much lower efficiency (Supplementary Information S1 and S2). To warrant complete disambiguation of nucleobase call and correctly identify samples containing the mutated allele, HRMA was performed directly on the obtained ARMS products (closed tube). By doing this, a more intense fluorescence signal is retrieved when in the presence of the mutation that provided for correct priming in ARMS reaction, resulting in a higher efficiency of amplification and amplicon yield. Consequently, the ARMS-HRMA approach is based on the difference of the melting profile (melting bands and corresponding intensity) after ARMS reaction, when compared to control genotypes (mutated or wild type).
ARMS-HRMA assay development
First, we used DNA retrieved from cell lines with known genotypes SW480 (homozygous for G12V), LS174T (wt/G12D), and SW48 (wt/wt) to optimize the ARMS-HRMA approach. A critical aspect was the definition of the criteria for allele discrimination, dependent on the melting temperature and fluorescence peak intensity. Following ARMS amplification using the G12V forward primer, the melting profile analysis, HRMA, revealed the presence of a melting peak at approximately the same temperature for both SW480 and SW48 cell lines (80 °C), which indicates that the amplification occurred in both samples (Fig. 2A). However, the melting peaks are clearly distinguishable as they present variations in intensity (fluorescence signal), with the highest peak corresponding to the homozygous G12V cell line, indicating that the degenerated forward primer (allele specific against G12V mutation) is allowing the amplification of DNA containing the G12V allele at a much higher efficiency than the amplification of wt phenotype, yielding more amplification product, which translates to a higher intensity of the melting peak. Overall, this shows a successful identification of samples bearing the KRAS G12V mutation via the proposed ARMS-HRMA approach (Fig. 2A).

HRMA derivative plot for KRAS mutation detection. (A1) Derivative plot generated after ARMS amplification for the detection of KRAS G12V and (B1) KRAS G12D mutations. (A2) Average intensity of the melting peak generated after ARMS-HRMA reaction for the detection of G12V and (B2) G12D mutations in G12V/G12V (SW480) (green line), wt/wt (SW48) (gray line), and G12D/wt (LS174T) (yellow line). ****P value < 0.0001 using Mann–Whitney test. Error bars represent the standard error mean of the average result of at least 8 replicates
We then evaluated the potential of ARMS-HRMA to detect the KRAS G12D mutation (GGT > GAT), using a G12D-specific primer. This primer differs from that specific for the G12V mutation in the 3′ nucleotide, which is allele specific. As for the G12V mutation, the detection of G12D was optimized using genomic DNA from cell line SW48 (wt/wt) as wild-type control and LS174T (wt/G12D) as G12D positive control. The derivative plot in Fig. 2B shows two distinct melting peaks, one around 75.5 °C, independent of the sample genotype, and a second around 79.5 °C, exclusive for the G12D mutant DNA (Fig. 2B). Overall, using ARMS-HRMA, it was possible to attain distinct melting profiles that correlate to the distinct genotypes, allowing for correct scoring of the wt/G12D genotype due to the presence of a melting peak at about 79.5 °C.
Our concept relies on a simple and rapid approach to detect the presence of KRAS G12D and/or G12V mutations directly in samples, and not to genotype, i.e., to provide a quick response of “the mutation is present” rather than to characterize the sample as wild-type (wt) homozygous or wt heterozygous. Nevertheless, a wt primer was designed to amplify the wt allele (Figure S2 panel D). SW48 (wt/wt) and LS174T (G12D/wt) cell lines were also used to validate these wt primers. The results show a higher peak intensity of the melting peak (~ 78.5 °C) for the SW48 cell line (wt/wt) (gray line in Figure S2 panel C). Furthermore, for the LS174T cell line (G12D/wt), a melting peak also appears in the same melting temperature but with a much lower intensity (yellow line in Figure S2 panel C), as expected due to the heterozygous state of the cell sample. Finally, the specificity for the wt allele can be observed with the homozygotic mutated cell line, SW480 (G12V/G12V), where the correspondent melting peak (~ 78.5 °C) does not appear (blue line in Figure S2 panel C).
