
Abstract
Background
Neuroblastoma is the most common extracranial solid tumor in children and accounts for 15% of pediatric cancer related deaths. Targeting neuroblastoma with immunotherapies has proven challenging due to a paucity of immune cells in the tumor microenvironment and the release of immunosuppressive cytokines by neuroblastoma tumor cells. We hypothesized that combining an oncolytic Herpes Simplex Virus (oHSV) with natural killer (NK) cells might overcome these barriers and incite tumor cell death.
Methods
We utilized MYCN amplified and non-amplified neuroblastoma cell lines, the IL-12 expressing oHSV, M002, and the human NK cell line, NK-92 MI. We assessed the cytotoxicity of NK cells against neuroblastoma with and without M002 infection, the effects of M002 on NK cell priming, and the impact of M002 and priming on the migratory capacity and CD107a expression of NK cells. To test clinical applicability, we then investigated the effects of M002 and NK cells on neuroblastoma in vivo.
Results
NK cells were more attracted to neuroblastoma cells that were infected with M002. There was an increase in neuroblastoma cell death with the combination treatment of M002 and NK cells both in vitro and in vivo. Priming the NK cells enhanced their cytotoxicity, migratory capacity and CD107a expression.
Conclusions
To the best of our knowledge, these investigations are the first to demonstrate the effects of an oncolytic virus combined with self-maintaining NK cells in neuroblastoma and the priming effect of neuroblastoma on NK cells. The current studies provide a deeper understanding of the relation between NK cells and neuroblastoma and these data suggest that oHSV increases NK cell cytotoxicity towards neuroblastoma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroblastoma, a pediatric solid tumor arising from neural crest cells, is the most common extracranial solid tumor in children and is responsible for nearly 15% of pediatric cancer related deaths [1]. Despite promising outcomes for children with low-risk tumors, those bearing high-risk disease have less than a 50% chance of event free survival and are often victim to refractory or relapsed disease [2,3,4]. High-risk disease is hallmarked by MYCN amplification which is known to promote an immunosuppressive tumor microenvironment (TME) [2, 3, 5].
Oncolytic viruses have seen success in other neural crest–derived malignancies such as melanoma, where treatment with the first FDA approved oncolytic herpes simplex virus (oHSV), talimogene laherparepvec (T-VEC, Imlygic), demonstrated decreased disease burden [4]. Oncolytic viruses are effective anti-cancer agents as they kill their targets through two mechanisms: (1) direct lysis of the cell through viral replication and (2) creation of an immunoreactive debris field that generates an antitumor immune response [6, 7]. Some oncolytic viruses, like the one used in the current study, have been genetically engineered to induce an even more robust anti-tumor immune response through the production of cytokines [8]. Our lab utilizes an oHSV known as M002, which has been engineered to express murine IL-12 (mIL-12) [9]. We have previously published data demonstrating the ability of M002 to target, replicate within, and kill neuroblastoma cells and human high-risk neuroblastoma patient-derived xenografts (PDXs) [10,11,12].
Neuroblastoma is an immunologically cold tumor, meaning there are few immune cells in the tumor microenvironment (TME) which has made the development of immunotherapies directed toward neuroblastoma challenging [13, 14]. NK cells are immune cells innately designed to attack tumor cells as well as cells infected with a virus [15]. Due to the immunoreactive properties of oncolytic viruses, we hypothesized that the addition of oHSV would create an immunologic tumor microenvironment favorable to NK cells and augment the overall cytotoxicity against neuroblastoma. Thus, in the current study, we aimed to dissect the interaction between neuroblastoma, oncolytic viruses, and NK cells.
Methods
Cell lines
All cell lines are of human origin. Neuroblastoma cell lines, SK-N-AS (MYCN non-amplified, CRL-2137) and SK-N-BE(2) (MYCN amplified, CRL-2271), and the self-sustaining human NK cell line, NK-92 MI (hereinafter referred to as NK, CRL, 2408), were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). SK-N-AS cells were cultured and tested in DMEM (30–2601, ATCC) containing 10% fetal bovine serum (FBS, HyClone, South Logan, UT, USA), 4 mmol/L l-glutamine (Thermo Fisher Scientific Inc., Waltham, MA, USA), 1 μmol/L non-essential amino acids, and 1 μg/mL penicillin/streptomycin (Sigma Aldrich, Burlington, MA, USA). SK-N-BE(2) cells were cultured and tested in a 1:1 mixture of minimum Eagle medium and Ham F-12 medium (30–2004, ATCC) with 10% FBS (HyClone), 2 mmol/L l-glutamine (Thermo Fisher Scientific Inc.), 1 μmol/L nonessential amino acids and 1 μg/mL penicillin/streptomycin (Sigma Aldrich). NK-92 MI cells were cultured and tested in Roswell Park Memorial Institute (RPMI) medium containing 20% FBS, 1 μg/mL penicillin/streptomycin, 2 mmol/L l-glutamine, and 1 M of HEPEs (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer. We validated all cell lines via short tandem repeats on an annual basis and tested for mycoplasma as determined by a universal mycoplasma detection kit (30-1012 K, ATCC).
Antibodies and reagents
The following antibodies and reagents were employed: anti-MYCN (D1V2A, Cell Signaling Technologies, Danvers, MA, USA), human PE-conjugated anti-CD107a (Miltenyi Biotec, Gaithersburg, MD, USA), human PE-conjugated anti-NKp46 (Miltenyi), and mouse monoclonal β-actin (A1978, Sigma Aldrich) antibodies; CellTracker Green CMFDA Dye (C2925, ThermoFisher Scientific); SYTOX Orange Dead Cell Stain (S38461, ThermoFisher Scientific).
Cytotoxicity assay
Neuroblastoma cells were counted as previously described [12] and NK cells were counted using a BioRad Cell Counter (BioRad, Hercules, CA, USA). Neuroblastoma cells were labeled with the fluorescent dye, CellTracker Green (CMFDA), at a 1:1000 dilution for 30 min under standard incubation conditions. Following labeling, cells were washed with phosphate buffered saline (PBS) and resuspended in respective media. Labeled neuroblastoma target cells (5 × 103) were co-incubated with expanded NK cells at effector-to-target (E:T) ratios of 10:1 (SK-N-AS) and 20:1 (SK-N-BE(2)) in triplicate in 96-well plates. NK cells were cultured with neuroblastoma cells for either 4 (SK-N-BE(2)) or 24 (SK-N-AS) hours. After this period, the contents of each well were collected, resuspended in 200 µL of PBS, stained with Sytox Orange (dilution of 1:1000) for detection of dead cells, and analyzed by flow cytometry on an Attune Next Su Flow Cytometer (Thermo Fisher Scientific). The flow cytometry data were analyzed using FlowJo v10.0.6 (Tree Star Inc., Ashland, OR, USA). Percent cytotoxicity calculated as (CMFDA + /Sytox +) / [(CMFDA + /Sytox +) + (CMFDA + /Sytox-)] * 100. Fold change was calculated off the baseline amount of neuroblastoma cell death in the untreated group by the formula (% cytotoxicity of treatment group) / (% cytotoxicity untreated neuroblastoma).
