
Abstract
It has been postulated that the changes in the molecular characteristics of the malignant clone(s) and the abnormal activation of JAK-STAT signaling are responsible for myeloproliferative neoplasm progression to more advanced disease phases and the immune escape of the malignant clone. The continuous JAK-STAT pathway activation leads to enhanced activity of the promoter of CD274 coding programmed death-1 receptor ligand (PD-L1), increased PD-L1 level, and the immune escape of MPN cells. The aim of study was to evaluate the PDL1 mRNA and JAK2 mRNA level in molecularly defined essential thrombocythaemia (ET) patients (pts) during disease progression to post-ET- myelofibrosis (post-ET-MF). The study group consisted of 162 ET pts, including 30 pts diagnosed with post-ET-MF. The JAK2V617F, CALR, and MPL mutations were found in 59.3%, 19.1%, and 1.2% of pts, respectively. No copy-number alternations of the JAK2, PDL1, and PDCDL1G2 (PDL2) genes were found. The level of PD-L1 was significantly higher in the JAK2V617F than in the JAK2WT, CALR mutation-positive, and triple-negative pts. The PD-L1 mRNA level was weakly correlated with both the JAK2V617F variant allele frequency (VAF), and with the JAK2V617F allele mRNA level. The total JAK2 level in post-ET-MF pts was lower than in ET pts, despite the lack of differences in the JAK2V617F VAF. In addition, the PD-L1 level was lower in post-ET-MF. A detailed analysis has shown that the decrease in JAK2 and PDL1 mRNA levels depended on the bone marrow fibrosis grade. The PDL1 expression showed no differences in relation to the genotype of the JAK2 haplotypeGGCC_46/1, hemoglobin concentration, hematocrit value, leukocyte, and platelet counts. The observed drop of the total JAK2 and PDL1 levels during the ET progression to the post-ET-MF may reflect the changes in the JAK2V617F positive clone proliferative potential and the PD-L1 level–related immunosuppressive effect. The above-mentioned hypothesis is supported by The Cancer Genome Atlas (TCGA) data, confirming a strong positive association between CD274 (encoding PD-L1), CXCR3 (encoding CXCR3), and CSF1 (encoding M-CSF) expression levels, and recently published results documenting a drop in the CXCR3 level and circulating M-CSF in patients with post-ET-MF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Activation of the Janus kinase, a signal transducer and activator of transcription (JAK-STAT) signaling pathway in myeloid progenitor cells, is a hallmark of Philadelphia negative myeloid neoplasms (MPNPh −). It resulted in the proliferative advantage of myeloid cells, leading to clinical and laboratory symptoms of MPN [1, 2]. In the majority of MPNPh- patients (pts), clonal proliferation of myeloid progenitor cells is the result of the mutation acquisition by hematopoietic stem cells in the gene coding proteins of the JAK-STAT signaling pathway [3]. In 2005, a gain-of-function point mutation in Janus 2 kinase gene (JAK2) was discovered and characterized by a few independent scientific groups [4,5,6]. The presence of another somatic activating mutation of the thrombopoietin receptor gene (MPL proto-oncogene) was confirmed by Pikman et al. and, independently, by Pardanani et al. in 2006 [7, 8]. The most frequent mutations affect the hotspot codon W515 (W515L/R/A/G) which is localized at the boundary of the transmembrane and the cytosolic domains of MPL [9]. W515 prevents spontaneous activation of MPL, but in the case of gain-off-function MPL mutations, the TPO-independent activation of the receptor takes place, resulting in the downstream JAK-STAT pathway signaling [10]. In 2013, the whole-exome sequencing studies confirmed the presence of recurrent frameshift mutation in the calreticulin gene (CALR) in ET pts negative for JAK2 and MPL mutation [11, 12]. Until now, more than 60 mutations in CALR in MPN pts have been identified. Eighty percent of them are type 1 (a 52-bp deletion; c.1099_1150del, p.Leu367Thrfs*46) or type 2 mutations (a 5-bp insertion; c.1154_1155insTTGTC, p.Lys385Asnfs*47). The rest of the CALR gene mutations can be categorized into type-1-like, type-2-like, or other types [13]. According to recent data, the distribution of CALR mutations varies dependently from the type of MPNPh − . In ET, most patients harbor a mutation in one of three genes: JAK2 V617F (in 60%), CALR (in 20%), or MPL in (3%), with the distribution of type 1 and type 2 mutations being similar (51 vs. 39% of pts, respectively) [14,15,16]. The CALR mutants form a molecular complex with thrombopoietin receptor, determining a cytokine-independent JAK-STAT activation with an increased megakaryocyte proliferation [17, 18]. Recently, it has been postulated that defective interactions of mutant CALR with store-operated calcium (Ca2 +) entry machinery (SOCE) are responsible for (i) constitutive activation of SOCE, (ii) MPL activation, and (iii) subsequent phosphorylation of STAT5, AKT, and ERK1/2 and (iv) megakaryocyte proliferation [15]. In the case of the MPL gene mutations, coding the thrombopoietin receptor structure, the constitutive activation of thrombopoietin receptor activation is responsible for JAK-STAT pathway activation [9].
The proliferation and differentiation of MPNPh − cells due to abnormal JAK-STAT pathway signaling results in an indirect paracrine secretion of inflammatory cytokines released by the bone marrow microenvironment cells and the cytokine storm. One of the consequences is bone marrow fibrosis associated with an increased level of interleukin 8 (IL-8), oncostatin-M, lipocalin-2, transforming growth factor β1, platelet derived growth factor (PDGF), fibroblast growth factor (FGF), venous endothelial growth factor (VEGF), and inhibitors of matrix metalloproteinases in the peripheral blood [19,20,21]. The frequency of fibrotic transformation of ET to post-essential thrombocythaemia myelofibrosis (post-ET-MF) was determined as 0.8–4.9% at 10 and 4–11% at 15 years, respectively [22]. According to the available data, the cumulative risk of post-ET-MF increases over time and is 0.3% at 5 years and 3.9% at 10 years, with the median follow-up time of 9.1 years [23]. Pre-fibrotic primary myelofibrosis bone marrow morphology, advanced age, and anemia were identified as factors predisposing to post-ET-MF. The presence of the JAK2 mutation was associated with a low fibrotic transformation risk of ET [24,25,26]. It has been also postulated that JAK2V617F variant allele frequency (VAF) is correlated with fibrotic progression [27].
