
Abstract
Fibroblast growth factor 23 (FGF23) overexpression has been identified as a causative factor for tumor-induced osteomalacia (TIO) characterized by hypophosphatemia due to increased renal phosphate wasting, low 1,25(OH)2D3 serum levels, and low bone density. The effects of long-lasting disturbed phosphate homeostasis on bone mineralization are still not well understood. We report on a patient with a 12-year history of TIO, treated with 1,25(OH)2D3 and phosphate, who finally developed hyperparathyroidism with gland hyperplasia before the tumor could be localized in the scapula and removed. During surgery a transiliac bone biopsy was obtained. FGF23 expression in the tumor cells was confirmed by in situ hybridization. Serum FGF23 levels as measured by ELISA were found to be extremely elevated before and decreased after removal of the tumor. Bone histology/histomorphometry and measurement of bone mineralization density distribution using quantitative backscattered electron imaging were performed on the bone biopsy. The data showed important surface osteoidosis and a slightly increased osteoblast but markedly decreased osteoclast number. The mineralized bone volume (−11%) and mineralized trabecular thickness (−18%) were low. The mean degree of mineralization of the bone matrix (−7%), the most frequent calcium concentration (−4.1%), and the amounts of fully mineralized bone (−40.3%) were distinctly decreased, while the heterogeneity of mineralization (+44.5%) and the areas of primary mineralization (+131.6%) were dramatically increased. We suggest that the elevated levels of FGF23 and/or low phosphate concentrations disturb the mineralization kinetics in vivo without affecting matrix mineralization of pre-existing bone packets.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Fibroblast growth factor 23 (FGF23) plays a central role as a hormone regulator of phosphate and vitamin D metabolism. It is a causative factor for tumor-induced osteomalacia (TIO), a rare acquired disorder associated with several different types of tumors [1, 2]. The clinical and radiological findings in TIO include muscle pain and weakness, spontaneous fractures, and/or osteomalacia. Biochemical characteristics are hypophosphatemia due to impaired renal reabsorption of phosphate and inappropriately low levels of circulating 1,25(OH)2D3 [3, 4]. Tumors causing TIO were recently shown to express abundant amounts of FGF23, a protein which has emerged to be a major phosphate-regulating molecule. FGF23 affects renal and intestinal phosphate uptake and parathyroid glands and plays an important role on overall ion homeostasis [5–7]. It is now well accepted that FGF23 acts as a negative regulator of phosphate and chronic increased levels of FGF23 result in phosphate deprivation and wasting. In the majority of cases, FGF23-producing tumors are of mesenchymal origin, benign, small, and often difficult to localize [4]. As a result, even thorough examination of patients with suspected TIO can fail to identify these neoplasms and it can take several years from diagnosis until tumor removal [8], which represents the definitive treatment of the disease [9–12]. Consequently, affected patients develop and maintain osteomalacia over a relative long time period.
Interestingly, the relationship among FGF23, phosphate, and osteomalacia is not fully understood [6]. In the last years transgenic mice models overexpressing FGF23 showing hypophosphatemia [13–15] and null mutants characterized by hyperphosphatemia [16–18] have been created. Confusion has arisen from the fact that both genotypes display accumulation of unmineralized osteoid layers. Although the precise mechanisms leading to impaired bone development and mineralization abnormalities are still not elucidated, there is increasing evidence that the skeletal disorders in FGF23 transgenic animals are mainly related to the concomitant profound alterations in 1,25(OH)2D3 metabolism, which appears to act as a FGF23 counter-regulatory hormone [19]. Moreover, beyond the systemic effects of FGF23 on phosphate metabolism, recent studies have also suggested an independent role of phosphate and FGF23 in mineralization in rodents [20, 21] and in humans [22]. It has been shown that, on one hand, hypophosphatemia may suppress osteoclastogenesis [23] and, on the other hand, FGF23 itself acts locally as a negative regulator of osteoblast differentiation and bone matrix mineralization [24, 25]. These results strengthen the concept that in vivo chronic elevation of circulating FGF23 and low phosphate might severely hamper bone cell homeostasis in addition to mineral homeostasis.
In TIO patients, osteomalacia is generally established by laboratory findings, radiographic methods, DEXA, and pathohistology, however, until now there is little knowledge of bone material quality. In fact, osteomalacia is related to the histological appearance of bone and is generally defined as accumulation of osteoid due to defective mineralization rather than to increased bone turnover caused by reactive hyperparathyroidism [26]. A limitation is that histology or quantitative bone histomorphometry represents only a snapshot at the timepoint when the biopsy was taken, and does not really reflect the history of bone metabolism of the patient. Using quantitative back-scattered electron imaging (qBEI), further insight can be gained by characterizing the bone mineralization density distribution (BMDD) at the microscopic level. The BMDD can be considered a fingerprint of bone mineralization reflecting bone turnover, mineralization kinetics, and tissue age. The variations/distribution observed in local bone matrix mineralization is generated, on one hand, by the bone turnover, the frequency of bone remodeling (resorption of old bone and formation of new bone) and, on the other hand, by the kinetics of bone mineralization of the newly formed bone matrix (osteoid). It can be shown that, once mineralization has been started in the osteoid, the mineral content increases rapidly, within a few days, up to 70% of its final value and is referred to as primary mineralization. The last 30% increase in mineral content occurs slowly, over a time scale of months to years, and is referred to as secondary mineralization. As a consequence, bone at the tissue level appears as a mosaic of individual bone packets with specific mineral contents depending on their age. The young bone packets have the lowest, and the old ones have the highest, mineral content. Thus, changes in bone turnover as well as disturbances in the mineralization process will have a profound impact on the shape of the BMDD [27, 28]. Taking advantage of the fact that the BMDD of healthy individuals shows remarkably little biological variance [29], the method has been widely used in the past as a diagnostic tool in diverse bone diseases and/or treatments [30]. The impact of a long-term excess of FGF23 and decreased phosphate levels on BMDD has not been investigated up to now.