NK cell migration
Neuroblastoma cells (5 × 104) were plated in a 24-well plate in 1 mL of their respective media and treated with M002 at a multiplicity of infection (MOI) of 0 or 1 plaque forming unit (PFU)/cell. Corning Transwell 3 μm pore inserts (Corning Inc., Corning, NY, USA) were washed in PBS and added to wells. NK cells (5 × 105) were labeled with CMFDA as described above, added to the top of the inserts, and allowed to migrate for 24 h at 37 °C with 5% CO2. Following this period, the media and NK cells were collected, centrifuged, resuspended in 200 μL of PBS, and analyzed with flow cytometry to count the number of CMFDA + cells indicating the number of NK cells migrating through the membrane.
CD107a expression
NK cells (2 × 105) were co-cultured with target neuroblastoma cells at E:T ratios of 10:1 (SK-N-AS) or 20:1 (SK-N-BE(2)) in a 96 well plate for stimulation. An unstimulated control group was created by incubating NK cells alone without neuroblastoma cells. Phycoerythrin (PE)-conjugated anti-CD107a antibody (Miltenyi) was added at the beginning of the co-culture. A negative control was created by not adding antibody to a stimulated group. After one hour of incubation, Golgi Stop (Thermo Fisher Scientific) was added to all samples. NK cells were co-cultured for 4 h to allow for NK cell degranulation and for the CD107a to be expressed on the cell surface. NK cells were collected, washed, resuspended in PBS and analyzed with flow cytometry. FlowJo v10.0.6 (Tree Star Inc.) was used to calculate the percentage of CD107a positive NK cells within the total NK cell population for each experimental condition. Fold change was calculated off the baseline CD107a expression of non-stimulated NK cells.
NK cell priming
Neuroblastoma cells (5 × 105) were plated in a 6 well plate and infected with M002 (0.1 or 0.5 PFU/cell) or mock infected with vehicle (10% glycerol in PBS, vehicle) for 48 h. The media was collected and saved (conditioned media). These infected neuroblastoma cells (5 × 104) were then co-cultured with NK cells at 10:1 (SK-N-AS) or 20:1 (SK-N-BE(2)) E:T ratios in the conditioned media in 12 well plates for 24 h. Following co-culture, NK cells were collected from the media. These primed NK cells were washed with PBS and reserved for studies.
MYCN overexpression
SK-N-AS cells (2 × 105) were plated in a 6-well plate and allowed to grow overnight. A MYCN plasmid and empty vector (EV) plasmid (pcDNA3.1D/V5-His-TOPO, Invitrogen) were utilized [16]. A transfection complex was made by diluting the Qiagen SuperFect (Qiagen, Germantown, MD, USA) transfection reagent in serum-free medium at a ratio of 20 μL of SuperFect per 2 μg of plasmid DNA. A control group was created using SuperFect in serum-free medium. The mixture was incubated at room temperature for 10 min. Media from the plated SK-N-AS cells was removed and the cells were washed with PBS prior to the addition of 600 μL of serum containing media. Transfection media (100 μL) was added to each well and cells incubated at 37 °C with 5% CO2 for 3 h. The transfection mixture was removed and 2 mL of growth medium with serum was added to the wells. Cells were allowed to grow for 48 h, collected for validation of MYCN overexpression with immunoblotting, and used for studies.
In vivo studies
SK-N-AS tumors were established in athymic nude mice. In accordance with national, international, NIH guidelines, and University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee (IACUC) approval (IACUC 09363). SK-N-AS cells (1.5 × 106 cells) in 25% Cultrex BME (R&D Systems, Minneapolis, MN, USA)/RPMI were injected into the subcutaneous space of the flank. Treatment was initiated when tumors reached 200 mm3. All treatments were administered under inhalational anesthesia using 3% isoflurane. M002 (106 PFU/50 μL) was administered via intratumoral injection using 28-gauge needle and syringe. NK-92 MI cells (5 × 106) were administered via peritumoral injection using 30-gauge needle and syringe. Control mice were treated with sterile PBS in lieu of virus or NK cells. Tumors were measured using a caliper and tumor volumes were determined using the formula: V = (length × width2) / 2 [17]. Mice were humanely euthanized when tumors reached 2000 mm3 or the animal met IACUC parameters. Tumor specimens were allocated to be flash frozen or paraffin embedded.
NKp46 staining
The presence of human NK cells in established xenograft tumors was assessed by NKp46 expression via flow cytometry. Tumors were dissociated as previously described [17]. A 100 μL sample of dissociated tumor cells was collected then resuspended in 70 μL (80 μL for a negative control) of autoMACS running buffer (Miltenyi Biotec). All samples were blocked for 15 min on ice with 20 µL FcR Blocker (Miltenyi Biotec) followed by the addition of 10 μL of PE-conjugated human anti-NKp46 antibody (R&D Systems). Cells only given FcR blocker served as negative controls. After 20 min on ice in the dark, the stained cells were centrifuged, washed twice with autoMACS buffer (Miltenyi Biotec), reconstituted in 200 μL of PBS, and analyzed on an Attune Next Su Flow Cytometer (Thermo Fisher Scientific). Data were analyzed with FlowJo v10.0.6 (Tree Star Inc.).
Data analysis
At least three biologic replicates were used for each experiment and data reported as mean ± SEM. Statistical analysis was performed with Graphpad Prism 9 using a Student’s t-test, ANOVA, chi-squared, and Kaplan–Meier with log rank statistics as appropriate with p ≤ 0.05 considered significant.
Results
NK Cells are cytotoxic and migrate toward neuroblastoma infected with oncolytic virus
Previous studies have demonstrated that NK-92 cells, another human natural killer cell line, effectively kill neuroblastoma cells; however, the cytotoxic effect of NK-92 MI cells against neuroblastoma cells has not been investigated. We initially tested the cytotoxicity of NK-92 MI (NK) and found with an E:T of 10:1, the MYCN non-amplified line, SK-N-AS, was significantly more susceptible to NK cell killing than the MYCN amplified line, SK-N-BE(2) (16.5 ± 2.0% vs. 11.6 ± 1.1%, SK-N-AS vs. SK-N-BE(2), p ≤ 0.05, Fig. 1A). When SK-N-BE(2) cells were treated with NK cells at an E:T of 20:1, cytotoxicity in SK-N-BE(2) cells reached an average of 18.9 ± 1.4%, levels similar to those seen with SK-N-AS cells at a 10:1 ratio (Supplemental Fig. 1). Since NK cells not only attack tumor cells but innately target virus infected cells, we assessed the cytotoxic effect of NK cells toward neuroblastoma cells infected with virus. In both the SK-N-AS (Fig. 1B) and SK-N-BE(2) (Fig. 1C) cell lines, the combination of NK cells and M002 resulted in a significantly greater increase in neuroblastoma cell death than oncolytic virus alone. In the SK-N-BE(2) cell line, treatment with NK cells alone was equally cytotoxic as NK cells with oHSV.