ARMS-HRMA application to tumor DNA
The successful mutation identification via the ARMS-HRMA scheme, with sensitivity to differentiate between a homozygous wild-type sample from a homozygous or heterozygous mutated sample, was then applied to the analysis of DNA extracted from tumor biopsies and matching plasma samples. ARMS-HRMA was performed in a total of 30 tumor samples from patients diagnosed with PDAC. All the DNA samples with sufficient material were also analyzed by SS and ddPCR. Due to the small amount of patient material available from biopsy, samples P3, P10, P13, P15, P17, and P19 were not assessed by SS (Supplementary Information S3), and samples T9, T10, T13, T24, P1, P10, P15, and P19 were not assessed by ddPCR (Supplementary Information S4).
A first observation was that the fluorescence intensity signals showed some variation between samples, which may be attributed to different efficiencies in amplification and, thus, of the amount of amplified product that is then available for the HRMA step. Therefore, the melting plot of the SW48 cell line (negative for G12V and G12D mutations) was used as reference and, consequently, its melting peak as the threshold. Applying this criterion, the analyzed samples can be divided into two groups: mutated for G12V (G12V positive), which continues to exhibit a melting peak with high fluorescence signal, and not mutated for G12V (G12V negative), showing a melting curve below or equal to the reference line—Fig. 3. To illustrate this issue, a set of four tumor samples (T1–T4) are shown, all analyzed by ARMS-HRMA for presence of the G12V mutation. Figure 3A shows that all samples present a melting temperature consistent with that observed for SW480 or SW48 cell lines, G12V/G12V and wt/wt, respectively. Samples T1 and T3 may be easily scored as mutated for G12V, since a higher melting peak is present, like that of the SW480 cell line, and T2 and T4 samples are not mutated for G12V, due to the low-intensity melting peak (like that of SW48) (Fig. 3B). These results were corroborated by SS, in which the KRAS G12V mutation was present in T1 and T3 and not detected for T2 (G12D mutation) and T4 (wt) samples (Fig. 3C).

ARMS-HRMA for the detection of KRAS G12V mutation. A HRMA derivative plot for KRAS G12V mutation analysis for the tumor samples T1 (blue green line), T2 (yellow line), T3 (blue line), and T4 (dark gray line). Cell lines SW480 (turquoise line) and SW48 (gray line) as G12V positive and negative controls, respectively. B Difference plot. The difference plot shows the melting curve of each tumor sample subtracted from the SW48 cell line. C Chromograms for SS results for KRAS codon 12 (wt: GGT). Sequence obtained with forward primer. Arrows indicate the mutations at positions 2 of codon 12 of KRAS. Sanger sequencing showed a G to T transversion at position 2 of codon 2 (G12V: GGT > GTT) in tumors T1 and T3, and a G to A transition (G12D: GGT > GAT) in tumor T2. T4 has a wt KRAS codon 12
Figure 4 shows the output of the ARMS-HRMA approach for G12V scoring in all 30 tumor samples. As shown, all tumor samples positive for G12V mutation by ARMS-HRMA also scored as G12V by ddPCR indicating a concordance of 100%, and only one tumor was not scored as G12V via direct SS (T12 in Fig. 4 and sample 12 in Table 1). It should be mentioned that the described reactions were performed in quadruplicate with the same result, which highlights the high reproducibility of the ARMS-HRMA methodology.

ARMS-HRMA for the detection of KRAS G12V mutation in tumor samples. A G12V mutation scoring for the set of 30 tumor samples, based on the intensity of the melting peak at 79.5 °C. The dark blue bars represent tumor samples scored as G12V non-mutated. B Statistical analysis of the average intensity of the melting peak in each sample group. Four asterisks, P value < 0.0001 using Mann–Whitney test. The blue green bar represents tumor samples scored as G12V mutated; the light gray bar represents the SW48 cell line as the control for G12V non-mutated samples; the turquoise bar represents the SW480 cell line as the control for G12V mutated samples. The dagger represents samples scored as G12V mutated with non-concordant result based on SS. Error bars represent the standard error mean of the average result of at least 4 replicates
Analysis of tumor samples by ARMS-HRMA for G12D mutation showed similar performance to that of G12V. Figure 5 depicts the typical ARMS-HRMA output from tumor samples for G12D mutation detection using 4 different tumor samples (T2 to T5) (Fig. 5A). Since the melting peak around 79.5 °C is exclusive for G12D mutated samples, there is no need to perform a difference plot for G12D scoring. The analysis of the melting profile allowed scoring T2 and T5 as mutated for G12D (due to the presence of a similar melting peak to the LS174T cell line; Table 1), whereas T1, T3, and T4 were scored as not mutated for G12D. All these mutation profiles were corroborated by direct SS results (Table 1). As for G12V, the same set of 30 tumor samples was screened for the presence of the G12D mutation by ARMS-HRMA (Fig. 4B and Supplementary Information S5, S6, and S7; Table 1).