In the present investigation, we report on a male patient who presented with a 12-year medical history of bone pain, hypophosphatemia, and skeletal changes consistent with osteomalacia as determined by radiological findings before a mesenchymal tumor with a hemangiopericytoma-like morphology in the scapula was detected, then excised, and a transiliac bone biopsy was taken. In addition to measurements of serum levels, FGF23 expression in tumor tissue was confirmed by in situ hybridization (ISH). Bone histomorphometry and BMDD data from the transiliac biopsy were related to normative reference data [29], giving new insights into the material quality of bone in TIO.
Materials and Methods
Subject
Biopsies of the scapula and of the iliac crest were obtained during surgery with informed consent of the patient, a 54-year-old male patient with severe bone pain, low serum phosphate, and calcium and alkaline phosphatase levels at the upper limit of normal range. Considering the tentative diagnosis of idiopathic, acquired, hypophosphatemic osteomalacia, treatment with 1,25(OH)2D3 and oral phosphate was initiated. Due to the fact that the patient had no family history of bone disease, and his stature and development had been normal, autosomal dominant hypophosphatemic rickets, X-linked hypophosphatemia, autosomal recessive hypophosphatemic rickets, and hypophosphatemic hereditary rickets with hypercalcuria were clinically excluded. Eleven years later, following a routine checkup, increased levels of serum PTH and serum calcium were found. MIBI scan resulted in enlarged parathyroid glands and a discrete accumulation of tracer in the margo medialis of the right scapula. Consequently, a total parathyroidectomy was performed and half of the right cranial parathyroid gland was autotransplanted onto the upper pole of the thyroid gland. During this surgical procedure, biopsies of the scapula and of the iliac crest were taken. The histopathological examination of the scapula biopsy showed a mesenchymal tumor with hemangiopericytoma-like morphology, which consequently was totally excised together with a portion of the scapula.
Detection of Serum FGF23
Serum samples obtained at the time of parathyroidectomy and 5 years after tumor removal were stored at −80°C before chemical analysis. Serum FGF23 levels were determined by a commercially available sandwich ELISA to the C-terminal fragments of FGF23, according to the manufacturer’s protocol (Immunotopics Inc., USA).
FGF23 Expression in Excised Tumor Tissue
Construction of mRNA Probes for In Situ Hybridization
Part of the third exon of human FGF23 DNA was amplified by PCR from genomic DNA comprising AA 98-221 (forward primer, 5′-GAC CGG GAG AAC TGC AGG TT-3′; reverse primer, 5′-CTG GCC ATC GGG CTG TTG TC-3′). The PCR product was cloned into the pGEM©-T Easy vector (Promega, USA). The sequence of the cloned DNA fragment was verified by automatic sequencing. The plasmid was linearized with SpeI or SphI to generate antisense and sense biotin-labeled transcripts using either T7 or SP6 RNA polymerase, respectively (Biotin RNA Labeling Kit, Roche).
In Situ Hybridization of Tumor Tissue
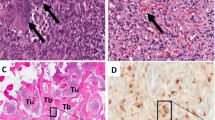
After surgical resection, the tumor from the right scapula and the removed parathyroid glands were fixed in 4% paraformaldehyde for 24 h and embedded in paraffin. For ISH, sections of 5-μm thickness were mounted on silan-coated slides, air-dried, incubated for 2 h at 37°C, and dewaxed with xylene. The deparaffinized sections were rehydrated by sequential 2-min immersions in 30%, 50%, 70%, and 96% ethanol and fixed again in 4% paraformaldehyde for 20 min at room temperature. The fixed sections were dehydrated by sequential 2-min immersions in graded series of ethanol and, finally, chloroform, then air-dried. The transcripts (RNA probes) were freshly diluted in a hybridization mixture containing 1 μg/ml probe, 50% deionized formamide, 10% dextransulfate, 2× SSC, 1.1 μg/ml denatured, sheared salmon sperm DNA (Sigma), and 0.02% sodium dodecylsulfate. For hybridization 100 μl of hybridization mixture was directly applied to the sections and they were covered with coverslips. To ensure permeabilization and denaturation of probes and targets, slides were heated for 10 min at 80°C and then incubated overnight in a humid chamber at 50°C. After hybridization, the sections were washed with 2× SSC for 30 min. To remove any single-stranded (unbound) RNA probe, slides were incubated with 1× SSC with deionized formamide (50%) at 40°C for 1 h. Afterward, slides were incubated with ExtrAvidin (Sigma) conjugated with horseradish peroxidase diluted 1:50 in blocking solution (Roche) in TBS (50 mM Tris-Cl, 150 mM NaCl, pH 8.0) for 1 h at room temperature. After rinsing the slides with TBS, the sections were developed with diaminobenzidine/H2O2 (DAB substrate kit for peroxidase; Vector Laboratories) for about 5 min at room temperature. The reaction was stopped with water, and the slides were counterstained with hemalaun, mounted in Aquatex (Merck), and imaged with a light microscope (Axiophot; Zeiss, Oberkochen, Germany) equipped with a videocamera (AxioCam; Zeiss) (Fig. 1).