NK cells are cytotoxic and migrate toward neuroblastoma cells infected with oncolytic virus. A NK-92 MI (NK) cells (10:1, effector: target ratio, E:T) were added to SK-N-AS and SK-N-BE(2) neuroblastoma cells (5 × 105) and cytotoxicity assessed as described in Supplemental Fig. 1. NK-92 MI cells were significantly more cytotoxic toward SK-N-AS than SK-N-BE(2) cells (16.5 ± 2.0% vs. 11.6 ± 1.1%, SK-N-AS vs. SK-N-BE(2), p ≤ 0.05). B NK cells (10:1, E:T) were added to M002 (0.1 PFU/cell) infected or non-infected SK-N-AS cells and the fold change (FC) in cytotoxicity was compared to cells infected with virus alone. NK cell cytotoxicity was significantly greater in the SK-N-AS cells that were infected with M002 than those cells treated with NK cells alone. Viability was not affected by virus treatment. C NK cells (20:1, E:T) were added to M002 (0.5 PFU/cell) infected or non-infected SK-N-BE(2) cells (5 × 105) for 24 h and cytotoxicity measured. NK cell cytotoxicity was significantly increased in the SK-N-BE(2) cells with and without M002 infection. Virus infection did not significantly affect viability. D To study NK cell migration, NK cells were stained with CMFDA allowed to migrate through 3 µm pore inserts for 24 h toward plated neuroblastoma cells. The NK cells were collected from the bottom well and quantified with flow cytometry. There was no significant difference in NK cell migration between the SK-N-AS and SK-N-BE(2) cells (612 ± 296 vs. 504 ± 224 cells, SK-N-AS vs. SK-N-BE(2), p = 0.78). E Neuroblastoma cells were not infected (-, white bar) or infected (+ , black bar) with 1 PFU/cell of M002 for 24 h, and NK cell migration was measured as described in (D). Cell migration is reported as fold change. There was a significant increase in NK cells attracted to M002 infected SK-N-AS (left) and SK-N-BE(2) (right) cells compared to non-infected cells. F A diagram depicting NK cell-mediated cytotoxicity directed at neuroblastoma, demonstrates the rationale for using CD107a expression as a surrogate measure for granzyme B degranulation of NK cells. (G, H) Neuroblastoma cells were infected with M002 (0.1 PFU/cell for SK-N-AS, 0.5 PFU/cell for SK-N-BE(2)) for 24 h, co-cultured with NK cells (10:1, E:T) with CD107a for one hour and analyzed with flow cytometry. Negative controls included samples without anti-CD107a antibody and a sample of NK cells not in co-culture with neuroblastoma for baseline CD107a expression. There was no significant difference between CD107a expression in NK cells exposed to neuroblastoma or neuroblastoma infected with M002 for both SK-N-AS (G) or SK-N-BE(2) (H). Data represent results of at least three biologic replicates reported as mean ± SEM. Student’s t-test was used for statistical analysis. NB = neuroblastoma; NK = NK-92 MI natural killer cells; FC = fold change; ns = not significant. *p ≤ 0.05. Cartoon created with BioRender.com
We examined whether oncolytic virus increased NK cell attraction towards neuroblastoma cells as a potential explanation for the increase cytotoxicity seen with the addition of virus. First, we examined whether NK cells were preferably attracted to the MYCN non-amplified SK-N-AS or MYCN amplified SK-N-BE(2) neuroblastoma cells. There was no significant difference in NK attraction using an E:T ratio of 10:1 between the two cell lines (Fig. 1D). Following 24 h of M002 infection with a MOI of 1 PFU/cell, there was a significant increase in NK cell attraction to both neuroblastoma cell lines that were infected with virus (Fig. 1E).
Another plausible reason for the increase in cytotoxicity after viral infection is an increase in activity of the NK cells. Upon activation from the binding of a target cell ligand to an NK cell activating receptor, NK cells release vesicles containing granzyme B which induces apoptosis in the target cell. CD107a is a glycoprotein on these vesicles that will be transiently expressed on the NK cell surface upon granzyme degranulation. Thus, the expression of CD107a may serve as a surrogate measure for granzyme B release, with an increase in CD107a (Fig. 1F) suggesting an increase in NK cell activity [18]. We observed no statistically significant difference in NK cell CD107a expression following coculture with neuroblastoma cells not infected compared to those that were infected with oncolytic virus (Fig. 1G, H).
Priming enhances NK cell cytotoxicity towards neuroblastoma
Exposure to cancer cells with cytokines increases the cytotoxicity of NK cells [19,20,21]. Additionally, viruses have been shown to generate “memory-like” NK cells [22]. We sought to determine whether neuroblastoma cells might prime the NK-92 MI (NK) cell line, and if the addition of an oHSV could enhance this priming effect. Neuroblastoma cells were treated with vehicle or infected with M002 for 48 h (Fig. 2A). NK cells were added at a 10:1 (SK-N-AS, MYCN non-amplified) or 20:1 (SK-N-BE(2), MYCN amplified) E:T for 24 h and the resulting primed NK cells collected (Fig. 2A). The primed NK cells were cocultured with the same neuroblastoma cell line with which they were primed, and cytotoxicity was assessed 24 (SK-N-AS) or 4 (SK-N-BE(2)) hours later. In the SK-N-AS cells, NK cells primed with SK-N-AS cells were significantly more cytotoxic than naïve (non-primed) NK cells. NK cells primed with SK-N-AS cells infected with M002 were not more cytotoxic than those primed with SK-N-AS cells or naïve (non-primed) NK cells (Fig. 2B). Conversely, with the SK-N-BE(2) cells, NK cells that were primed with M002 infected SK-N-BE(2) cells were significantly more cytotoxic than naïve (non-primed) NK cells or NK cells primed only with SK-N-BE(2) cells (Fig. 2C).