ARMS-HRMA for KRAS G12D mutation detection. A Derivative plot generated after ARMS amplification for the detection of KRAS G12D mutation in LS174T (wt/G12D) (yellow line) and SW48 (wt/wt) (gray line) cell lines and four tumor samples (T2 (dark yellow line), T3 (blue line), T4 (dark blue line), and T5 (brown line)). B G12D mutation scoring for the set of 30 tumor samples, based on the intensity of the melting peak at 79.5 °C; the dagger represents samples scored as G12D mutated with non-concordant result based on SS, and the asterisk represents samples with non-concordant results by both ddPCR and SS. C Statistical analysis of the average intensity of the melting peak in each sample group. Four asterisks, P value < 0.0001 using Mann–Whitney test. The dark gray bar represents tumor samples scored as G12D non-mutated; the dark yellow bar represents tumor samples scored as G12D mutated; the light gray SW48 cell line as control for G12D non-mutated samples; the light yellow bar represents the LS174T cell line as the control for G12D mutated samples. Error bars represent the standard error mean of the average result of at least 4 replicates
Interestingly, two samples positive for G12D by ARMS-HRMA (T6 and T7) were not scored as mutated by SS (Table 1 and Fig. 5), which might be attributed to the higher sensitivity of the combined techniques [53]. However, ddPCR on these samples showed that T6 was indeed G12D mutated but T7 did not harbor this mutation. The ARMS-HRMA result for T7 might be considered as a false positive or a higher sensitivity of the technique or even the result of genetic heterogeneity of the tumor sample. This is a common aspect when assessing biopsies where the proportion of normal/mutant cells is very high. Indeed, the limit of detection of both PCR-based techniques (ARMS-HRMA and ddPCR) is often much lower (single cell detection) than that of direct SS (requiring at least 10% of mutated cells) [54, 55].
In summary, apart from T7, all KRAS G12V or G12D mutated tumor samples identified by ARMS-HRMA were confirmed by ddPCR, and all these KRAS G12V or G12D mutated tumor samples, except for T6 and T7, also scored mutated for SS (which disagreed with the scoring for T6 against ddPCR). These results demonstrate that different techniques have different sensitivities, which is of relevance when assessing liquid biopsies where the amount of available template and the level of genetic heterogeneity might lead to disparate results. A 17% increase in the detection of mutant alleles in colon cancer tissue samples by a stand-alone ARMS-based approach in comparison to SS has been already reported [43]. Indeed, several studies have conveyed that SS is less sensitive when analyzing tumor samples with less than 30% of neoplastic cells, where ARMS-based approaches increased the mutant detection rate by 12% [56, 57].
ARMS-HRMA for mutation detection in liquid biopsies
We then assessed the performance of the proposed ARMS-HRMA approach in the context of liquid biopsies. ARMS-HRMA was performed on DNA extracted from plasma samples collected from the same set of 30 patients whose tumor samples had also been characterized in Table 1 (Supplementary Information S5 and S6 and Table 2). It should be noted that for some plasma samples, some techniques could not be performed due to the low amount of genetic material (DNA). Foremost, obtaining informative SS results of DNA extracted from plasma samples proved to be a challenge, most likely due to the low amount of genetic material retrieved from each extraction. In fact, it was only possible to obtain enough genetic material from 23 plasma samples (out of the 30 plasma samples). From these, it was not possible to obtain a readable SS for 10 of them; for three other samples, sequencing was only possible with one of the primers. This is in line with what has been observed for liquid biopsy characterization of patients with solid tumors and highlights the need for the development of improved approaches for the recovery of ctDNA from these samples and for mutation detection [56, 57].