Hao et al. and, independently, Barrett et al. postulated a possible relation between the expression of JAK2, and the PD-1 ligand genes [PD-L1 (CD274) and PD-L2 (CD273)], because all of them are located on chromosome 9p24.1, and an amplification of chromosome 9p24.1 upregulated the JAK2 expression and activated the JAK2-STAT3 pathway [28, 29]. It was found that JAK2V617F oncogenic activity resulted in an increased phosphorylation of STAT3 and STAT5 and enhanced PD-L1 promotor activity and PD-L1 protein level [30]. The upregulation of PD-L1 resulted in a reduced cytotoxic T cell activity, cell cycle progression, and T cell exhaustion [31, 32]. Recently, Milosevic-Feenstra et al. documented a higher PD-L1 mRNA expression in granulocytes in both JAK2V617F positive ET and primary myelofibrosis (PMF) patients, compared to CALR-mutated MPN patients. Moreover, they showed that MPN cells in JAK2-V617F-positive patients expressed higher levels of PD-L1, if 9p uniparental disomy (UPD) was present [33]. The aforementioned mechanism may confer a potent escape mechanism of MPN cells from the host immune system and disease progression. Therefore, our study aimed to evaluate the PD-L1 and JAK2 mRNA expression in molecularly defined ET patients, dependently from the disease phase, to answer the question of the mutual relation between the PD-L1mRNA and JAK2 mRNA expression and disease progression to post-ET- MF.
Materials and methods
Study group characteristics
The study group consisted of 132 pts with ET and 30 persons with a confirmed diagnosis of post-ET-MF according to the WHO criteria published in 2016 [34]. All patients were reevaluated for the presence of symptoms suggesting pre-fibrotic myelofibrosis or primary myelofibrosis, according to the criteria proposed by Barosi G et al. at the final step of the qualification process for the study [35]. The pts were recruited from two academic centers — the Department of Hematology and Bone Marrow Transplantation of Poznan University of Medical Sciences in Poznan, Poland, and the Department of Hematology and Department of General Pathology of Pomeranian Medical University in Szczecin, Poland. The duration of the follow-up and the type of cytoreductive treatment used in patients with ET and subjects with ET transforming to the post-ET-MF is presented in Table 1. The clinical patient workup included physical examination, ultrasonography, magnetic resonance, and computed tomography imaging. The diagnostic algorithm also took into consideration peripheral blood and bone marrow biopsies analysis, trephine bone marrow biopsy assessment, and molecular testing for the BCR-ABL fusion gene, as well as JAK2, CALR, and MPL mutation screening. All patients studied have been monitored regularly (every 1–3 months) in Outpatient Department. In the case of symptoms suggesting ET transformation to post ET-MF, a trephine biopsy was performed to confirm the fibrotic transformation. In all these patients, repeated analysis of the initial trephine biopsy specimen was performed (if available).
The criteria for the diagnosis of post-ET-MF was based on the consensus statement from the International Working Group for Myelofibrosis Research and Treatment and included the documentation of a previous diagnosis of ET, as defined by the WHO criteria, as well as the confirmation of bone marrow fibrosis grades 2–3 (on 0–3 scale), and at least two of the following criteria: anemia and a ≥ 2-g/dL decrease from baseline hemoglobin level, a leucoerythroblastic peripheral blood picture, increasing splenomegaly, defined as either an increase in palpable splenomegaly of ≥ 5 cm (the distance of the tip of the spleen from the left costal margin), or the appearance of newly palpable splenomegaly, increased lactic dehydrogenase activity above the reference level, the development of ≥ 1 of three constitutional symptoms: > 10% weight loss in 6 months, night sweats or unexplained fever (> 37.5 °C) [36, 37]. The grade of the bone marrow fibrosis was assessed according to the European consensus on grading bone marrow fibrosis and the assessment of cellularity [38].
Methods
DNA and RNA were extracted from whole-blood leukocytes at the time of the initial evaluation due to the clinical suspicion of ET or disease evolution to the fibrotic phase. Genomic DNA was isolated using the QIAamp® DNA Blood Mini Kit (QUIAGEN). Total RNA was extracted with TRIzol™ (Invitrogen). Purity and quantity of DNA and RNA were assessed with NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific).
The assessment for the presence of the JAK2V617F mutation was conducted by a quantitative allele-specific PCR (ASO-PCR) according to Larsen et al. [39], and standardized in cooperation with MPN&MPNr EuroNet [40]. A high-resolution melting analysis (HRMA) was used to detect mutations in CALR (exon 9), MPL (exon 10), SRSF2 (exon 1), and U2AF1 (exons 2 and 6) genes, as previously described by Klampfl et al. [12], Boyd et al. [41], Lin et al. [42], and Qian et al. [43], respectively. For the identification of the mutation type identified by HRMA, the Sanger sequencing was applied using the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Thermo Fisher Scientific). The sequence of exon 13 (range Ile574 to Glu727) of the ASXL1 gene (a region covering at least 83% of all known ASXL1 mutations), was analyzed by Sanger sequencing [44, 45]. To determine the JAK2 haplotypeGGCC_46/1, the rs12343867 SNP was genotyped [46].
The analysis of the JAK2WT and JAK2V617F mRNA level and PD-L1 mRNA level was done with the help of methods presented in the Supplementary File Methods. The gene copy number analysis (CNA) was performed with the use of the in-house developed multiplex ligation-dependent probe amplification (MLPA) test (Supplementary File Methods), designed and executed according to the well-established protocol, described before [47, 48].
Statistical analysis
All basic statistical analyses were performed in Statistica 13 [TIBCO Software Inc. (2017). Statistica (data analysis software system), version 13, www.statsoft.pl/statistica-i-tibco-software/. Depending on the distribution of the variables, a proper parametric (ANOVA F) or non-parametric (Mann–Whitney, Kruskal–Wallis) test was used. In the case of variance and correlation study, an analysis was performed in R ver. 4.0.4 and R Studio ver. 1.4.1106, with the following R packages: base, ggcorrplot, dplyr, and Hmisc. For the association of gene expression with the mutation status and clinical features, ANOVA and MANOVA tests were applied, for univariate and multivariate analysis of variance, respectively. Because the data did not follow a normal distribution, Spearman’s rank correlation coefficients were calculated to estimate the correlation between the studied gene expression levels and blood parameters. Information on protein–protein interactions and the co-expression score, based on the RNA expression pattern, was obtained from the STRING database v. 11.5 (https://string-db.org).