Detection of FGF23 mRNA expression on tumor tissue by ISH. a Tumor tissue resembling fibroblasts and thin-walled blood vessels. Only a portion of the tumor cells was positively labeled. c Tumor tissue with bone particle of the scapula. Osteoblasts (OB and osteocytes (OC) showed FGF23 expression (arrows). b, d No expression signals were visible in cells hybridized with sense FGF23 probe. Counterstaining of the cells was done with hemalaun. Bars represent 20 μm
Bone Histomorphometric Evaluation
The transiliac bone biopsy was fixed in 70% ethanol, dehydrated in a graded series of ethanol, and embedded undecalcified in polymethylmethacrylate. Consecutively, 3-μm-thick sections were cut from the tissue block with a Jung K microtome (Reichert-Jung, Heidelberg, Germany). The sections were deplastified with 2-methoxyethyl-acetate before being stained with a modified Goldner’s Trichrome method. Histological analysis was performed according to Parfitt et al. [31] on the whole area of the bone sections. A light microscope (Axiophot; Zeiss) equipped with a Zeiss AxioCam videocamera was used to obtain digital images of the section (Fig. 2). The images were analyzed using standard procedures (NIH Image software versions 1.62 and 1.63; Wayne Rasband, National Institutes of Health, Bethesda, MD, USA) on a Power Macintosh G4.

Light microscopic analysis of transiliac bone sections (Goldner′s Trichrome staining). a Surface osteoidosis. OS, enlarged osteoid without adherent osteoblasts; rims of lipocytes surrounding the trabeculae (boldface arrows). b A single trabecular area showing endosteal fibrosis (EF)
Quantitative Backscattered Electron Imaging
Residual block samples containing cancellous and cortical bone were prepared to planoparallel surfaces by grinding and polishing and coated with carbon by vacuum evaporation for qBEI analysis in a scanning electron microscope (SEM). Trabecular BMDD was determined by qBEI according to previously published procedure [32]. Briefly, a digital scanning electron microscope (DSM 962; Zeiss) equipped with a four-quadrant semiconductor backscattered electron (BE) detector was used. The accelerating voltage of the electron beam was adjusted to 20 kV, the probe current to 110 pA, and the working distance to 15 mm. The entire cancellous bone areas were imaged at ×50 nominal magnification, corresponding to a pixel resolution of 4 μm/pixel using a scan speed of 100 s/frame, resulting in digital calibrated BE images of 512 × 512 pixels. From the digital images, gray-level histograms were deduced, displaying the percentage of bone area occupied by pixels of a certain gray level. The transformation of these into calcium weight percentage (wt%) histograms led to a bin width of 0.17 wt% calcium. A technical precision of 0.3% was achieved. BMDD parameters such as the mean and the most frequent calcium concentration in the sample (CaMean and CaPeak), the width of the distribution (CaWidth) reflecting the heterogeneity in matrix mineralization, the amount of lowly mineralized bone (CaLow; 5th percentile of reference BMDD), and the amount of fully mineralized bone (CaHigh; 95th percentile of reference BMDD) [29, 30] were derived from the histogram (Fig. 3).

Bone mineralization density distribution (BMDD) parameters as derived from reference BMDD of normal adult trabecular bone published previously [29, 30]. Parameters characterizing the BMDD: CaMean, weighted mean Ca concentration (integrated area under BMDD curve); CaPeak, most frequent Ca concentration (BMDD peak position); CaWidth, heterogeneity of mineralization (peak width at half-maximum); CaLow, amount of low mineralized bone (bone less mineralized than the 5th percentile of the reference BMDD); CaHigh, amount of fully mineralized bone (bone mineralized above the 95th percentile of the reference BMDD)
Derivation of the Mineralization Law
The connection between the BMDD and the processes of remodeling and mineralization can be understood qualitatively using the example displayed schematically in Fig. 4. While the remodeling process is described by the new bone volume formed per year, the mineralization process is characterized by the mineralization law, which describes the increase in Ca content in a newly formed bone packet with time (upper right graph in Fig. 4). For normal turnover and normal mineralization, the resulting BMDD equals the reference BMDD for healthy humans defined in Ref. 29 (light-gray BMDD in Fig. 4, corresponding to case [1 + A]). An increase in turnover together with a normal mineralization reduces the time available for each bone packet to mineralize (Fig. 4; left image, which shows darker bone packets with respect to the right image, corresponding to normal bone). Therefore the peak of the BMDD is shifted to lower values of Ca concentration (dark-gray BMDD in Fig. 4; case [2 + A]). A normal bone turnover, but a disturbance in the mineralization process, causes a reduction in the Ca incorporated in the bone packet (case (1 + B) and will, therefore, also lead to a shift of the BMDD toward lower Ca values (again, the dark-gray BMDD in Fig. 4; case [1 + B]). Extraction of information about the mineralization process has recently been translated in a mathematical model and the kinetics of the mineralization process can now be quantitatively obtained from the measured BMDD curve [27, 28]. The mathematical details of how the transformation from the BMDD to the mineralization law is performed are given in Refs. 27 and 28. To obtain the mineralization law for the TIO patient (Fig. 5), equation (B6) from Ref. 27 was applied to the measured BMDD (Fig. 5b). The main assumption for the applicability of the transformation is that the BMDD histogram reaches roughly a steady state, i.e., does not change significantly with time. Since phosphate homeostasis is disturbed in the TIO patient for longer than a decade, the assumption seems well satisfied.