Priming enhances natural killer cell directed cytotoxicity towards neuroblastoma. A Schema depicting methods of NK cell priming with neuroblastoma cells ± infection with M002. (B, C). Neuroblastoma cells were treated with either PBS or M002 (0.1 PFU/cell, SK-N-AS) (0.5 PFU/cell, SK-N-BE(2)) for 48 h then co-cultured with NK-92 MI cells (10:1, E:T, SK-N-AS) (20:1, E:T, SK-N-BE(2)) for 24 h. After priming, NK cells were collected and then co-cultured with neuroblastoma cells (24 h, SK-N-AS) (4 h, SK-N-BE(2)). B NK cells that were primed with SK-N-AS cells had a significant increase in cytotoxicity compared to naïve, non-primed NK cells. NK cells primed with M002 infected SK-N-AS cells were not significantly more cytotoxic than naïve or SK-N-AS primed NK cells. C NK cells primed with SK-N-BE(2) cells were not more cytotoxic than naïve, non-primed NK cells. NK cells primed with M002 infected SK-N-BE(2) were more cytotoxic than naïve, non-primed NK cells or NK cells primed with non-infected SK-N-BE(2) cells. (D, E) We investigated the ability of primed NK cells to attack neuroblastoma cells infected with M002. NK cells were primed as previously described (B, C) but NK cells were co-cultured with neuroblastoma cells that had been infected with M002 (0.1 PFU/cell, SK-N-AS) (0.5 PFU/cell, SK-N-BE(2)) for 24 h. D SK-N-AS primed and SK-N-AS + M002 primed NK cells demonstrated more cytotoxicity towards SK-N-AS infected with M002 than naïve NK cells. E SK-N-BE(2) primed NK cells did not have significantly increased cytotoxicity towards M002 infected SK-N-BE(2) cells. NK cells primed with SK-N-BE(2) + M002 demonstrated an increase in cytotoxicity towards M002 infected SK-N-BE(2) cells. Data represent at least three biologic replicates reported as mean ± SEM. Student’s t-test was used for statistical analysis. NB = neuroblastoma; NK = NK-92 MI natural killer cells; FC = fold change; ns = not significant. *p ≤ 0.05, **p ≤ 0.01. Cartoon created with BioRender.com
We next examined whether primed NK cells were better at killing oHSV infected neuroblastoma cells. Under these conditions, NK cells primed by SK-N-AS cells that were and were not infected with M002 both demonstrated significantly greater cytotoxicity towards oHSV infected SK-N-AS cells (Fig. 2D). Similar to cytotoxicity against non-oHSV infected SK-N-BE(2) cells (Fig. 2C), cytotoxicity only significantly increased after NK cells were primed with oHSV infected SK-N-BE(2) cells (Fig. 2E). Thus, virus primed NK cells are more cytotoxic towards M002 infected neuroblastoma than naïve NK cells.
Priming increases NK cell migration and CD107a expression
With the data demonstrating that SK-N-AS cells alone were sufficient to prime NK cells but SK-N-BE(2) cells required an infection with oHSV to prime NK cells, we investigated potential explanations for these findings. We first studied the migratory capacity of primed NK cells. Since oHSV infection did not improve NK cytotoxicity over priming with SK-N-AS cells alone in SK-N-AS but did improve cytotoxicity in SK-N-BE(2) cells, for the next studies we employed those conditions; NK cells that were primed with SK-N-AS cells alone, or with oHSV infected SK-N-BE(2) cells. Migration was increased in primed NK cells but did not reach statistical significance in the SK-N-AS cell line (Fig. 3A, left panel). In the SK-N-BE(2) cells, migration of NK cells was significantly increased following priming (Fig. 3A, right panel).

Priming increases NK cell migration and CD107a expression. A NK cells (10:1, E:T) were primed with SK-N-AS cells for 24 h, collected, and stained.These stained NK cells were added at an E:T of 10:1 in 100 μL of the media in which they had been primed, placed in the top of 3 µm pore inserts, and allowed to migrate for 24 h to SK-N-AS cells plated in the bottom of the well. The collected samples were analyzed for the number of migrated NK cells via flow cytometry. There was no significant increase in migration of NK cells that were primed compared to naïve, unprimed NK cells (left panel). For SK-N-BE(2) NK migration, NK cells (20:1, E:T) were primed with M002 infected (0.5 PFU/cell for 48 h) SK-N-BE(2) cells for 24 h and migration assessed as described. There was a significant increase in migration of NK cells primed with SK-N-BE(2) + M002 towards SK-N-BE(2) versus naïve NK cells (271 ± 61 cells vs. 175 ± 69 cells, M002 Primed NK vs. Naïve NK, p ≤ 0.05) (right panel). We next investigated whether there were changes in CD107a expression following priming. B NK cells were primed with SK-N-AS as previously described. NB primed NK and naïve (non-primed) NK cells (E:T, 10:1) were plated with SK-N-AS cells. After 1 h, CD107a cell surface expression was assessed with flow cytometry. Significantly more primed NK cells had cell surface expression of CD107a compared to naïve NK cells (left panel). NK cells were primed with SK-N-BE(2) + M002 as previously described. CD107a expression was assessed as per SK-N-AS, with the exception that NK cells were co-cultured at an E:T of 20:1. Significantly more primed NK cells expressed CD107a on their surface compared to naïve NK cells (right panel). Data represent at least three biologic replicates reported as mean ± SEM. Student’s t-test was used for statistical analysis. NB = neuroblastoma; NK = NK-92 MI natural killer cells; FC = fold change; ns = not significant. *p ≤ 0.05
Since primed NK cells migrated towards neuroblastoma in greater numbers, we wished to understand if these cells were more active once they encountered the tumor cells. The CD107a expression was compared between naïve and primed NK cells. For both SK-N-AS primed (Fig. 3B, left panel) and oHSV infected SK-N-BE(2) primed (Fig. 3B, right panel) NK cells, there was significant increase in the CD107a expression compared to naïve NK cells (Fig. 3B), indicating that primed NK cells have increased activity over non-primed NK cells.
MYCN Does not affect NK priming
We wanted to determine if observed differences in NK cell migration and cytotoxicity were potentially attributable to MYCN amplification. MYCN was overexpressed in the non-amplified SK-N-AS cell line. NK cell-mediated cytotoxicity was examined and there was no significant difference in NK cell cytotoxicity between the empty vector (EV, MYCN -) and overexpressing (OE, MYCN +) cells (Fig. 4A).