Regarding G12V mutation, from the 10 tumor samples previously characterized as mutated for G12V (T1, T3, T8, T12, T13, T18, T19, T21, T27, T29), it was only possible to obtain SS data for 4 plasma samples (P1, P9, P18, and P27), from which ARMS-HRMA and ddPCR identified three as G12V positive (P8, P9, and P18) (see Fig. 6), whereas SS only pointed P9 as G12V mutated. Regarding the two other samples, one was scored as wt (P18) by SS and the other one without a readable sequence (P8). These results underline the higher sensitivity of ARMS-HRMA in comparison to SS, since the proposed approach attained accurate mutation discrimination for samples with the concentration of genetic material below SS working range. Furthermore, samples P8, P9, and P18 belong to patients with G12V-positive tumors detected by SS (T8, T9, and T18), ARMS-HRMA (T8 and T18), and ddPCR (T8 and T18) (Tables 1 and 2). Sample T9 genetic material (DNA) only allowed performing SS genotyping, and the lack of additional tumor material did not allow its genotyping by ARMS-HRMA or ddPCR.

ARMS-HRMA detection of KRAS G12V mutation in plasma samples. A G12V mutation scoring for the set of 30 plasma samples, based on the intensity of the melting peak at 79.5 °C. The dark blue bars represent tumor samples scored as G12V non-mutated. B Statistical analysis of the average intensity of the melting peak in each sample group. Four asterisks, P value < 0.0001 using Mann–Whitney test. The dark blue bar represents plasma samples scored as G12V mutated; the dark gray bar represents the SW48 cell line as the control for G12V non-mutated samples; the blue bar represents the SW480 cell line as the control for G12V mutated samples. The dagger represents samples scored as G12V mutated with non-concordant result based on SS. Error bars represent the standard error mean of the average result of at least 4 replicates
Regarding KRAS G12D, the plasma samples correspondent to the 12 patients whose tumor samples were determined as G12D mutated by ARMS-HRMA (P2, P5, P6, P7, P11, P17, P20, P22, P23, P24, P28, and P30) were analyzed. Only sample P7 was scored as positive by ARMS-HRMA (Fig. 7), while SS and ddPCR did not score any plasma sample as G12D mutated (Table 2). Interestingly, P7 plasma corresponds to the tumor sample (T7) that was also scored as positive for G12D by ARMS-HRMA but not by SS and ddPCR (see discussion above). The fact that ARMS-HRMA detected the presence of the G12D mutation in both the plasma and tumor of this patient supports the high sensitivity of the ARMS-HRMA detection method, where, again, the disparate scoring might be due to sample condition/integrity or tumor heterogeneity or even be a false positive. Interestingly, this patient presented a high level of metastases.

ARMS-HRMA for KRAS G12D mutation detection on plasma samples. A Derivative plot generated after ARMS amplification for the detection of KRAS G12D mutation in plasma sample P7. B Statistical analysis of the average intensity of the melting peak in each sample group. Four asterisks, P value < 0.0001 using Mann–Whitney test. C Mutation scoring for the set of 24 plasma samples, based on the average intensity of the melting peak at 79.5 °C/80 °C. The dark gray bar represents plasma samples scored as G12D non-mutated; the dark yellow bar represents plasma samples scored as G12D mutated; the light gray bar represents the SW48 cell line as the control for G12D non-mutated samples; the yellow bar represents the LS174T cell line as the control for G12D mutated samples. The asterisk represents samples scored as G12D mutated with non-concordant result based on SS and ddPCR
In summary, the KRAS G12D mutation was detected in one plasma sample by ARMS-HRMA (P7), but not detected by SS or ddPCR, in either plasma or tumor samples of patient 7. However, since both tumor and plasma samples revealed the presence of the mutated allele by ARMS-HRMA, the KRAS G12D genotype may be inferred as a true-positive result. In fact, the presence of ctDNA fragments with mutated KRAS in the blood circulation is by itself a validating factor for the presence of mutated cells in a tumor [58].
Altogether, data show that ARMS-HRMA detected all G12V and G12D mutant alleles also assessed and detected by ddPCR in tumor and plasma samples. When compared to SS, the proposed ARMS-HRMA seems to have a higher sensitivity.