Schematic of the connection between the processes of remodeling and mineralization resulting in a BMDD. Remodeling leads to newly formed nonmineralized bone packets. The mineralization process is characterized by the mineralization law, which describes the increase in Ca concentration as a function of time in a newly formed bone packet (top right). This leads to a mosaic structure of bone packets of different ages (middle left and right). The effect of three different scenarios on the BMDD is sketched (bottom): (1 + A) normal turnover and normal mineralization; (2 + A) high turnover and normal mineralization; (1 + B) normal turnover and altered mineralization

qBEI analysis of transiliac biopsy from a TIO patient. a Biopsy (overview) with zoomed-in trabecular detail (250 × 200 pixels; 4 μm per pixel) displaying a high heterogeneity in mineral content. The white arrow points to a highly mineralized bone packet (whitish) corresponding to older bone, and the black arrow points to a lowly mineralized bone packet (dark gray) corresponding to younger bone. b Corresponding bone mineralization density distribution (BMDD). N, normal reference [30] (Fig. 3). c Mineralization kinetics derived from the BMDD histograms via 29,eq. (B6) of Ruffoni et al. [27]. The thick line corresponds to the mineralization law of the TIO patient; the thin line, to the normal population (N) [27]. Both curves are numerical transforms of the BMDDs in B. Inset: The same data but plotted only for the first 2 years for better visibility of the primary mineralization phase at early times. To obtain time in units of years, a turnover time of 5 years for trabecular bone was assumed [27]
Results
Normalization of Circulating Bone Markers After Tumor Excision
As reported in Table 1, the patient displayed slightly elevated calcium and alkaline phosphatase serum levels and a high PTH value shortly before tumor removal. All values were normalized within 1 week after surgery, and in particular, the low serum phosphate was increased to normal range. Remarkably, the very high FGF23 serum level measured before surgery was significantly decreased also 5 years after tumor resection (Table 1).
FGF23 was Strongly Expressed in Tumor Cells and in Osteoblasts and Osteocytes of the Excised Scapula
Examinations of tumor tissue by ISH showed strong expression of FGF23 (Fig. 1a). Brownish signals were scattered between the spindled tumor cells, which resembled activated fibroblasts and thin-walled blood vessels. Remarkably, some cells of the tumor tissue did not express FGF23 (Fig. 1a). Additionally, high expression was observed in osteoblasts and osteocytes within the neighboring bone tissue of the remaining scapula (Fig. 1c). No positive labeling was found using the sense probe as a negative control (Fig. 1b and d). ISH on the removed hyperplastic parathyroid glands revealed no detectable expression of FGF23 in parathyroid tissue (not shown).
Histomorphometric Analyses of Bone Biopsy Revealed Osteomalacia and Decreased Bone Turnover
Cancellous bone volume (BV/TV) and trabecular thickness (Tb.Th) of the patient appeared to be within normal range compared to normals (see Table 2). In this context, it should be emphasized that in conventional histomorphometric analyses, tissue volume includes mineralized tissue and nonmineralized osteoid tissue, which under normal conditions is rather small. However, in the biopsy of the TIO patient a striking surface osteoidosis (Fig. 2a) was apparent, resulting in a dramatic increase in all three osteoid indexes: volume (OV/BV; +546%), surface (OS/BS; +492%), and thickness (O.Th; +124%). Therefore, we took advantage of the fact that BEI does not detect unmineralized tissue and further determined structural parameters from back-scattered images and found a slightly decreased mineralized bone volume (mdBV/TV; −11%) and mineralized trabecular thickness (mdTb.Th; −18%) (Table 2). Interestingly, Goldner's Trichrome staining revealed, in addition to the large seam of unmineralized osteoid, only a single area with endosteal fibrosis (Fig. 2b). Evaluation of static parameters of bone formation and bone resorption revealed only a slight increase in osteoblast surface (Ob.S/BS; +38.3%) but a significant reduction in osteoclast surface (Oc.S/BS; −85.7%) compared to normal, indicating a situation of low bone turnover.