Results
Study group characteristics
A detailed characteristic of the studied ET pts (n = 162) is presented in Table 1. According to our data, the frequency of the fibrotic transformation in patients over 60 years old was 20% (17/85), in the group of individuals between 40 and 60 years old 14.6% (7/48), and in patients < 40 years old 20.7% (6/29). In reported individuals experiencing ET progression to the fibrotic phase, the median follow-up duration was as long as ~ 10 years, with the exception of patients > 60 years old (~ 5 years). For ET diagnosis confirmation, trephine biopsy was performed at the time of diagnosis in 132 out of 162 cases (in the other cases, only aspiration biopsy was performed). Repeated biopsy was performed in 32 patients with clinical and/or laboratory symptoms suggesting a possible disease transformation to post-ET-MF. In thirty of them, post-ET-MF was diagnosed. In our resources, there was no data concerning the reclassification of patients diagnosed with ET to pre-fibrotic primary myelofibrosis or primary myelofibrosis.
The comparison of the different molecularly defined subgroups showed that the hemoglobin blood concentration was significantly increased in the JAK2V617F-positive pts (ANOVA test, p = 0.0165; Supplementary File Results). On the contrary, there was no difference in the hemoglobin blood concentration between ET and post-ET-MF pts (Kruskal–Wallis test, p = 0.0843; Supplementary file Results). An analysis of the WBC count in relation to the JAK2V617F VAF (< 50% and ≥ 50%) confirmed increased WBC count in pts with VAF ≥ 50% (Kruskal–Wallis test, p = 0.0018; Supplementary file Results). The comparison of the platelet count in molecularly defined ET subgroups showed that the platelet count was significantly higher only in the CALR-mutation-positive vs. CALR-mutation-negative pts (Mann–Whitney test, p = 0.0219; Supplementary file Results). JAK2V617F mutation was present in 96 (59.3%), CALR mutation in 31 (types 1–12, types 2–15, others 4, overall 19.1%), and MPL mutations in two (1.2%) out of the 162 pts. The co-occurrence of JAK2V617F and either CALR or MPL mutation was identified in three (1.9%) and 1 (0.6%) of the pts, respectively. Twenty-nine out of the 162 (17.9%) analyzed pts were triple-negative. The ASXL1, SRSF2, and U2AF1 mutations co-occurred in 7/96 (7.3%) pts with JAK2V617F and 3/31 (9.7%) pts with the CALR mutations. Thirteen non-driver mutations (ASXL1, SRSF2, U2AF1) were present in 11/162 (6.8%) of the pts. The distribution of the non-driver gene mutations and molecular characteristics of ET and post-ET-MF pts harboring two or more mutations are shown in Supplementary file Results.
ET and JAK2 haplotype GGCC_46/1, driving mutations, and JAK2 variant allele frequency
The study of the JAK2 haplotypeGGCC_46/1 (tagged by the C allele of rs12343867 SNP) showed the C/C genotype in 31 (19%), C/T in 73 (45%), and T/T in 58 (36%) pts. An analysis of the frequency of different JAK2 haplotypeGGCC_46/1 genotypes in JAK2V617F positive ET pts showed a higher frequency of the mutation in the C/C vs. C/T and T/T haplotype carriers (Fisher exact test, p = 0.0228). No association of the JAK2 haplotypeGGCC_46/1 was observed with the CALR mutations (data not shown). The JAK2V617F VAF distribution between different JAK2 haplotypeGGCC_46/1 groups showed that the allelic burden was significantly higher in the C/C than in other genotype carriers (p = 0.0198; Supplementary file Results). Moreover, a significant increase of C/C genotype percentage was observed in the ET pts with JAK2V617F VAF > 50% (p = 0.0033; Supplementary file Results). No distribution differences were noticed in terms of the WBC, platelet count, hemoglobin concentration, and age at diagnosis in different JAK2 haplotypeGGCC_46/1 groups.
Gene copy number aberrations
No copy-number alternations of the JAK2, and CD274 and PDCDL1G2 gene (encoding proteins PD-L1 and PD-L2, respectively) located in close proximity to JAK2 at the 9p24 chromosome locus was detected with the use of the in-house designed MLPA assay (Supplementary File Methods).
JAK2V617F variant allele frequency and expression
JAK2V617F VAF was determined by the ASO-PCR and MLPA technique (r = 0.9217). Low and high JAK2V617F allele burden was found in 81/96 (84.4%) and 15/96 (10.4%) of the pts, respectively. In the JAK2V617F positive ET pts, the JAK2V617F allelic mRNA level was positively correlated with the JAK2V617F VAF (r = 0.6337; Supplementary file Results). The analysis of the JAK2V617F mRNA showed the presence of a low-level JAK2V617F allele in four additional individuals in which the mutation was not detected by a DNA analysis with ASO-PCR and MLPA. The total JAK2 level in JAK2V617F positive pts was higher than in JAK2WT individuals (p = 0.0130; Supplementary file Results).
JAK2V617F VAF was higher in pts with leukocytosis at the time of evaluation (p = 0.0015). In addition, the WBC count was higher in ET pts with high JAK2V617F VAF (Supplementary file Results). An analysis performed in pts with low (≤ 50%) and high (> 50%) JAK2V617F VAF did not show any differences in the mean values of hemoglobin concentration and platelet count in the blood. Pts carrying the CALR mutation had significantly higher median platelet count in comparison to the CALR mutation-negative pts (ANOVA test, p = 0.028; Supplementary file Results). A similar analysis was not performed for the MPL mutated pts due to an insufficient number of positive pts (n = 3).
The total JAK2 level did not differ between the different genotypes of the JAK2 haplotypeGGCC_46/1. No male/female differences in the JAK2 level (both total, in all pts, and JAK2V617F positive pts) were observed.
PD-L1 and JAK2 expression in the molecularly defined essential thrombocythaemia dependently from the bone marrow fibrosis grade
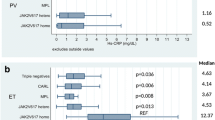
The total JAK2 expression level (JAK2V617F + wild type) showed no differences depending on the types of driving mutation (Kruskal–Wallis test, p = 0.1085; Supplementary file Results). The expression of PD-L1 was significantly higher in the JAK2V617F mutation-positive vs. JAK2WT ET pts (Kruskal-Willis test, p = 0.0051) (Fig. 1). A similar analysis performed in the CALR mutation-positive and CALRWT pts showed no differences (Kruskal-Willis test, p = 0.1908; Fig. 1). There were no differences in the expression of PD-L1 in the JAK2/CALR/MPL mutation-positive and triple-negative ET pts. The PD-L1 expression in the JAK2V617F positive pts was higher than in the CALR mutation-positive and triple-negative pts (Kruskal-Willis test, p = 0.0439 and p = 0.0485, respectively; Fig. 1).