Conclusions
We developed, optimized, and validated an ARMS-HRMA methodology to screen the two most common KRAS mutations associated to PDAC—KRAS G12V and G12D. The proposed approach is based on the allele-specific amplification by ARMS followed by HRMA to increase the sensitivity and specificity of the detection, thus enhancing the screening potential in small samples. A critical aspect for the development of this approach was the fast run time, specificity, and sensitivity to allow the support of the clinical decision. When analyzed by ARMS-HRMA, the detection of KRAS mutations improved compared to that attained by direct sequencing in tumor and plasma samples, respectively. It has been reported that SS requires at least 15% of mutant copies in a background of wild-type DNA to yield a conclusive identification of a given mutation [41], which becomes a critical limiting factor when analyzing liquid biopsies and ctDNA in plasma and blood samples [36, 37]. Moreover, depending on the stage, disease burden, and treatment response of patients, the percentage of ctDNA in the total cfDNA background can range from less than 0.1% to over 10% [28], and thus, SS might not be sufficient for screening liquid biopsies and is surpassed in sensitivity without loss of specificity by HRMA or ARMS [41] separately. The synergy attained by ARMS-HRMA shows the potential for detecting low levels of mutated alleles, which is of utmost relevance in assessing liquid biopsies. In fact, even when the amount of ctDNA in plasma is not below the limit of detection of the chosen method, the frequency of the mutated allele is affected by the tumor’s heterogeneity, which, combined with non-tumor cfDNA, can result in a high background signal for the wild-type allele that might suppress the detection of the mutated allele [36, 37], ultimately impacting the result.
Despite the capability of ARMS-HRMA to detect the mutant alleles in plasma, it was only possible to detect G12V or G12D alterations in 4 plasma samples, performing “better” than SS and in line with ddPCR. This might be due to the quality of the sample material and highlights that there is still plenty to be done before the widespread use of liquid biopsies in clinical settings, namely in what relates to the molecular heterogeneity of tumors, the shedding and release of genetic material, and the efficiency of the extraction and purification of genetic material. In fact, one of the major challenges associated with ctDNA extraction concerns the high degree of fragmentation of DNA, which then impacts subsequent operations, and the isolation of small DNA fragments is tremendously important to prevent the loss of fragments that harbor tumor-specific mutations [39]. In the present study, it was not possible to perform ARMS-HRMA and/or SS for some samples due to lack of viable material.
Finally, besides sensitivity and specificity, other factors need to be considered when selecting an assay for mutation detection, particularly turnaround time, technical difficulty, and ease of interpretation of results. In fact, the proposed ARMS-HRMA methodology requires only a single laboratory procedure, ARMS amplification followed by a denaturation step, performed in a closed system using the same apparatus; i.e., in terms of laboratory work, ARMS-HRMA consists exclusively of the preparation of an ARMS reaction mix, which constitutes a clear advantage for the timely response in the clinics. What is more, the ease of operation and the requirement of a standard apparatus in current molecular biology labs are clear advantages of this approach. This is particularly relevant since, considering the efforts to find inhibitors targeting the different KRAS mutant proteins, ARMS-HRMA might be applied to help guide therapy decision by identifying which patients will be susceptible to the available targeted drugs. KRAS codon 2 mutation analysis is not only useful to identify cancer patients with specific KRAS mutations, but also patients with no mutations in that codon, for whom different therapeutic options may be available, namely direct surgery if the tumor is resectable. The present approach may be extended to target any relevant single-point mutation in the genome.
References
Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. https://doi.org/10.1016/j.cell.2017.06.009.RAS.
Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, Roszik J. Targeting KRAS in cancer: promising therapeutic strategies. Cancers (Basel). 2021;13:1–14. https://doi.org/10.3390/cancers13061204.
Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther. 2021;6:1–20. https://doi.org/10.1038/s41392-021-00780-4.
Zhu G, Pei L, Xia H, Tang Q, Bi F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol Cancer. 2021;20:1–17. https://doi.org/10.1186/s12943-021-01441-4.
Xie M, Xu X, Fan Y. KRAS-mutant non-small cell lung cancer: an emerging promisingly treatable subgroup. Front Oncol. 2021;11:1–11. https://doi.org/10.3389/fonc.2021.672612.
Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17:153–68. https://doi.org/10.1038/s41575-019-0245-4.
Pan W, Yang Y, Zhu H, Zhang Y, Zhou R, Sun X. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: a meta-analysis of 41 studies. Oncotarget. 2016;7:8373–88. https://doi.org/10.18632/oncotarget.7080.