MYCN does not affect NK priming. To determine if observed differences between SK-N-AS and SK-N-BE(2) cells could be attributed to MYCN amplification, we overexpressed MYCN in SK-N-AS cells. SK-N-AS cells (2 × 105) were transfected with an empty vector (EV) plasmid or a MYCN plasmid (OE) for 48 h. A SK-N-AS EV (MYCN (-)) and OE (MYCN ( +)) cells were stained, co-cultured for 24 h with NK cells (10:1, E:T), and cytotoxicity assessed with flow cytometry. There was no difference in NK cell cytotoxicity between MYCN (-) versus MYCN ( +) cells. B SK-N-AS EV (MYCN (-)) and OE (MYCN ( +)) cells were used to prime NK-92 MI cells (10:1, E:T) for 24 h. Primed NK cells were co-cultured for 24 h with non-transfected SK-N-AS cells, and cytotoxicity assessed with flow cytometry. There was no significant difference in cytotoxicity. C NK cells were primed with SK-N-BE(2) cells, co-cultured (10:1, E:T) with SK-N-AS cells, cytotoxicity assessed via flow cytometry. There was no significant change in cytotoxicity of SK-N-BE(2) primed NK cells compared to naïve NK cells towards SK-N-AS cells. D NK cells were primed with SK-N-AS cells, co-cultured (20:1, E:T) with SK-N-BE(2) cells, and cytotoxicity assessed via flow cytometry. There was no significant difference in cytotoxicity of SK-N-AS primed NK cells compared to naïve NK cells towards SK-N-BE(2) cells. Data represent at least three biologic replicates reported as mean ± SEM. Student’s t-test was used for statistical analysis. NB = neuroblastoma; NK = NK-92 MI natural killer cells; FC = fold change; ns = not significant
The effect of MYCN on NK cell priming was examined. NK cells were primed with either MYCN EV (MYCN -) or OE (MYCN +) SK-N-AS cells. These primed NK cells were tested against wild type SK-N-AS cells. There was no significant difference in cytotoxicity suggesting MYCN may not play a direct role in the priming of NK cells (Fig. 4B).
Since there was not a difference in cytotoxicity between MYCN EV and OE cell lines, we hypothesized NK priming may be cell line dependent in which primed NK cells would only be cytotoxic when primed by the target cells. NK cells primed with SK-N-BE(2) were not more cytotoxic against SK-N-AS cells compared to the naïve NK cells (Fig. 4C). When NK cells were primed with SK-N-AS and then co-cultured with SK-N-BE(2), again, there was no significant increase in cytotoxicity compared to non-primed NK cells (Fig. 4D). These data would suggest that priming may be tumor cell specific.
M002 in combination with NK cells inhibits neuroblastoma growth in vivo
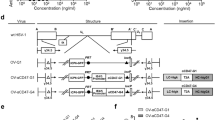
Based on the in vitro data, we progressed to in vivo studies. SK-N-AS tumors were established in the flank of athymic nude mice. When tumors reached a volume of 200 mm3, animals were randomized to receive (i) intratumoral injection of vehicle (glycerol 10% in PBS, n = 8), (ii) intratumoral injection of M002 (106 PFU/50 μL, n = 16), or (iii) peritumoral injection of NK cells (5 × 106, n = 8). Of those that received M002, 8 mice received (iv) peritumoral injections of NK cells 3 days following M002 injection to allow for adequate viral replication. Mice were monitored for tumor growth for 22 days post initial treatment (Fig. 5A). The animals treated with the combination of M002 with NK cells (Fig. 5B, red solid line, circle markers) demonstrated significantly less tumor growth than those that received vehicle (Fig. 5B, solid black line, square markers), NK cells (Fig. 5B, dotted black line, circle markers) or M002 (Fig. 5B, dashed black line, triangle markers). To investigate the relative abundance of NK-92 MI (NK) cells in the tumor, five days following the last treatment, tumors from three random animals from each treatment group were excised, dissociated, and stained with anti-NKp46 antibody for analysis via flow cytometry. Only the groups receiving NK cells had detectable NKp46 expression (Fig. 5C). Those tumors that were treated with NK cells and M002, had a significant increase in percentage of NKp46 positive cells (Fig. 5C).

The combination of M002 and NK-92 MI significantly inhibits tumor growth in vivo. A A diagram of the in vivo experiment timeline. Mice were injected with SK-N-AS cells (1.5 × 106 cells in 25% Cultrex/RPMI). When tumors reached 200 mm3 each group (n = 8 animals per group) received intratumoral injection of (i) vehicle (50 μL of 10% glycerol in PBS) or (ii) M002 (106 PFU/50 μL), or (iii) peritumoral injection of NK cells (5 × 106 cells/100 μL of PBS). The combination treatment group (iv) received an initial intratumoral injection of M002 (106 PFU/50 μL) followed 72 h later with a peritumoral injection of NK cells (5 × 106 cells/100 μL of PBS). B Mice that received combination treatment with M002 and NK cells had significantly smaller tumors compared to mice that received monotherapy with NK cells (†p ≤ 0.05) or M002 (*p ≤ 0.05). C From each group, a random selection of mice (n = 3) was euthanized 5 days after the last treatment to evaluate NKp46 expression as a measure of NK cells within the tumor. Tumors were dissociated, a 100 μL sample from each tumor was stained anti-NKp46 antibody and samples analyzed via flow cytometry. Compared to the group receiving NK cells only (other groups correctly had no NKp46 signal), there was a significant increase in NKp46 expression of the group that received both M002 and NK cells. D Mice were injected with SK-N-BE(2) cells (1.5 × 106 cells in 25% Cultrex/RPMI). When tumors reached 200 mm3, mice were randomized to receive intratumoral injection of (i) vehicle (50 μL of 10% glycerol in PBS, n = 7) or (ii) M002 (106 PFU/50 μL, n = 6), or (iii) peritumoral injection of NK cells (5 × 106 cells/100 μL of PBS, n = 6). The combination treatment group (iv) received an initial intratumoral injection of M002 (106 PFU/50 μL) followed 72 h later with a peritumoral injection of NK cells (5 × 106 cells/100 μL of PBS, n = 6). Mice that received combination treatment with M002 and NK cells had significantly smaller tumors compared to mice that received monotherapy with NK cells (†p ≤ 0.05, ††p ≤ 0.01, †††p ≤ 0.001, ††††p ≤ 0.0001) or M002 (*p ≤ 0.05, **p ≤ 0.01) E Kaplan–Meier graph of animal survival. By day 54 post-treatment, all mice in control, NK, and M002 groups had expired. Animals that received M002 + NK cells had significantly higher odds of survival compared to those that received vehicle, M002, or NK cells alone. Data reported as mean ± SEM and a Student’s t-test was used for statistical analysis. NK = NK-92 MI natural killer cells; FC = fold change; *p ≤ 0.05
We repeated the in vivo studies using SK-N-BE(2) cells. When tumors reached a volume of 200 mm3, animals were randomized to receive (i) intratumoral injection of vehicle (glycerol 10% in PBS, n = 6), (ii) intratumoral injection of M002 (106 PFU/50 μL, n = 6), (iii) peritumoral injection of NK cells (5 × 106, n = 6), or (iv) an initial intratumoral injection of M002 (106 PFU/50 μL) and then peritumoral injection of NK cells (5 × 106) 3 days later (n = 6). Animals receiving the combination of M002 and NK cells (Fig. 5D, red solid line, circle markers) had significantly delayed tumor growth in comparison to those that were given vehicle (Fig. 5D, solid black line, square markers), NK cells alone (Fig. 5D, dotted black line, circle markers), or M002 alone (Fig. 5D, dashed black line, triangle markers). By day 28, the average tumor volume in the animals treated with the combination was no longer significantly different than the M002 group, suggesting the tumors may have overcome the treatment intervention. However, when we compare survival, animals receiving both M002 and NK cells had significantly improved survival over those treated with M002 or NK cells alone or vehicle control, suggesting a benefit of using NK cells and M002 in combination (Fig. 5E).