The mRNA expression of programmed death ligand 1 (PD-L1) and Janus tyrosine kinase 2 (JAK2 (WT + V617F)), dependently from the ET driver mutation status (A) and the grade of myelofibrosis (B)
An analysis performed independently from the driver mutation status showed that the PD-L1 mRNA level was significantly lower in the post-ET-MF than ET pts (Kruskal-Willis test, p = 0.0176; Fig. 1).
In addition, the total JAK2 level in post-ET-MF pts was lower than in ET pts without fibrotic transformation (Kruskal Willis test, p = 0.0003). It was evident, despite the lack of differences between the JAK2V617F VAF in ET and post-ET-MF pts (Kruskal–Wallis test, p = 0.3785; Supplementary file Results). A detailed analysis has shown that the decrease in JAK2 and PD-L1 mRNA level was gradual, depending on the bone marrow fibrosis grade (MF grade 0 vs 1 vs 2 p < 0.001, respectively; Fig. 1). A weak correlation was observed in the case of the PD-L1 and JAK2 total (JAK2V617F + JAK2WT) mRNA level (r = 0.1259, data not shown). Moreover, a low correlation between the PD-L1 mRNA level and JAK2V617F mutation VAF or the JAK2V617F allele mRNA level was found (r = 0.204; p = 0.049 and r = 0.232; p = 0.024, respectively). The correlation between the expression of PDL1 and JAK2 (both, mutated and WT allele) (Fig. 2B) observed here corresponds with the data retrieved from the STRING database v. 11.5 (https://string-db.org) [49]. According to STRING, the predicted JAK2 and PDL1 protein interaction is supported by the co-expression of the genes encoding both proteins (Fig. 2A; RNA co-expression score 0.097).

The association between the expression of PDL1 and JAK2 (both, V617F mutated and wild type allele (wt)) and complete blood count results in the studied cohort of ET patients. A The analysis of relations between genes encoding JAK2 and CD274 (PDL1) proteins. The network of five proteins interacting with JAK2 and CD274 (PDL1) generated by STRING v. 11.5 (https://string-db.org). RNA co-expression score observed in Homo sapiens for JAK2 and CD274 was 0.097. B A graphical presentation of Spearman correlation of JAK2 and PDL1 gene expression levels and blood parameters measured for patients included into the study. The color intensities reflect the correlation coefficients according to the legend on the right side of the graph (blue circles mean positive correlations, and red circles mean negative ones). The size of the circles is proportional to the level of significance (only statistically significant correlations are presented, with the p value threshold set as 0.05). Abbreviations: JAK2_mut_VAF, JAK2 V617F variant allele frequency; JAK2_mut, expression of mutated JAK2 allele; JAK2_wt, expression of wild type JAK2 allele; JAK2_mut_vs_wt, the ratio of expression of mutated JAK2 allele versus wild-type JAK2 allele; JAK2_total, total JAK2 expression (the sum of both alleles); PDL1, expression of PDL1 (CD274) gene; HB, hemoglobin; HCT, hematocrit; PLT, platelets; WBC, white blood count
The study of the relationship between the PD-L1 expression and JAK2 haplotypeGGCC_46/1 genotype, hemoglobin concentration, hematocrit value, leukocyte, and platelet count showed no differences (data not shown).
Discussion
In 2008, Pardanani et al. based on an analysis of SNPs in four candidate genes (EPOR, MPL, GCSFR, JAK2) confirmed a significant association between the specific SNPs in the JAK2 gene (46/1 haplotype or GGCC haplotype) and the onset of sporadic MPNs [50]. The allele frequency of the JAK2 haplotypeGGCC_46/1 in the healthy population is about 24%. Its presence is significantly increased (40 to 80%) in JAK2V617F positive MPN pts [51,52,53,54,55].
In our study, we showed that the JAK2 haplotypeGGCC_46/1 was significantly more frequent in JAK2V617F-positive than in CALR-positive ET pts. The latter observation is in agreement with the data previously reported by others [56].
Our results confirmed a significantly higher JAK2V617F VAF in homozygous carriers of JAK2 haplotypeGGCC_46/1 (C/C genotype) and a significant increase of the C/C genotype in ET pts with JAK2V617F VAF > 50%.
The total JAK2 mRNA level did not significantly differ between pts defined by JAK2 haplotypeGGCC_46/1, but it was significantly increased in JAK2V617F positive pts. In our opinion, the increased total JAK2 expression in ET pts may result from different factors, including the allelic expression imbalance of JAK2 V617F mutation, MPN-associated chronic inflammation, the presence of other non-coding SNP affecting the JAK2 expression, or mutations of epigenetic genes regulators (DNMT3A, TET2, EZH2, ASXL1, and IDH1/2 (via effects on TET2-mediated methylation)).
In 2013, Kim et al. demonstrated the JAK2V617F allelic imbalance leads to an increase of mutant allele, especially in ET (threefold increase) and polycythaemia vera (PV) (twofold increase) pts. Another possibility of an increased total JAK2 mRNA expression in ET pts is abnormal JAK-STAT signaling. The potential link between a chronic inflammation and the development of myeloproliferative neoplasm has been postulated by Hasselbalch in 2012 [57]. Later, it was shown that MPN driver mutations, such as JAK2V617F and MPL, were responsible for continuous, increased signaling via the JAK2-STAT pathway and the promotion of cytokine production by malignant and non-malignant cells [58,59,60,61]. The above-mentioned hypothesis may be supported by the data concerning another JAK2 co-expressed gene–programmed death-ligand 1 (PD-L1). In 2019, Guru et al. showed that JAK2 V617F mutation was accompanied by an increased PD-L1 expression. An increased expression of PD-L1 may be caused by excessive activation of STAT3/5 which are the regulators of PD-L1. It was also shown that in the case of JAK2V617F, the PD-L1 expression was mainly mediated by STAT3 [62]. The overexpression of PD-L1 may also be caused by the acquisition of 9q UPD, which was confirmed in 6–18% of cases with ET [33, 63].
It should be noted that the PD-L1 mRNA level is not affected by the PD-L1 or JAK2 gene copy number variations, which has been shown in our study.