qBEI Analyses Revealed an Increase in Mineralization Heterogeneity and a Slowdown of Mineralization
The qBEI image showed the presence of well-delimited differently mineralized bone packets throughout the trabeculae. Areas of fully mineralized bone packets in the central part of the trabeculae coexisted with broad “seams” of low-mineralized bone packets on trabecular surfaces (Fig. 5a). The mineralized trabecular bone matrix exhibited a significant reduction in mean and most frequent mineral concentration (CaMean, −7.0%; CaPeak, −4.1%) while the heterogeneity of mineralization was substantially increased (CaWidth, +44.5%) (Fig. 5b). The amount of low-mineralized bone packets, corresponding to areas of primary mineralization, was dramatically increased (CaLow, +131.6%), whereas the amount of fully mineralized bone packets was found to be markedly decreased (CaHigh, −40.3%) compared to normal (Table 3). When the BMDD histogram was numerically transformed into a mineralization law (time course of mineralization of bone packets), a reduced slope in the primary mineralization phase (slowed-down mineralization) compared to a normal reference BMDD was found (Fig. 5c). Also, the secondary mineralization phase deviated from the time course of normal adults, but the curve was slowly approaching the normal level (Fig. 5c).
Discussion
The present study addresses for the first time the effects of long-term tumor-induced osteomalacia (TIO) on bone mineralization density distribution (BMDD), a powerful tool to characterize bone material quality in individuals with and without bone diseases [30]. For better characterization of the rare disorder we also measured FGF23 in serum and by in situ hybridization (ISH) in the tumor tissue.
The reported case of TIO had a typical long history (12 years) of hypophosphatemia, treated with 1,25(OH)2D3 and oral phosphate, without substantial clinical improvement. Serum levels of phosphate were abnormally low, while FGF23 protein levels were extremely elevated. Despite the treatment, normal phosphate serum levels were not achieved, nor was osteomalacia prevented. Subsequently, the patient developed reactive hyperparathyroidism with gland hyperplasia, a feared complication of the treatment [33–36], shortly before the responsible tumor could be localized and removed. After surgery serum markers normalized and remained stable, and in particular, FGF23 values were found to be much lower also 5 years postsurgery. It should be noted that, in our patient, the extremely high serum levels of FGF23 were found to be reduced (−92%) compared to presurgical values but were still markedly above the published reference range [37, 38]. Indeed, other studies report a dramatic drop in FGF23 from circulation to undetectable levels after tumor excision for about 10 days before they increase to a normal concentration [38]. This can be explained by the fact that in TIO the tumor becomes the main source of circulatory FGF23 and endogenous production of FGF23 from other tissues is suppressed due to a negative feedback mechanism until the tumor is removed [39, 40]. Unfortunately, we did not have the chance to measure the FGF23 levels in our patient immediately after surgery. We also cannot fully rule out the possibility that the tumor was incompletely removed and therefore was still active in FGF23 production. However, all other laboratory and diagnostic parameters of the patient remained normalized and the patient was asymptomatic from the time after surgery onward. The concentration of serum FGF23 measured 5 years after excision of the tumor could also corroborate the hypothesis that serum FGF23 increases with age [41]. Moreover, it is also known that in some patients, FGF23 was elevated without apparent abnormalities of phosphate homeostasis or renal function [41–43].
Analysis of the tumor material using ISH showed expression of FGF23 mRNA on mesenchymal tumor tissue, but not in all cells in the tumor, which confirms earlier findings [44, 45]. This indicates that only a portion of the total number of tumor cells was responsible for FGF23 production. In phosphaturic tumor cells, FGF23 expression and metabolism are regulated locally at the single-cell level [46], which may lead to the observed mosaicism of FGF23 expression. The importance of local regulators in the tumor tissue is further highlighted by the fact that, in TIO, circulating high levels of FGF23 are based on the excessive production of this factor by a relatively small number of cells. This pattern of patchy expression of FGF23 in tumor tissue is in striking contrast to the case of fibrous dysplasia, where hypophosphatemia is caused by a severe excess of osteogenic cells that produce FGF23 at normal levels [46, 47]. A few cells expressing FGF23 in the neighborhood of the tumor were morphologically classified as osteoblasts and osteocytes of the remnants of the scapula. The possibility that hyperplastic parathyroid gland tissue might be contributing to the elevated serum FGF23 expression was explored, but we were unable to detect FGF23 mRNA transcripts by ISH (data not shown). This negative result is in line with an expression analysis of FGF23 in parathyroid glands of a patient with primary hyperparathyroidism that demonstrated the absence of expression in this tissue [48].