Discussion
In the current study, we investigate the hypothesis that oncolytic virotherapy may be utilized to augment NK cell cytotoxicity. One of the main concerns with combining NK cells and oncolytic virotherapy is that NK cells will clear the virus, depleting formation of virions and limiting infectivity. The data regarding this issue are mixed [23]. In glioblastoma, Han et al. gave a single dose of TGFβ prior to administering oHSV to inhibit the NK cell immune response. They found the oHSV had a more significant tumor suppressing effect and demonstrated increased animal survival in syngeneic mouse models with TGFβ administration [24]. Altomonte and colleagues found that reducing NK cell activating ligands on tumor cells increased oncolytic virus function [25]. Although these studies that inhibited NK cell function to allow for uninhibited oncolytic virus replication were promising, it seems ill advised to remove a strong defensive player in the anti-tumor response. As such, other researchers have demonstrated the benefit of using NK cells and oHSV together in cancer. Studies evaluating the combination of EGFR-CAR NK cells with oHSV showed significantly decreased glioblastoma tumor burden and breast cancer metastasis in vivo [26, 27]. Researchers have utilized mathematical models to achieve a synergistic tumor killing effect with NK cells and oncolytic virus. These models demonstrated that an adequate viral load is needed prior to the influx of NK cells [28, 29]. For the current experiments, we found 24 h in vitro and 72 h in vivo to be adequate time for M002 infection to prevent the NK cells from making the virus effete. These findings were based on prior viral recovery studies with M002 in neuroblastoma that showed substantial viral titers at 24 h post-infection [10, 12]. In our cytotoxicity, priming, and in vivo experiments, the addition of NK cells significantly increased cytotoxicity compared to the oHSV alone, suggesting a benefit to a combinatorial approach of oncolytic virus and NK cells. For future studies, and to confirm the virus is not being depleted by the NK cells, we intend to investigate tumors following combination treatments via PCR and viral recovery assays to determine if NK cells affect viral load.
Previous preclinical data with M002 and unpublished clinical trial data with M032, the same parent virus genetically engineered to express human IL-12, suggest that intratumoral injections of the oHSV attract NK cells to the TME of solid tumors [10, 12, 30], but to our knowledge, the current report is the first to demonstrate the ability of M002 to increase NK cell attraction to neuroblastoma infected tumor cells in vitro. Our in vivo data similarly suggest that the virus attracts NK cells to the tumor, with an increase in NKp46 staining in tumors given M002. However, as discovered in both animal experiments, the effect of M002 and NK cells in combination is not sustained indefinitely. We hypothesized the effects of the combination therapy were driven by the virus. We anticipate future studies where both virus and NK cells are repeatedly readministered.
The ability to increase NK cell cytotoxicity and generate memory-like NK cells through cytokines has been well documented, but data regarding the use of cancer cells as a priming agent remain conflicting [20, 21, 31]. Sabry et al. found the transcriptome of NK cells exposed to CTV-1 leukemia cancer cells was more representative of dysfunctional NK cells, despite an increased release of pro-inflammatory molecules from these NK cells [32]. Pal et al. took irradiated cell fragments from the NALM-16 leukemia cell line and developed memory-like NK cells that had higher tumor directed cytotoxicity in vitro and in vivo, and the enhanced cytotoxicity was cell specific [19]. Like Sabry et al. we used live cancer cells as priming agents, but unlike Pal et al., we found that priming with only one of the tumor cell lines, SK-N-AS, enhanced NK cell cytotoxicity and this effect was only directed towards the cells with which the NK cells were primed. These current data and those of other investigators call into question whether priming represents true “memory”.
We found that effective priming of NK cells toward SK-N-BE(2) tumor cells required the addition of an oHSV. We believe this finding may be attributed to the virus sensitizing the SK-N-BE(2) cells to the NK cells, similar to the benefit seen by other investigators who demonstrated that the addition of anti-GD2 antibody rendered neuroblastoma cells more sensitive to NK cell antibody-dependent cell killing [33]. Other investigators have noted similar findings with viral priming. Ogbomo et al. took NK cell resistant prostate cancer cells (DU145) and found NK cells were only cytotoxic following priming with virus infected DU145 cells [34]. In a 3D spheroid model of lung cancer, Varudkar found an oncolytic parvovirus, as well as the cellular factors released from viral infection (e.g. DAMPS and proinflammatory cytokines) into the surrounding media of the 3D spheroids, improved NK cell cytotoxicity toward infected tumor cells as well as the inner core cells of the tumor, which had yet to be infected with oncolytic virus [35]. Our results corroborate this finding as NK cells primed by SK-N-BE(2) infected with M002 were better killers of non-infected SK-N-BE(2) than unprimed NK cells. Additionally, similar to Varudkar, we identified significant virus product (e.g., mIL-12) in the surrounding media of 3D bioprinted models of neuroblastoma PDXs after treatment with M002, which could potentially augment NK cell directed cytotoxicity [12]. We believe this is occurring with our in vivo studies, where priming of NK cells is occurring within the TME following administration of the virus. This could explain the decrease in tumor growth of SK-N-BE(2) tumors with the combination of NK cells and virus as the combination alone was not enough to increase cytotoxicity in vitro but with priming, cytotoxicity was increased. Thus, future investigations will employ the 3D model to determine if the by-products of virus infection are enough to prime NK cells.
A major difference between SK-N-AS and SK-N-BE(2) cell lines is MYCN amplification. We hypothesized the noticeable differences in NK cell cytotoxicity between the two cell lines could be attributed to MYCN. Our reasoning extended from findings by other investigators. Bao and colleagues demonstrated that MYCN amplification correlates to a lower immune cell infiltrate [36]. Brandetti found that MYCN amplification decreased the NK cell activating ligands on the cell surface of neuroblastoma [5, 36]. When we overexpressed MYCN in the SK-N-AS MYCN non-amplified cell line, we found no significant differences in overall NK cytotoxicity. These findings could be due to several factors, one being that induced MYCN expression does not guarantee a similar increase or decrease of downstream protein activity that is seen in wild type MYCN amplified neuroblastoma. For example, MYCN amplified tumors have been found to secrete less monocyte chemoattractant protein-1 (MCP-1, CCL2), a chemokine that promotes NK cell cytolytic activity, whereas MYCN non-amplified tumors have high amounts of secreted MCP-1 [36, 37]. Due to the scope of this study, we did not test whether induced MYCN expression suppressed CCL2 secretion, in what could have contributed to the lack of noticeable difference. Additionally, sequencing and pathway analysis comparing the immune interactions between MYCN amplified and non-amplified cell lines revealed 16 unique gene clusters that could be responsible for immunosuppression in MYCN amplified lines [37], which we did not investigate in the OE MYCN cells. It is also possible the differences are cell line dependent or due to differences in another protein such as TRAIL-2, which is not correlated to MYCN status but positively correlates with NK cell cytotoxicity resistance [38]. Thus, further testing with wild type MYCN non-amplified and amplified cell lines, as well as MYCN non-amplified and amplified PDX lines, is needed to fully elucidate the role of MYCN in NK cell cytotoxicity and priming.