In 2020, Jacquelin et al. confirmed the mutational cooperation between the JAK2V617F expression and the loss of DNA methyltransferase 3A in hematopoietic cells due to monoallelic or biallelic mutations of the DNMT3A gene. The coexistence of the above-mentioned mutations resulted in an aberrant self-renewal, inflammatory signaling, driven by increased accessibility at enhancer elements, and finally the progression of PV to the fibrotic phase [64]. Another possibility includes the presence of other JAK2 gene mutations affecting the mRNA splicing machinery. mRNA investigations showed no splicing defects around exon 14, and a constant level of mRNA accumulation per JAK2 gene copy, regardless of the presence or absence of the exon 14 JAK2V617F mutation [65].
Another important question concerns the fluctuation of the total JAK2 mRNA level during a natural disease outcome and evolution into the fibrotic phase (post-ET-MF). The results of our study showed a lower total JAK2 mRNA level in post-ET-MF, in comparison to ET pts. It is in agreement with previously published data documenting lower the PD-L1 mRNA expression in JAK2V617F positive primary myelofibrosis in comparison to JAK2V617F positive ET and PV patients [33]. The detailed analysis has shown that the decrease in JAK2 and PD-L1 mRNA expression depends on the bone marrow fibrosis grade. A similar analysis concerning JAK2V617F VAF in ET and post-ET-MF pts did not reveal significant differences. The interpretation of the obtained data is difficult due to the fact that the fibrotic transformation in ET is rarely observed and occurs in 9–15% of pts during a long-term follow-up [66, 67]. Our data indicates that after a median time of 10 years, in patients < 60 years old, the frequency of fibrotic transformation is 8.0% (13/162 pts) which is slightly higher than reported by others (0.8–4.9%, respectively [24, 22]. An association with the applied therapy cannot be ruled out because 11.7% (19/162) of our patients were treated with anagrelide and 2.3% with PEG-interferon α [68, 69].
On the other hand, it should be noted that the clinical MPN manifestation is not only related to the type of the driver mutation but also depends on the profile of other coexisting mutations modifying the disease phenotype. The frequency and VAF of specific coexisting mutation(s) differ between pts, contributing to a specific disease phenotype in individual cases [1, 11, 70]. Among others, the mutations involved in the DNA methylation (ASXL1, TET2, DNMT3A, IDH1, IDH2), histone modification (EZH2, ASXL1), and splicing (SF3B1, SRSF2, U2AF1, and ZRSR2), and mutations in the transcription factors genes (RUNX1, NFE2, PPM1D, and TP53) are most frequently found in ET pts [71, 72]. An adverse prognostic relevance of some of them (SH2B3/LNK, SF3B1, U2AF1, TP53, IDH2, and EZH2) on overall, leukemia-free and myelofibrosis-free survival of ET pts was recently demonstrated [70, 73,74,75]. Furthermore, the ASXL1 mutations (most frequently found in post-ET-MF pts) have been also identified as a genetic risk factor for the fibrotic transformation of ET [22]. Similarly, the co-occurrence of variants/mutations of SRSF2 and U2AF1 increased the risk of myelofibrotic evolution in PV and ET pts, respectively [74]. The results of our study confirmed the co-occurrence of ASXL1, SRSF2, and U2AF1 mutations in the studied ET pts group. Their frequency was below 10% and was similar to that reported by others [73, 76]. However, it should be mentioned that its frequency in our JAK2V617F positive cases was 2 times higher in post-ET-MF than in ET individuals.
It is, however, important to note that MPNs evolve in a biological continuum from early cancer-stages (ET, PV) to the advanced myelofibrosis stage, and other factors, including age, may also play a role [77]. Our data also indicates that the frequency of post-ET MF is age-dependent, lowest in the group of 40–60-year-old patients. Published data revealed that the C-X-C motif chemokine receptor (CXCR3) expression decreases over the MPN-continuum which may confirm the hypothesis of the reduction of the JAK-STAT induced inflammatory reaction intensity in the fibrotic disease phase [78]. The CXCR3-axis has been shown to have angiostatic and antifibrotic properties and via its chemokines’ ligands (CXCL9, CXCL10, and CXCL11) participate in the recruitment of immune/inflammatory cells: Th1 cells, CD8 + central memory T cells and effector-memory T cells, natural killer cells, natural killer T cells, plasmacytoid dendritic cells, B cells, regulatory T cells, and follicular helper T cells [79]. Recently, it was discovered that the circulating cytokine levels of B lymphocyte chemoattractant (BLC or CXCL13), TIMP metallopeptidase inhibitor 1 (TIMP-1), eotaxin-2, and macrophage colony-stimulating factor (M-CSF, previously CSF-1) reflected inflammation inside the bone marrow niche (BMN) [80, 81]. A detailed analysis of published data showed that M-CSF concentration decreases in the fibrotic phase of MPNPh- (MF > 1 vs MF ≤ 1) [81]. The mentioned changes may explain the drop in PD-L1 expression in the fibrotic phase of MPN because The Cancer Genome Atlas (TCGA) data documents a strong positive association between M-CSF, CXCR3 expression, and PD-L1 expression in another myeloid malignancy-acute myeloid leukemia (Fig. 3).

Relation between CD274 (encoding PD-L1), CXCR3 (encoding CXCR3), and CSF1 (encoding M-CSF) expression levels in AML. A Heatmap graphs showing the expression (normalized mRNA levels; RNAseq log2(norm_count + 1)) of CD274, CXCR3, and CSF1 genes in 173 AML samples from The Cancer Genome Atlas (TCGA) cohort (LAML). The samples were sorted according to CD274 levels, from the highest (top, labeled in red) to the lowest (bottom, labeled in blue). The graphs visualize the overall positive relationship between levels of the three genes. B Scatter plots showing pairwise correlations between levels (RNAseq log2(norm_count + 1)) of the free genes; (from the top) CC274 vs. CXCR3, CD274 vs. CSF1, and CXCR3 vs. CSF1. Each dot represents an individual AML sample. On each graph, the trend line, correlation coefficients (r), and p values for the correlations are indicated. The data were extracted, and the graphs were prepared with the use of the UCSC XENA oncogenomic portal (102)
The interpretation of the simultaneous lowering of the PD-L1 and JAK2 mRNA expression detected in our study in ET patients transforming to the fibrotic phase is difficult. The bone marrow failure in the advanced ET phase is likely associated with bone marrow fibrosis, reduction of bone marrow cellularity, bone marrow myeloid metaplasia, and diversity in molecular characteristics of the emerging subclones. The latter may result in a distinct pattern of expression of JAK2 and PD-L1 genes [82,83,84]. The association between PD-L1 expression and JAK2V617F mutation was recently documented by Hara et al. in a patient with a coexisting JAK2V617F-positive ET and lung carcinoma in whom pembrolizumab (a drug directly blocking the interaction between PD-1 and its ligands, PD-L1, and PD-L2) treatment resulted in simultaneous normalization of the platelet count and a decrease of JAK2V617F VAF [85].