Jones RP, Sutton PA, Evans JP, Clifford R, McAvoy A, Lewis J, Rousseau A, Mountford R, McWhirter D, Malik HZ. Specific mutations in KRAS codon 12 are associated with worse overall survival in patients with advanced and recurrent colorectal cancer. Br J Cancer. 2017;116:923–9. https://doi.org/10.1038/bjc.2017.37.
Marabese M, Ganzinelli M, Garassino MC, Shepherd FA, Piva S, Caiola E, Macerelli M, Bettini A, Lauricella C, Floriani I, et al. KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy. Oncotarget. 2015;6:34014–22. https://doi.org/10.18632/oncotarget.5607.
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. https://doi.org/10.1200/JCO.2007.14.7116.
United European Gastroenterology (UEG) Pancreatic cancer across Europe. Taking a united stand; 2018. Available at https://ueg.eu/p/80.
Heredia-Soto V, Rodríguez-Salas N, Feliu J. Liquid biopsy in pancreatic cancer: are we ready to apply it in the clinical practice? Cancers (Basel). 2021;13:1986. https://doi.org/10.3390/cancers13081986.
Dillon DA, Johnson CC, Topazian MD, Tallini G, Rimm DL, Costa JC. The utility of Ki-Ras mutation analysis in the cytologic diagnosis of pancreatobiliary neoplasma. Cancer J. 2000;6:294–301.
Tada M, Tateishi K, Kawabe T, Sasahira N, Isayama H, Komatsu Y, Shiratori Y, Omata M. Quantity of mutant K-Ras gene in pancreatic secretions for diagnosis of pancreatic carcinoma with different assays: analysis of 100 patients. Clin Chim Acta. 2002;324:105–11.
Däbritz J, Hänfler J, Preston R, Stieler J, Oettle H. Detection of Ki-Ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridisation probes. Br J Cancer. 2005;92:405–12. https://doi.org/10.1038/sj.bjc.6602319.
Bournet B, Souque A, Senesse P, Assenat E, Barthet M, Lesavre N, Aubert A, O’Toole D, Hammel P, Levy P, et al. Endoscopic ultrasound-guided fine-needle aspiration biopsy coupled with KRAS mutation assay to distinguish pancreatic cancer from pseudotumoral chronic pancreatitis. Endoscopy. 2009;41:552–7. https://doi.org/10.1055/s-0029-1214717.
Morandi L, de Biase D, Visani M, Cesari V, de Maglio G, Pizzolitto S, Pession A, Tallini G. Allele specific locked nucleic acid quantitative PCR (ASLNAqPCR): an accurate and cost-effective assay to diagnose and quantify KRAS and BRAF mutation. PLoS One. 2012;7:e36084. https://doi.org/10.1371/journal.pone.0036084.
Chen CL, Chen CK, Ho CL, Chi WM, Yeh CH, Hu SP, Friebe P, Palmer S, Huang CS. Clinical evaluation of IntelliPlex™ KRAS G12/13 mutation kit for detection of KRAS mutations in codon 12 and 13: a novel multiplex approach. Mol Diagn Ther. 2019;23:645–56. https://doi.org/10.1007/s40291-019-00418-w.
Adams JA, Post KM, Bilbo SA, Wang X, Sen JD, Cornwell AJ, Malek AJ, Cheng L. Performance evaluation comparison of 3 commercially available PCR-based KRAS mutation testing platforms. Appl Immunohistochem Mol Morphol. 2014;22:231–5. https://doi.org/10.1097/PDM.0b013e3182a127f9.
Udar N, Lofton-Day C, Dong J, Vavrek D, Jung AS, Meier K, Iyer A, Slaughter R, Gutekunst K, Bach BA, et al. Clinical validation of the next-generation sequencing-based extended RAS panel assay using metastatic colorectal cancer patient samples from the phase 3 PRIME study. J Cancer Res Clin Oncol. 2018;144:2001–10. https://doi.org/10.1007/s00432-018-2688-3.
Piña-Sánchez P, Chávez-González A, Ruiz-Tachiquín M, Vadillo E, Monroy-García A, Montesinos JJ, Grajales R, Gutiérrez de la Barrera M, Mayani H. Cancer biology, epidemiology, and treatment in the 21st century current status and future challenges from a biomedical perspective. Cancer Control. 2021;28:1–21. https://doi.org/10.1177/10732748211038735.
Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med. 2020;12:1–19. https://doi.org/10.1186/s13073-019-0703-1.
Kamel HFM, Al-Amodi HSAB. Exploitation of gene expression and cancer biomarkers in paving the path to era of personalized medicine. Genomics Proteomics Bioinformatics. 2017;15:220–35. https://doi.org/10.1016/j.gpb.2016.11.005.
Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: treating cancer with specificity. Eur J Pharmacol. 2018;834:188–96. https://doi.org/10.1016/j.ejphar.2018.07.034.
Millner LM, Strotman LN. The future of precision medicine in oncology. Clin Lab Med. 2016;36:557–73. https://doi.org/10.1016/j.cll.2016.05.003.
Goossens N, Nakagawa S, Sun X, Hoshida Y. Cancer biomarker discovery and validation. Transl Cancer Res. 2015;4:256–69. https://doi.org/10.3978/j.issn.2218-676X.2015.06.04.
Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–86. https://doi.org/10.1200/JCO.2012.45.2011.
Siravegna G, Mussolin B, Venesio T, Marsoni S, Seoane J, Dive C, Papadopoulos N, Kopetz S, Corcoran RB, Siu LL, et al. How liquid biopsies can change clinical practice in oncology. Ann Oncol. 2019;30:1580–90. https://doi.org/10.1093/annonc/mdz227.
Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501:355–64. https://doi.org/10.1038/nature12627.
Endesfelder D, Math D, Gronroos E, Ph D, Martinez P, Ph D, Matthews N, Sc B, Stewart A, Sc M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92.
McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. https://doi.org/10.1016/j.ccell.2014.12.001.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. National Institutes of Health. 2014;6:69–122. https://doi.org/10.1126/scitranslmed.3007094.Detection.
Lee CK, Kim S, Lee JS, Lee JE, Kim SM, Yang IS, Kim HR, Lee JH, Kim S, Cho BC. Next-generation sequencing reveals novel resistance mechanisms and molecular heterogeneity in EGFR-mutant non-small cell lung cancer with acquired resistance to EGFR-TKIs. Lung Cancer. 2017;113:106–14. https://doi.org/10.1016/j.lungcan.2017.09.005.
Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DWY, Marass F, Gale D, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015;6:1–6. https://doi.org/10.1038/ncomms9760.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;19:224a. https://doi.org/10.1093/nar/gkq421.
Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Goodman SN, David KA, Juhl H, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005;102:16368–73. https://doi.org/10.1073/pnas.0507904102.
Gorgannezhad L, Umer M, Islam MN, Nguyen NT, Shiddiky MJA. Circulating tumor DNA and liquid biopsy: opportunities, challenges, and recent advances in detection technologies. Lab Chip. 2018;18:1174–96. https://doi.org/10.1039/c8lc00100f.
Devonshire AS, Whale AS, Gutteridge A, Jones G, Cowen S, Foy CA, Huggett JF. Towards standardisation of cell-free DNA measurement in plasma: controls for extraction efficiency, fragment size bias and quantification. Anal Bioanal Chem. 2014;406:6499–512. https://doi.org/10.1007/s00216-014-7835-3.
Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, Gligorich KM, Rostomily RC, Bronner MP, Shendure J. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12:1–24. https://doi.org/10.1371/journal.pgen.1006162.
Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, del Rio M, Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010;38:6159–75. https://doi.org/10.1093/nar/gkq421.
Shackelford RE, Whitling NA, McNab P, Japa S, Coppola D. KRAS testing: a tool for the implementation of personalized medicine. Genes Cancer. 2012;3:459–66. https://doi.org/10.1177/1947601912460547.
Tsiatis AC, Norris-Kirby A, Rich RG, Hafez MJ, Gocke CD, Eshleman JR, Murphy KM. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: diagnostic and clinical implications. J Mol Diagn. 2010;12:425–32. https://doi.org/10.2353/jmoldx.2010.090188.
Franklin WA, Haney J, Sugita M, Bemis L, Jimeno A, Messersmith WA. KRAS mutation: comparison of testing methods and tissue sampling techniques in colon cancer. J Mol Diagn. 2010;12:43–50. https://doi.org/10.2353/jmoldx.2010.080131.