The iliac bone biopsy displayed marked surface osteoidosis with marginal endosteal fibrosis. Endosteal fibrosis is a typical skeletal feature of severe hyperparathyroidism where resorbed bone is replaced by vascular fibrous tissue and has been described in patients with chronic renal failure [26, 49]. The fact that just one area with fibrosis was viewed in our biopsy indicates that the emerging hyperparathyroidism of the described patient had not altered the bone phenotype of osteoidosis and low bone cell activity. Indeed, the most obvious histological feature of the bone sample was accumulation of unmineralized osteoid. It is widely accepted that osteomalacia develops either following increased bone turnover [50] or, in contrast, in situations of decreased bone cell activity due to prolongation of mineralization lag time and/or inhibition of osteoblast function [26]. In our sample, the osteoblast surface was slightly increased, whereas the osteoclast surface was clearly decreased. It has to be emphasized that in our TIO patient the increased amount of osteoblasts did not lead to an increased amount of mineralized matrix, since mineralized bone volume and mineralized trabecular thickness were found to be markedly reduced. The osteoblastic activity was also reflected by slightly increased levels of alkaline phosphatase, which is consistent with other reports in TIO patients [11, 39]. Indeed, alkaline phosphatase is considered a key enzyme for mineralization [21], and it has been suggested that, in situations of severe hypophosphatemia, enhanced expression of alkaline phosphatase could be part of a compensatory mechanism to increase mineralization-promoting proteins [51] and/or reflect an autonomous cellular dysfunction [52]. Moreover, also the excess of circulating FGF23 itself might have hampered bone formation in our patient. According to recent findings, FGF23 negatively regulates osteoblast differentiation and matrix mineralization in an autocrine/paracrine manner independently of its systemic effects of phosphate homeostasis [24, 25]. The reduced number of osteoclasts observed in our patient could be the result of impaired bone turnover but might also be more directly related to phosphate metabolism. Indeed, a significant reduction in number of osteoclasts has been reported in hypophosphatemic mice, and concomitantly a suppression of osteoclast differentiation and function has been observed in bone marrow cell cultures with a low phosphate concentration [23]. Taken together, these findings indicate a situation of marked prolongation in mineralized matrix formation associated with decreased bone resorptive activity [26].
The BMDD measurements in our patient revealed a decrease in the mean degree of mineralization and an increase in the heterogeneity of mineralization compared to the reference population [29]. Normally, high bone turnover is the cause of such an undermineralization and increased heterogeneity, because of the generation of high amounts of new bone packets, which are not yet fully mineralized [53, 54]. In contrast, low bone turnover generally leads to an increased amount of higher or fully mineralized bone packets concomitant with a transient increase in the homogeneity of mineralization density [55, 56]. Since the histomorphometric data on the TIO patient did not reveal a situation of high bone turnover, the increase in CaWidth and CaLow has to be related to a disturbance in mineralization kinetics.
The observation of a generally reduced mineral content in the bone matrix in a situation where the turnover is not enhanced could indicate modified mineralization kinetics of the bone matrix. Numerical transformation of the TIO BMDD into a curve indicating the time course of mineralization confirmed this assumption. Because the turnover rate is not higher in the TIO patient than in normal individuals, this analysis shows that the primary mineralization kinetics is slowed down. This difference would be even larger for a reduced turnover in the TIO patient. It is also interesting to note that the secondary mineralization kinetics shows much less difference between the patient and controls. This is most likely because the relevant bone packets were formed 10 years or more previously, which corresponds to the time before the breakout of the disease. Compared to healthy individuals, the BMDD of the patient showed about half of the fully mineralized bone packets coexisting with more than double the amount of low-mineralized bone matrix. Keeping in mind that fully mineralized bone corresponds to bone formed before the outbreak and spared by the remodeling process, it must be assumed that these bone areas correspond to remnants of normal mineralized matrix. In contrast, the newly formed bone matrix, which normally reaches 70% of complete mineralization within several days, seems to mineralize more slowly than expected, leading to an increase in the inhomogeneous mineral content and to the observed increase in the BMDD peak width. Taken together, these data suggest strongly that our patient had a normal BMDD most likely due to normal bone turnover and mineralization kinetics before the phosphaturic tumor appeared. As a consequence of this disease, bone turnover decreased and primary mineralization was slowed down, leading to osteomalacia and decreased bone mineral content in the newly formed bone matrix without an effect on the older one.
In summary, we have shown that, during long-lasting TIO, the mineralization kinetic in the newly formed bone matrix is profoundly disturbed, whereas pre-existing nonresorbed bone remains unaffected. The formation of bone mineral (hydroxyapatite) requires calcium and phosphate at a certain ratio, and the role of extracellular phosphate as a prime determinant of mineralization is widely accepted. Beyond its crucial role in the maintenance of a physiologic phosphate balance, there exists increasing evidence that FGF23 also modulates, in an autocrine/paracrine manner, osteoblast differentiation and matrix mineralization. Further investigations are necessary to elucidate the interrelationship among FGF23, calcium, phosphate, 1,25(OH)2D3, and PTH and their role in bone metabolism and mineralization.