Despite the progress in the last years in the treatment of ET, none of the available therapies can change the outcome of the disease [86]. For these reasons, there is a need to identify a new potential molecular mechanism affecting the drug resistance to improve the ET outcome. One of them is the PD1/PD-L1 axis. In 2018, Holmström et al. documented that PD-L1 specific T cell response was stronger in pts with ET and PV and weaker and rarer in pts with pre-PMF and PMF MPN [87].
The results of our study can explain, at least in part, the lack of efficacy (clinical or bone marrow pathologic response) of the pembrolizumab treatment in patients with advanced primary, post-ET-MF, and post-PV myelofibrosis [88] and shed more light on the relationship between the types of driver mutations, the PD-L1 expression, and the ET progression to the fibrotic phase.
Study limitations
We realize that a proper distinction between ET and pre-fibrotic MF is difficult and sometimes impossible at the initial diagnosis step. We hope that the relatively long time of the follow-up and detailed monitoring of the appearance of new symptoms, characteristic for the pre-fibrotic phase of primary myelofibrosis, allowed us to omit the diagnostic pitfall in most of the ET cases. Moreover, the impact of the others factors influencing the individual ET outcome, fibrotic transformation process, and PD-L1 expression should be taken into consideration in the final interpretation of the data.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Vainchenker W, Kralovics R (2017) Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 129(6):667–679
Grinfeld J, Nangalia J, Green AR (2017) Molecular determinants of pathogenesis and clinical phenotype in myeloproliferative neoplasms. Haematologica 102(1):7–17
Jang MA, Choi CW (2020) Recent insights regarding the molecular basis of myeloproliferative neoplasms. Korean J Intern Med 35(1):1–11
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP et al (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7(4):387–97. https://pubmed.ncbi.nlm.nih.gov/15837627/
Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR et al (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352(17):1779–90. https://pubmed.ncbi.nlm.nih.gov/15858187/
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S et al (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365(9464):1054–61. https://pubmed.ncbi.nlm.nih.gov/15781101/
Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M et al (2006) MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 3(7):1140–1151
Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M et al (2006) MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 108(10):3472–3476
Defour JP, Chachoua I, Pecquet C, Constantinescu SN (2016) Oncogenic activation of MPL/thrombopoietin receptor by 17 mutations at W515: implications for myeloproliferative neoplasms. Leukemia 30(5):1214–6
Staerk J, Lacout C, Sato T, Smith SO, Vainchenker W, Constantinescu SN (2006) An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood 107(5):1864–1871
Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC et al (2013) Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 369(25):2391–405. https://pubmed.ncbi.nlm.nih.gov/24325359/
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD et al (2013) Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 369(25):2379–90. https://pubmed.ncbi.nlm.nih.gov/24325356/
Pietra D, Rumi E, Ferretti VV, Di Buduo CA, Milanesi C, Cavalloni C et al (2016) Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 30(2):431–438
Cabagnols X, Defour JP, Ugo V, Ianotto JC, Mossuz P, Mondet J et al (2015) Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: relevance for disease evolution. Leukemia 29(1):249–52
Di Buduo CA, Abbonante V, Marty C, Moccia F, Rumi E, Pietra D et al (2020) Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 135(2):133–144
Tefferi A, Pardanani A (2022) Essential thrombocythemia. NEJM 381(22):2135–44. https://doi.org/10.1056/NEJMcp1816082
Masubuchi N, Araki M, Yang Y, Hayashi E, Imai M, Edahiro Y et al (2020) Mutant calreticulin interacts with MPL in the secretion pathway for activation on the cell surface. Leukemia 34(2):499–509
Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C et al (2016) Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 127(10):1325–35. https://pubmed.ncbi.nlm.nih.gov/26668133/
Lussana F, Rambaldi A (2017) Inflammation and myeloproliferative neoplasms. J Autoimmun 85:58–63
Bock O, Höftmann J, Theophile K, Hussein K, Wiese B, Schlué J et al (2008) Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am J Pathol 172(4):951–960
Lu M, Xia L, Liu YC, Hochman T, Bizzari L, Aruch D et al (2015) Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 126(8):972–982
Cerquozzi S, Tefferi A (2015) Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors, vol 5. Nature Publishing Group, Blood Cancer Journal
Passamonti F, Rumi E, Arcaini L, Boveri E, Elena C, Pietra D et al (2008) Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica 93(11):1645–1651
Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M et al (2011) Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: a international study. J Clin Oncol 29(23):3179–3184
Passamonti F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L et al (2010) A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 24(9):1574–1579
Barbui T, Thiele J, Ferrari A, Vannucchi AM, Tefferi A (2020) The new WHO classification for essential thrombocythemia calls for revision of available evidences. Blood Cancer J 10(2):22
Latagliata R, Polverelli N, Tieghi A, Palumbo GA, Breccia M, Sabattini E et al (2018) Comparison of JAK2V617F-positive essential thrombocythaemia and early primary myelofibrosis: the impact of mutation burden and histology. Hematol Oncol 36(1):269–75. https://pubmed.ncbi.nlm.nih.gov/28509339/
Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA (2014) Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and Mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res 20(10):2674–2683
Barrett MT, Anderson KS, Lenkiewicz E, Andreozzi M, Cunliffe HE, Klassen CL et al (2015) Genomic amplification of 9p24.1 targeting JAK2 PD-L1 and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget 6(28):26483–93
Prestipino A, Emhardt AJ, Aumann K, O’Sullivan D, Gorantla SP, Duquesne S et al (2018) Oncogenic JAK2 V617F causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci Transl Med 10(429):eaam7729. https://pubmed.ncbi.nlm.nih.gov/29467301/