Unlike T and B cells, the mechanism behind the adaptive phenotype of NK cells has yet to be fully determined. What has been shown by both Sabry et al. and Pal et al., is that primed NK cells have a significantly different transcriptome and epigenetic profile [19, 32]. Sabry et al. noted that inhibitory receptors on the cell surface of NK cells increased upon priming. In the current studies, we employed the NK-92 MI cell line which lacks many cell receptors, including the inhibitory killer cell immunoglobulin-like receptors (KIRs), making it difficult to attribute a change in NK cell surface receptors as an explanation for priming in the current studies. The increase in CD107a expression on the cell surface supports the notion that primed NK cells are more active, potentially explaining improved cytotoxicity [18]. We recognize that a limitation to these studies is the inability to elucidate the factor driving the NK cell cytotoxicity and memory-like effects. For example, further experiments with additional treatment groups, such as UV inactivated viruses and exogenous IL-12, could assist in determining whether the increased cytotoxic effects from the virus are pathogen-associated molecular patterns or pro-inflammatory cytokines, respectively. However, since we did not know if viruses could even prime NK cells, these specifics were not investigated as it was outside the scope of the hypothesis.
We employed the NK-92 MI cell line for the current studies. This cell line is a derivative of the NK-92 cell line that has been stably transfected with an interleukin-2 (IL-2) plasmid to eliminate the need for addition of IL-2 to culture media [39]. The parent line, NK-92, has been heavily studied as a tool to better understand NK cell biology and due to the lack of KIRs on its cell surface, was found as a strong anti-cancer weapon [40]. Previous research has shown NK-92 cells cytotoxic to neuroblastoma cells and has been used as a construct for developing CAR-NK cells against neuroblastoma [41,42,43]. No studies have reported the cytotoxicity of NK-92 against the cell lines used in this study, and to our knowledge, the use of NK-92 MI cells has not been reported in neuroblastoma, lending a novel aspect to the data. NK-92 cells have a promising potential for use in the clinical arena as they are more easily expanded in culture than donor NK cells. In children receiving myeloablative chemotherapeutics, the NK cell population is significantly depleted making administration of exogenous NK cells an attractive treatment option for these patients. NK-92 cells have undergone phase I clinical trials and graft versus host disease or other safety concerns were not identified [44, 45]. With regard to our data, the minimal side effects of the NK-92 cells combined with the minimal side effects seen with the oncolytic virus preclinically [9] and other oHSVs in clinical trials [46], support the potential for clinical translation using combinatorial administration of oncolytic virus with exogenous NK cells. Since a limitation to oncolytic virotherapy is the need for intratumoral injection [47], we believe the addition of the NK cells could significantly augment its success as primed NK cells could go and target metastatic sites that would not be able to be injected by the virus. The data showing primed NK cells can kill non-infected neuroblastoma cells support this theory, however, further experiments are necessary and are a current area of future investigation.
We utilized athymic nude mice for the in vivo studies to focus on understanding the impact of oHSV and the exogenous administration of NK cells. These immunocompromised animals allowed us to remove the confounding variable of other immune cells, such as CD8 + T cells, when evaluating the effects of exogenous NK cells. We acknowledge that this model does not exactly replicate the human condition as these animals do have some NK cells. Future investigations should include immune competent mice using a syngeneic model of neuroblastoma such as the Neuro-2a cell line. Importantly though, the transgenic lines of neuroblastoma still harbor an immunosuppressive phenotype [48]. Furthermore, we postulate that NK cell priming is occurring in vivo, but to adequately test true memory, in vivo experiments administering primed NK cells followed by tumor cell challenge will be required.
Conclusion
In this study, we provide evidence for the role of oncolytic virotherapy in augmenting NK cell killing in neuroblastoma. We demonstrate that the cytotoxicity of NK-92 MI (NK) cells is increased when the NK cells are primed and combined with oHSV. From our data, we believe a benefit of the oHSV is related to the increased attraction in vitro and maintenance of NK cells in vivo while the cytotoxic effect of priming may be a result of increased activity of the primed NK cells. Further studies are needed to fully determine why differences exist between MYCN non-amplified and amplified cell lines. The current findings contribute to the documented benefit of combining oncolytic viruses with NK cells but are novel in that they demonstrate the ability of an oncolytic virus to induce a memory-like phenotype against neuroblastoma. The current studies broaden our knowledge of the interaction between NK cells and neuroblastoma, potentially providing a translational pathway for clinical application of this approach.
Data availability
No datasets were generated or analysed during the current study.