References
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 98:6500–6505
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429–435
Ryan EA, Reiss E (1984) Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases. Am J Med 77:501–512
Jan de Beur SM (2005) Tumor-induced osteomalacia. JAMA 294:1260–1267
White KE, Larsson TE, Econs MJ (2006) The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev 27:221–241
Liu S, Quarles LD (2007) How fibroblast growth factor 23 works. J Am Soc Nephrol 18:1637–1647
Fukumoto S (2008) Physiological regulation and disorders of phosphate metabolism-pivotal role of fibroblast growth factor 23. Intern Med 47:337–343
Drezner MK (2001) Tumor-induced osteomalacia. Rev Endocr Metab Disord 2:175–186
Seufert J, Ebert K, Muller J, Eulert J, Hendrich C, Werner E, Schuuze N, Schulz G, Kenn W, Richtmann H, Palitzsch KD, Jakob F (2001) Octreotide therapy for tumor-induced osteomalacia. N Engl J Med 345:1883–1888
Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, Fukumoto S (2002) Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87:4957–4960
Nasu T, Kurisu S, Matsuno S, Tatsumi K, Kakimoto T, Kobayashi M, Nakano Y, Wakasaki H, Furuta H, Nishi M, Sasaki H, Suzuki H, Ito N, Fukumoto S, Nanjo K (2008) Tumor-induced hypophosphatemic osteomalacia diagnosed by the combinatory procedures of magnetic resonance imaging and venous sampling for FGF23. Intern Med 47:957–961
Hannan FM, Athanasou NA, Teh J, Gibbons CL, Shine B, Thakker RV (2008) Oncogenic hypophosphataemic osteomalacia: biomarker roles of fibroblast growth factor 23,1,25-dihydroxyvitamin D3 and lymphatic vessel endothelial hyaluronan receptor 1. Eur J Endocrinol 158:265–271
Bai X, Miao D, Li J, Goltzman D, Karaplis AC (2004) Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145:5269–5279
Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, Juppner H, Jonsson KB (2004) Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 145:3087–3094
Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2004) FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun 314:409–414
Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568
Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Juppner H, Lanske B (2004) Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol 23:421–432
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD (2006) Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab 291:E38–E49
Sitara D, Razzaque MS, St-Arnaud R, Huang W, Taguchi T, Erben RG, Lanske B (2006) Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol 169:2161–2170
Stubbs JR, Liu S, Tang W, Zhou J, Wang Y, Yao X, Quarles LD (2007) Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol 18:2116–2124
Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G (2005) Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev 19:1093–1104
Geller JL, Khosravi A, Kelly MH, Riminucci M, Adams JS, Collins MT (2007) Cinacalcet in the management of tumor-induced osteomalacia. J Bone Miner Res 22:931–937
Hayashibara T, Hiraga T, Sugita A, Wang L, Hata K, Ooshima T, Yoneda T (2007) Regulation of osteoclast differentiation and function by phosphate: potential role of osteoclasts in the skeletal abnormalities in hypophosphatemic conditions. J Bone Miner Res 22:1743–1751
Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, Aubin JE, Maeda N (2008) Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res 23:939–948
Sitara D, Kim S, Razzaque MS, Bergwitz C, Taguchi T, Schuler C, Erben RG, Lanske B (2008) Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet 4:e1000154
Parfitt AM (2003) Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 12:387–403
Ruffoni D, Fratzl P, Roschger P, Klaushofer K, Weinkamer R (2007) The bone mineralization density distribution as a fingerprint of the mineralization process. Bone 40:1308–1319
Ruffoni D, Fratzl P, Roschger P, Phipps R, Klaushofer K, Weinkamer R (2008) The effect of temporal changes in bone turnover on the bone mineralization density distribution: a computer simulation study. J Bone Miner Res 23:1905–1914
Roschger P, Gupta HS, Berzlanovich A, Ittner G, Dempster DW, Fratzl P, Cosman F, Parisien M, Lindsay R, Nieves JW, Klaushofer K (2003) Constant mineralization density distribution in cancellous human bone. Bone 32:316–323
Roschger P, Paschalis EP, Fratzl P, Klaushofer K (2008) Bone mineralization density distribution in health and disease. Bone 42:456–466
Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2:595–610
Roschger P, Fratzl P, Eschberger J, Klaushofer K (1998) Validation of quantitative backscattered electron imaging for the measurement of mineral density distribution in human bone biopsies. Bone 23:319–326
Roetzer KM, Varga F, Zwettler E, Nawrot-Wawrzyniak K, Haller J, Forster E, Klaushofer K (2007) Novel PHEX mutation associated with hypophosphatemic rickets. Nephron Physiol 106:8–12
White KE, Jonsson KB, Carn G, Hampson G, Spector TD, Mannstadt M, Lorenz-Depiereux B, Miyauchi A, Yang IM, Ljunggren O, Meitinger T, Strom TM, Juppner H, Econs MJ (2001) The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab 86:497–500
Bielesz B, Klaushofer K, Oberbauer R (2004) Renal phosphate loss in hereditary and acquired disorders of bone mineralization. Bone 35:1229–1239
Jan de Beur SM (2006) Tumor-induced osteomalacia. In: Favus M (ed) Primer on the metabolic bone diseases and disorders of mineral metabolism. American Society of Bone and Mineral Research, Washington, DC, pp 345–351