Tsushima F, Yao S, Shin T, Flies A, Flies S, Xu H et al (2007) Interaction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood 110(1):180–5. https://pubmed.ncbi.nlm.nih.gov/17289811/
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I et al (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2(3):261–8. https://pubmed.ncbi.nlm.nih.gov/11224527/
Milosevic Feenstra JD, Jäger R, Schischlik F, Ivanov D, Eisenwort G, Rumi E et al (2022) PD-L1 overexpression correlates with JAK2-V617F mutational burden and is associated with 9p uniparental disomy in myeloproliferative neoplasms. Am J Hematol 97(4):390–400. https://onlinelibrary.wiley.com/doi/full/10.1002/ajh.26461
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127(20):2391–405
Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S et al (2008) Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the international working group for myelofibrosis research and treatment. Leukemia 22(2):437–8. https://www.nature.com/articles/2404914
Passamonti F, Mora B, Barraco D, Maffioli M (2018) Post-ET and post-PV myelofibrosis: updates on a distinct prognosis from primary myelofibrosis. Curr Hematol Malig Rep 13(3): 173–82
Barosi G, Mesa RA, Thiele J, Cervantes F, Campbell PJ, Verstovsek S et al (2008) Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 22(2):437–8. https://pubmed.ncbi.nlm.nih.gov/17728787/
Thiele J, Kvasnicka HM, Facchetti F, Franco V, Van Der Walt J, Orazi A (2005) European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 90(8):1128–1132
Larsen TS, Christensen JH, Hasselbalch HC, Pallisgaard N (2007) The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br J Haematol 136(5):745–751
Jovanovic JV, Ivey A, Vannucchi AM, Lippert E, Oppliger Leibundgut E, Cassinat B et al (2013) Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: a joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) stu. Leukemia 27(10):2032–2039
Boyd EM, Bench AJ, Goday-Fernández A, Anand S, Vaghela KJ, Beer P et al (2010) Clinical utility of routine MPL exon 10 analysis in the diagnosis of essential thrombocythaemia and primary myelofibrosis: Research paper. Br J Haematol 149(2):250–257
Lin J, Yang J, Wen XM, Yang L, Deng ZQ, Qian Z et al (2014) Detection of SRSF2-P95 mutation by high-resolution melting curve analysis and its effect on prognosis in myelodysplastic syndrome. PLoS One 9(12):e115693
Qian J, Yao DM, Lin J, Qian W, Wang CZ, Chai HY et al (2012) U2AF1 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome Navarro A editor. PLoS One 7(9):e45760
Pratcorona M, Abbas S, Sanders MA, Koenders JE, Kavelaars FG, Erpelinck-Verschueren CAJ et al (2012) Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica 97(3):388–392
Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N et al (2009) Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 145(6):788–800
Pardanani A, Lasho TL, Finke CM, Gangat N, Wolanskyj AP, Hanson CA et al (2010) The JAK2 46/1 haplotype confers susceptibility to essential thrombocythemia regardless of JAK2V617F mutational statusclinical correlates in a study of 226 consecutive patients. Leukemia 24(1):110–114
Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H et al (2007) Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet 121(3–4):389–400. https://pubmed.ncbi.nlm.nih.gov/17287951/
Marcinkowska M, Wong KK, Kwiatkowski DJ, Kozlowski P (2010) Design and generation of MLPA probe sets for combined copy number and small-mutation analysis of human genes: EGFR as an example. Scientific World Journal 10:2003–18
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J et al (2019) STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47(D1):D607–D613. https://pubmed.ncbi.nlm.nih.gov/30476243/
Pardanani A, Fridley BL, Lasho TL, Gilliland DG, Tefferi A (2008) Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood 111(5):2785–2789
Tefferi A, Lasho TL, Patnaik MM, Finke CM, Hussein K, Hogan WJ et al (2010) JAK2 germline genetic variation affects disease susceptibility in primary myelofibrosis regardless of V617F mutational status: nullizygosity for the JAK2 46/1 haplotype is associated with inferior survival. Leukemia 24(1):105–109
Andrikovics H, Nahajevszky S, Koszarska M, Meggyesi N, Bors A, Halm G et al (2010) JAK2 46/1 haplotype analysis in myeloproliferative neoplasms and acute myeloid leukemia. Leukemia 24(10):1809–13
Matsuguma M, Yujiri T, Yamamoto K, Kajimura Y, Tokunaga Y, Tanaka M et al (2019) TERT and JAK2 polymorphisms define genetic predisposition to myeloproliferative neoplasms in Japanese patients. Int J Hematol 110(6):690–698
Olcaydu D, Harutyunyan A, Jäger R, Berg T, Gisslinger B, Pabinger I et al (2009) A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet 41(4):450–454
Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL et al (2009) JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet 41(4):446–449
Gau JP, Chen CC, Chou YS, Liu CJ, Bin YuY, Hsiao LT et al (2015) No increase of JAK2 46/1 haplotype frequency in essential thrombocythemia with CALR mutations: functional effect of the haplotype limited to allele with JAK2V617F mutation but not CALR mutation. Blood Cells Mol Dis 55(1):36–9
Hasselbalch HC (2012) Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 119(14):3219–25
Koschmieder S, Mughal TI, Hasselbalch HC, Barosi G, Valent P, Kiladjian JJ et al (2016) Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Vol. 30, Leukemia 30(5):1018–24
Geyer HL, Kosiorek H, Dueck AC, Scherber R, Slot S, Zweegman S et al (2017) Associations between gender, disease features and symptom burden in patients with myeloproliferative neoplasms: an analysis by the MPN QOL international working group. Haematologica 102(1):85–93
Lubberich RK, Walenda T, Goecke TW, Strathmann K, Isfort S, Brümmendorf TH et al (2018) Serum of myeloproliferative neoplasms stimulates hematopoietic stem and progenitor cells. PLoS One 13(5):e0197233
Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L et al (2015) JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov 5(3):316–331
Guru SA, Alpana MP, Mir R, Najar IA, Zuberi M, Gupta N et al (2019) JAK2 (V617F) positively regulates PD-L1 mRNA expression via STAT3/5 activation in MNP (PV and ET) patients. Ann Oncol 30(suppl. 6):VI110
Wang L, Wheeler DA, Prchal JT (2015) Acquired uniparental disomy of chromosome 9p in hematologic malignancies. Exp Hematol 44(8):644–52. https://pubmed.ncbi.nlm.nih.gov/26646991/