References
Siegel RL et al (2022) Cancer statistics, 2022. CA Cancer J Clin 72(1):7–33
Irwin MS et al (2021) Revised neuroblastoma risk classification system: a report from the children’s oncology group. J Clin Oncol 39(29):3229–3241
Yogev O et al (2019) In vivo modeling of chemoresistant neuroblastoma provides new insights into chemorefractory disease and metastasis. Cancer Res 79(20):5382–5393
Senzer NN et al (2009) Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 27(34):5763–5771
Brandetti E et al (2017) MYCN is an immunosuppressive oncogene dampening the expression of ligands for NK-cell-activating receptors in human high-risk neuroblastoma. Oncoimmunology 6(6):e1316439
Takasu A et al (2016) Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Ther 23(4):107–113
Lin D, Shen Y, Liang T (2023) Oncolytic virotherapy: basic principles, recent advances and future directions. Signal Transduct Target Ther 8(1):156
Inoue T et al (2021) Oncolytic vaccinia virus gene modification and cytokine expression effects on tumor infection, immune response, and killing. Mol Cancer Ther 20(8):1481–1494
Markert JM et al (2012) Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. J Virol 86(9):5304–5313
Gillory LA et al (2013) Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PLoS ONE 8(10):e77753
Megison ML et al (2014) Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of pediatric solid tumors. PLoS ONE 9(1):e86843
Quinn CH et al (2022) Targeting high-risk neuroblastoma patient-derived xenografts with oncolytic virotherapy. Cancers 14(3):762
Raffaghello L et al (2005) Multiple defects of the antigen-processing machinery components in human neuroblastoma: immunotherapeutic implications. Oncogene 24(29):4634–4644
Verhoeven BM et al (2022) The immune cell atlas of human neuroblastoma. Cell Rep Med 3(6):100657
Trinchieri G (1989) Biology of natural killer cells. Adv Immunol 47:187–376
Beierle EA et al (2007) N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem 282(17):12503–12516
Quinn CH et al (2021) Downregulation of PDGFRss signaling overcomes crizotinib resistance in a TYRO3 and ALK mutated neuroendocrine-like tumor. Transl Oncol 14(7):101099
Alter G, Malenfant JM, Altfeld M (2004) CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods 294(1–2):15–22
Pal M et al (2017) Tumor-priming converts NK cells to memory-like NK cells. Oncoimmunology 6(6):e1317411
Cooper MA et al (2009) Cytokine-induced memory-like natural killer cells. Proc Natl Acad Sci U S A 106(6):1915–1919
Romee R et al (2016) Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 8(357):357ra123
Foley B et al (2012) Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol 189(10):5082–5088
Alvarez-Breckenridge CA et al (2012) Deciphering the multifaceted relationship between oncolytic viruses and natural killer cells. Adv Virol 2012:702839
Han J et al (2015) TGFbeta treatment enhances glioblastoma virotherapy by inhibiting the innate immune response. Cancer Res 75(24):5273–5282
Altomonte J et al (2009) Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells. Cancer Gene Ther 16(3):266–278
Chen X et al (2016) A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 7(19):27764–27777
Ma R et al (2021) An oncolytic virus expressing IL15/il15ralpha combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res 81(13):3635–3648
Kim Y et al (2018) Complex role of NK cells in regulation of oncolytic virus-bortezomib therapy. Proc Natl Acad Sci U S A 115(19):4927–4932
Senekal NS et al (2021) Natural killer cells recruitment in oncolytic virotherapy: a mathematical model. Bull Math Biol 83(7):75
Parker JN et al (2000) Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A 97(5):2208–2213
Uppendahl LD et al (2019) Cytokine-induced memory-like natural killer cells have enhanced function, proliferation, and in vivo expansion against ovarian cancer cells. Gynecol Oncol 153(1):149–157
Sabry M et al (2019) Tumor- and cytokine-primed human natural killer cells exhibit distinct phenotypic and transcriptional signatures. PLoS ONE 14(6):e0218674
Iguchi M et al (2023) Development of anti-GD2 antibody-producing mesenchymal stem cells as cellular immunotherapy. Anticancer Res 43(6):2417–2424
Ogbomo H et al (2010) Tumor cells infected with oncolytic influenza A virus prime natural killer cells for lysis of resistant tumor cells. Med Microbiol Immunol 199(2):93–101
Varudkar, N., et al. (2021) Oncolytic parainfluenza virus combines with NK cells to mediate killing of infected and non-infected lung cancer cells within 3D spheroids: role of type I and type III interferon signaling. J Immunother Cancer, 9(6)
Bao R et al (2021) Immunogenomic determinants of tumor microenvironment correlate with superior survival in high-risk neuroblastoma. J Immunother Cancer 9(7):e002417
Kacher J et al (2022) Impaired antitumor immune response in MYCN-amplified neuroblastoma is associated with lack of CCL2 secretion and poor dendritic cell recruitment. Cancer Res Commun 2(7):577–589
Sheard MA et al (2013) Membrane-bound TRAIL supplements natural killer cell cytotoxicity against neuroblastoma cells. J Immunother 36(5):319–329
Tam YK et al (1999) Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther 10(8):1359–1373
Klingemann H (2023) The NK-92 cell line-30 years later: its impact on natural killer cell research and treatment of cancer. Cytotherapy 25(5):451–457
Dasgupta A, Shields JE, Spencer HT (2012) Treatment of a solid tumor using engineered drug-resistant immunocompetent cells and cytotoxic chemotherapy. Hum Gene Ther 23(7):711–721
Esser R et al (2012) NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med 16(3):569–581
Grote S et al (2021) CD276 as a novel CAR NK-92 therapeutic target for neuroblastoma. Advances in Cell and Gene Therapy 4(1):e105
Tonn T et al (2013) Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 15(12):1563–1570
Arai S et al (2008) Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy 10(6):625–632
Streby KA et al (2017) Intratumoral Injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin Cancer Res 23(14):3566–3574
Streby KA et al (2019) First-in-human intravenous seprehvir in young cancer patients: a phase 1 clinical trial. Mol Ther 27(11):1930–1938
Carlson LM et al (2013) Low-dose aspirin delays an inflammatory tumor progression in vivo in a transgenic mouse model of neuroblastoma. Carcinogenesis 34(5):1081–1088
Acknowledgements
The authors wish to thank Vidya Sagar Hanumanthu at UAB Comprehensive Flow Cytometry Core for his incredible assistance.
Funding
This project was made possible by funding from the National Cancer Institute of the National Institutes of Health under award numbers 5T32GM008361: Medical Scientist Training Program (CHQ), T32 CA229102 (JRJ), P30 AR048311 and P30 AI027767 (Flow Cytometry Core, UAB), and P30 CA013148 (Genomics Core UAB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Other funding sources include Kaul Pediatric Research Foundation, Rally Foundation for Childhood Cancer Research, and Hyundai Hope on Wheels (EAB).
Author information
Authors and Affiliations
Contributions
Conceptualization and design of the study were completed by Dr. Colin Quinn, Dr. Janet Julson, Dr. Elizabeth Beierle, Dr. Nazia Nazam, Dr. Jianmei Leavenworth, and Dr. Elizabeth Beierle. Material preparation were completed by all authors. Experiment completion and data collection were completed by Dr. Colin Quinn, Dr. Janet Julson, Hooper Markert, Dr. Nazia Nazam, Dr. Swatika Butey, and Jerry Stewart. Data analysis and interpretation completed by Dr. Colin Quinn, Dr. James Markert, and Dr. Elizabeth Beierle. First draft of the manuscript written by Dr. Colin Quinn and edited by all authors. All authors read and approved of the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Dr J.M. Markert holds equity in Aettis, Inc. (< 8%), a company that holds stocks of oncolytic virus and Treovir, Inc. (25%), a company holding intellectual property and funding clinical trials of oncolytic virus for pediatric brain tumors. A company that Dr J.M. Markert formerly held equity in (< 8%) Catherex, Inc., was purchased in a structured buyout. Dr J.M. Markert has served as a consultant for Imugene. He also holds a fraction of the IP associated with oncolytic virus C134, which is licensed by Mustang Biotech.
Ethics approval
All animal experiments were conducted in accordance with national, international, NIH guidelines, and University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee (IACUC) approval (IACUC 09363).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Quinn, C.H., Julson, J.R., Markert, H.R. et al. Oncolytic virotherapy augments self-maintaining natural killer cell line cytotoxicity against neuroblastoma. Cancer Immunol Immunother 73, 221 (2024). https://doi.org/10.1007/s00262-024-03818-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00262-024-03818-y