Olmedillas-López S, Olivera-Salazar R, García-Arranz M, García-Olmo D. Current and emerging applications of droplet digital PCR in oncology: an updated review. Mol Diagn Ther. 2022;26:61–87. https://doi.org/10.1007/s40291-021-00562-2.
Soverini S, Bernardi S, Galimberti S. Molecular testing in CML between old and new methods: are we at a turning point? J Clin Med. 2020;9:1–16. https://doi.org/10.3390/jcm9123865.
Chung HJ, Hur M, Yoon S, Hwang K, Lim HS, Kim H, Moon HW, Yun YM. Performance evaluation of the QXdX BCR-ABL %is droplet digital PCR assay. Ann Lab Med. 2020;40:72–5. https://doi.org/10.3343/alm.2020.40.1.72.
Reed GH, Kent JO, Wittwer CT. High-resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics. 2007;8:597–608. https://doi.org/10.2217/14622416.8.6.597.
Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154–60. https://doi.org/10.1006/abio.1996.9916.
Huang MM, Arnheim N, Goodman MF. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 1992;20:4567–73. https://doi.org/10.1093/nar/20.17.4567.
Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF. Analysis of any point mutation in DNA. The Amplification Refractory Mutation System (ARMS). Nucleic Acids Res. 1989;17:2503–16. https://doi.org/10.1093/nar/17.7.2503.
Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJM, Leitão CN, Fodde R, Smits R. The, “just-Right” signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002;11:1549–60. https://doi.org/10.1093/hmg/11.13.1549.
Morão B, Oliveira B, Costa B, Saraiva S, Fidalgo C, Faias S. Molecular profiling and prognostic value of KRAS gene mutations in tumor samples and liquid biopsies from patients with pancreatic ductal adenocarcinoma. Ann Gas-troenterol Dig Dis. 2022;5:1–15.
Moding EJ, Nabet BY, Alizadeh AA, Diehn M. Detecting liquid remnants of solid tumors: circulating tumor DNA minimal residual disease. Cancer Discov. 2021;11:2968–86. https://doi.org/10.1158/2159-8290.CD-21-0634.
Gomes A, Korf B. Chapter 5 Genetic testing techniques. In: Robin NH, Farmer MB, editors. Pediatric cancer geneticsm. Elsevier; 2018. pp. 47–64. https://doi.org/10.1016/b978-0-323-48555-5.00005-3.
Hagemann IS. Chapter 1 Overview of technical aspects and chemistries of next-generation sequencing. In: Kulkarni S, Pfeifer J, editors. Clinical genomics. Boston: Academic Press; 2015. p. 3–19 (ISBN 978-0-12-404748-8).
Malapelle U, Bellevicine C, Salatiello M, De Luca C, Rispo E, Riccio P, Sparano L, De Stefano A, Carlomagno C, Maiello FM, et al. Sanger sequencing in routine KRAS testing: a review of 1720 cases from a pathologist’s perspective. J Clin Pathol. 2012;65:940–4. https://doi.org/10.1136/jclinpath-2012-200773.
Tol J, Dijkstra JR, Vink-Börger ME, Nagtegaal ID, Punt CJA, van Krieken JHJM, Ligtenberg MJL. High sensitivity of both sequencing and real-time PCR analysis of KRAS mutations in colorectal cancer tissue. J Cell Mol Med. 2010;14:2122–31. https://doi.org/10.1111/j.1582-4934.2009.00788.x.
Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, Gillet B, Gongora C, Dechelotte P, Robert B, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20:430–5. https://doi.org/10.1038/nm.3511.
Acknowledgements
This work is financed by national funds from FCT—Fundação para a Ciência e a Tecnologia, I.P., in the scope of the project UIDP/04378/2020 and UIDB/04378/2020 of the Research Unit on Applied Molecular Biosciences—UCIBIO and the project LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy—i4HB. FCT-MCTES is also acknowledged for 2020.07660.BD for BBO.
Funding
Open access funding provided by FCT|FCCN (b-on).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Institutional review board statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of Hospital Beatriz Angelo (protocol code HBA 00184).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oliveira, B.B., Costa, B., Morão, B. et al. Combining the amplification refractory mutation system and high-resolution melting analysis for KRAS mutation detection in clinical samples. Anal Bioanal Chem 415, 2849–2863 (2023). https://doi.org/10.1007/s00216-023-04696-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-023-04696-6