Ito N, Fukumoto S, Takeuchi Y, Yasuda T, Hasegawa Y, Takemoto F, Tajima T, Dobashi K, Yamazaki Y, Yamashita T, Fujita T (2005) Comparison of two assays for fibroblast growth factor (FGF)-23. J Bone Miner Metab 23:435–440
Khosravi A, Cutler CM, Kelly MH, Chang R, Royal RE, Sherry RM, Wodajo FM, Fedarko NS, Collins MT (2007) Determination of the elimination half-life of fibroblast growth factor-23. J Clin Endocrinol Metab 92:2374–2377
Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y, Yamashita T, Miyamoto Y, Okazaki H, Nakamura K, Nakahara K, Fukumoto S, Fujita T (2004) Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab 89:3979–3982
Imel EA, Hui SL, Econs MJ (2007) FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res 22:520–526
Weber TJ, Liu S, Indridason OS, Quarles LD (2003) Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res 18:1227–1234
Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM, Shulman D, Kassem M, Rackoff P, Zimering M, Dalkin A, Drobny E, Colussi G, Shaker JL, Hoogendoorn EH, Hui SL, Econs MJ (2006) Sensitivity of fibroblast growth factor 23 measurements in tumor-induced osteomalacia. J Clin Endocrinol Metab 91:2055–2061
Marsell R, Grundberg E, Krajisnik T, Mallmin H, Karlsson M, Mellstrom D, Orwoll E, Ohlsson C, Jonsson KB, Ljunggren O, Larsson TE (2008) Fibroblast growth factor-23 is associated with parathyroid hormone and renal function in a population-based cohort of elderly men. Eur J Endocrinol 158:125–129
Larsson T, Zahradnik R, Lavigne J, Ljunggren O, Juppner H, Jonsson KB (2003) Immunohistochemical detection of FGF-23 protein in tumors that cause oncogenic osteomalacia. Eur J Endocrinol 148:269–276
Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, Econs MJ, Inwards CY, Jan de Beur SM, Mentzel T, Montgomery E, Michal M, Miettinen M, Mills SE, Reith JD, O’Connell JX, Rosenberg AE, Rubin BP, Sweet DE, Vinh TN, Wold LE, Wehrli BM, White KE, Zaino RJ, Weiss SW (2004) Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol 28:1–30
Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P (2003) FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 112:683–692
Kobayashi K, Imanishi Y, Koshiyama H, Miyauchi A, Wakasa K, Kawata T, Goto H, Ohashi H, Koyano HM, Mochizuki R, Miki T, Inaba M, Nishizawa Y (2006) Expression of FGF23 is correlated with serum phosphate level in isolated fibrous dysplasia. Life Sci 78:2295–2301
Kobayashi K, Imanishi Y, Miyauchi A, Onoda N, Kawata T, Tahara H, Goto H, Miki T, Ishimura E, Sugimoto T, Ishikawa T, Inaba M, Nishizawa Y (2006) Regulation of plasma fibroblast growth factor 23 by calcium in primary hyperparathyroidism. Eur J Endocrinol 154:93–99
Krempien B, Ritz E, Tschope W (1980) The effect of 1,25 (OH)2D3 on bone mineralization: ultrastructural studies in patients with renal osteodystrophy. Contrib Nephrol 18:122–134
Parisien M, Silverberg SJ, Shane E, de la Cruz L, Lindsay R, Bilezikian JP, Dempster DW (1990) The histomorphometry of bone in primary hyperparathyroidism: preservation of cancellous bone structure. J Clin Endocrinol Metab 70:930–938
Hollberg K, Marsell R, Norgard M, Larsson T, Jonsson KB, Andersson G (2008) Osteoclast polarization is not required for degradation of bone matrix in rachitic FGF23 transgenic mice. Bone 42:1111–1121
Xiao ZS, Crenshaw M, Guo R, Nesbitt T, Drezner MK, Quarles LD (1998) Intrinsic mineralization defect in Hyp mouse osteoblasts. Am J Physiol 275:E700–E708
Misof BM, Roschger P, Cosman F, Kurland ES, Tesch W, Messmer P, Dempster DW, Nieves J, Shane E, Fratzl P, Klaushofer K, Bilezikian J, Lindsay R (2003) Effects of intermittent parathyroid hormone administration on bone mineralization density in iliac crest biopsies from patients with osteoporosis: a paired study before and after treatment. J Clin Endocrinol Metab 88:1150–1156
Roschger P, Dempster DW, Zhou H, Paschalis EP, Silverberg SJ, Shane E, Bilezikian JP, Klaushofer K (2007) New observations on bone quality in mild primary hyperparathyroidism as determined by quantitative backscattered electron imaging. J Bone Miner Res 22:717–723
Roschger P, Fratzl P, Klaushofer K, Rodan G (1997) Mineralization of cancellous bone after alendronate and sodium fluoride treatment: a quantitative backscattered electron imaging study on minipig ribs. Bone 20:393–397
Zoehrer R, Roschger P, Paschalis EP, Hofstaetter JG, Durchschlag E, Fratzl P, Phipps R, Klaushofer K (2006) Effects of 3- and 5-year treatment with risedronate on bone mineralization density distribution in triple biopsies of the iliac crest in postmenopausal women. J Bone Miner Res 21:1106–1112
Freemont A (1995) Bone histomorphometry. In: Tovey F, Stamp T (eds) The measurement of metabolic bone disease. Parthenon, New York, pp 77–90
Acknowledgments
The authors thank Dr. Elefterios Paschalis for critical reading of the manuscript and Gerda Dinst, Phaedra Messmer, and Daniela Gabriel for careful sample preparations and qBEI measurements. This study was supported by the AUVA (research funds of the Austrian Workers Compensation Board), by the WGKK (Viennese Sickness Insurance Funds), and by the FWF (Austrian Science Fund; Project P20646-B11).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
K. Nawrot-Wawrzyniak and F. Varga contributed equally to this work.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nawrot-Wawrzyniak, K., Varga, F., Nader, A. et al. Effects of Tumor-Induced Osteomalacia on the Bone Mineralization Process. Calcif Tissue Int 84, 313–323 (2009). https://doi.org/10.1007/s00223-009-9216-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-009-9216-z