Jacquelin S, Straube J, Cooper L, Vu T, Song A, Bywater M et al (2018) Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 132(26):2707–2721
Nauroy P, Delhommeau F, Baklouti F (2014) JAK2V617F mRNA metabolism in myeloproliferative neoplasm cell lines. Blood Cancer J 4(6):e222
Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L et al (2014) Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124(16):2507–2513
Tefferi A (2021) Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am J Hematol 96(1):145–162
Birgegård G, Besses C, Griesshammer M, Gugliotta L, Harrison CN, Hamdani M et al (2018) Treatment of essential thrombocythemia in Europe: a prospective long-term observational study of 3649 high-risk patients in the Evaluation of Anagrelide Efficacy and Long-term Safety study. Haematologica 103(1):51–60. https://pubmed.ncbi.nlm.nih.gov/29079600/
O’Neill C, Siddiqi I, Brynes RK, Vergara-Lluri M, Moschiano E, O’Connell C (2016) Pegylated interferon for the treatment of early myelofibrosis: correlation of serial laboratory studies with response to therapy. Ann Hematol 95(5):733–8. https://pubmed.ncbi.nlm.nih.gov/26961933/
Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I et al (2014) Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 123(14):2220–2228
Leibundgut EO, Haubitz M, Burington B, Ottmann OG, Spitzer G, Odenike O et al (2021) Dynamics of mutations in patients with essential thrombocythemia treated with imetelstat. Haematologica 106(9):2397–2404
Loscocco GG, Guglielmelli P, Vannucchi AM (2020) Impact of mutational profile on the management of myeloproliferative neoplasms: a short review of the emerging data. Onco Targets Ther 13:12367–12382
Tefferi A, Guglielmelli P, Lasho TL, Coltro G, Finke CM, Loscocco GG et al (2020) Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol 189(2):291–302
Tefferi A, Lasho TL, Guglielmelli P, Finke CM, Rotunno G, Elala Y et al (2016) Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv 1(1):21–30
Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R et al (2018) Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med 379(15):1416–1430
Agarwal R, Blombery P, McBean M, Jones K, Fellowes A, Doig K et al (2017) Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: data from targeted next-generation sequencing in the diagnostic laboratory. Ann Hematol 96(5):725–732
Szuber N, Vallapureddy RR, Penna D, Lasho TL, Finke C, Hanson CA et al (2018) Myeloproliferative neoplasms in the young: Mayo Clinic experience with 361 patients age 40 years or younger. Am J Hematol 93(12):1474–84. https://pubmed.ncbi.nlm.nih.gov/30157297/
Liisborg C, Skov V, Kjaer L, Hasselbalch HC, Sørensen TL (2022) Lower CXCR3 expression in both patients with neovascular AMD and advanced stages of chronic myeloproliferative blood cancers. PLoS One 17(6):e0269960. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0269960
Ozga AJ, Chow MT, Luster AD (2021) Chemokines and the immune response to cancer. Immunity 54(5):859–74. https://pubmed.ncbi.nlm.nih.gov/33838745/
Fisher DAC, Fowles JS, Zhou A, Oh ST (2021) Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front Immunol 1(12):2034
Chen P, Wu B, Ji L, Zhan Y, Li F, Cheng L et al (2021) Cytokine consistency between bone marrow and peripheral blood in patients with Philadelphia-negative myeloproliferative neoplasms. Front Med 24(8):607
Sharma M, Bhavani C, Suresh SB, Paul J, Yadav L, Ross C et al (2021) Gene expression profiling of CD34(+) cells from patients with myeloproliferative neoplasms. Oncol Lett 21(3):1–1. http://www.spandidos-publications.com/http://www.spandidos-publications.com/10.3892/ol.2021.12465/abstract
Mead AJ (2019) Unravelling heterogeneity of stem and progenitor cells in myeloid neoplasms through single cell multi-omics. Blood 134(Suppl. 1):SCI-31
Kumar S, Sharawat SK (2018) Epigenetic regulators of programmed death-ligand 1 expression in human cancers. Transl Res 202:129–45. https://pubmed.ncbi.nlm.nih.gov/30401465/
Hara R, Kawada H, Kikuti YY, Kikkawa E, Harada K, Aoyama Y et al (2021) A case of JAK2V617F-positive essential thrombocythemia where allele burden was reduced by a PD-1 inhibitor. Int J Hematol 113(4):606–10. https://pubmed.ncbi.nlm.nih.gov/33389657/
Gerds AT (2019) Beyond JAK-STAT: Novel therapeutic targets in Ph-negative MPN. Hematol (United States) 2019(1):407–14. Available from: https://pubmed.ncbi.nlm.nih.gov/31808852/
Holmström MO, Riley CH, Skov V, Svane IM, Hasselbalch HC, Andersen MH (2018) Spontaneous T-cell responses against the immune check point programmed-death-ligand 1 (PD-L1) in patients with chronic myeloproliferative neoplasms correlate with disease stage and clinical response. Oncoimmunology 7(6):e1433521. https://pubmed.ncbi.nlm.nih.gov/29872567/
Hobbs G, Cimen Bozkus C, Moshier E, Dougherty M, Bar-Natan M, Sandy L et al (2021) PD-1 inhibition in advanced myeloproliferative neoplasms. Blood Adv 5(23):5086–5097
Acknowledgements
We would like to thank Katarzyna Lewandowska for editing and proofreading the final version of the manuscript.
Funding
The study was funded by the Poznań University of Medical Sciences statutory fund no. 2705 and National Science Centre grant 2016/22/A/NZ2/00184.
Author information
Authors and Affiliations
Contributions
Conception and design, K. L. and P. K.; methodology, K. L., P. K., M. G.; acquisition of the clinical data, K. L., E. P., M. J., M. M., P. M. N.; investigation, K. L., Z. K., M. G., M. W., P. M. N.; analysis and interpretation of the clinical and laboratory data, Z. K., M. G., M. W., E. P., P. M. N., K. L., P. K.; statistical analysis, M. G., L. H.; project administration, K. L.; formal analysis, K. L.; writing—original draft, K. L.; writing—review and editing; L. K., P. K., M. G, Z. K., L. H. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Ethics Committee of Poznan University of Medical Sciences (number 1056/16 and 181/18) and was conducted in accordance with the Declaration of Helsinki.
Informed consent
Informed consent was obtained from all the subjects involved in the study.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lewandowski, K., Kanduła, Z., Gniot, M. et al. Essential thrombocythaemia progression to the fibrotic phase is associated with a decrease in JAK2 and PDL1 levels. Ann Hematol 101, 2665–2677 (2022). https://doi.org/10.1007/s00277-022-05001-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-022-05001-8