
Abstract
2-Hydroxy-3-(1-naphthyloxy)propyl methacrylate (NOPMA) monomer was synthesized from reaction of 2-[(2-naphthyloxy)methyl]oxirane with methacrylic acid in the presence of pyridine. The polymerization of NOPMA was carried out by free radical polymerization method in the presence of AIBN at 60 °C. The structure of monomer and polymer was characterized by 1H-NMR, 13C-NMR and FT-IR spectroscopy techniques. The glass transition temperature and average-molecular weights of poly(NOPMA) were measured using differential scanning calorimetry and gel permeation chromatography, respectively. The thermal degradation behavior of poly(NOPMA) has been investigated by FT-IR studies of the partially degraded polymer and thermogravimetry. The cold ring fractions (CRFs) were collected at two different temperatures, initially fraction-1 (CRF1) is from room temperature to 320 °C, and the other fraction-2 (CRF2) is from 320 to 500 °C. The volatile products of the degradation were trapped at −195 °C (in liquid nitrogen). All the fractions were characterized by FT-IR, 1H and 13C-NMR spectroscopic techniques, and the cold ring fractions (CRFs) were also characterized by GC–MS. For the degradation of polymer, the major compound between products of CRFs is α-naphthol. The GC–MS, FT-IR and NMR data showed that depolymerization corresponding to monomer was not prominent below 320 °C in the thermal degradation of poly(NOPMA). The mode of thermal degradation containing formation of the major products was identified. The dielectric permittivity (ε′), the loss factor (ε″) and conductivity (σac) were measured using a dielectric analyzer in the frequency range of 50 Hz to 20 kHz.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Many studies on thermal behavior of methacrylate polymers have been reported for a long time. Because the degradation mechanism is very good comprehensible initiation of depropagation with first-order termination or depropagation by chain end and random chain breakdown, widely different rate constants and E a (activation energy) have been investigated [1–3]. The researches necessary to undertake on the thermal degradation of polymers are important in view of the many applications depending on their thermal stability. To accomplish this goal, many characterization methods including gas chromatography–mass spectrometry (GC–MS), thermal analysis and spectroscopic techniques, etc., [4] have been used. The basic degradation pathways of depolymerization and side group reaction for homopolymer are first outlined and some of the additional reaction types and behaviors for random copolymers have been illustrated [5]. The thermal behavior of methacrylate polymers having a polar functional side group such as C=O, O–H can change via interchain and intra-chain of the side groups which results in cyclization, cross-linking, and so on [6–8]. In the recent years, investigations on thermal degradation of methacrylate and acrylate polymers having different side groups [9–11] have also been recorded in the literature. The chemical structure of a polymer having a reactive side group may change through interchain and intra-chain of the side groups resulting in the formation of a cross-linked and cyclic ladder structure during degradation of the polymer [12–14].
Many investigations on electrical behaviors of various polymer methacrylates and styrenes have been reported [15–17]. In order to characterize the nature of the conduction process, ac conductivity measurements are very significant. When a dielectric material is inserted between electric charges, it decreases the force acting between them, just as if they had been moved away from each other. The dielectric constant of a material affects how electromagnetic signals move through the material [18]. So, the dielectric constant and dissipation factor provide more information on the physical or chemical state of the polymer. They are crucial quantities required in the design of certain electronic devices. The dielectric treatment of polymer is defined by the thermal motion and charge distribution of its polar groups. Poly(NOPMA) is a polymer containing hydroxyl side group. Polymers such as poly(2-hydroxy ethyl methacrylate) and poly(2-hydroxy propyl methacrylate), a major component in materials for contact lenses, drug delivery, and hydrogels are used for a variety of applications. Some polymers with OH group possess hydrogel properties. The swollen hydrogels display extensive properties such as high water content, good biocompatibility, soft consistency similar to natural tissue, and elasticity, structural integrity. In this context, there are expectations being important polymer in view of materials used for contact lenses, drug delivery, and hydrogels of poly(NOPMA) which is similar to hydroxylated homologs [19–21].
In this work, we report on the preparation of poly(NOPMA) and thermal degradation behavior from ambient temperature to 320 °C and then 500 °C. Thermal behavior of poly(NOPMA) has been investigated using thermal analysis techniques. The degradation products were analyzed using GC–MS technique. The degradation behavior of poly(NOPMA) was partially compared with those of poly(2-hydroxyethyl methacrylate) and poly(2-hydroxypropyl methacrylate). A mode of degradation by considering the formation of degradation products has been formulated based on the results of TGA, FT-IR, NMR and GC–MS analyses. Also, the dielectric properties of poly(NOPMA) were measured based on frequency and temperature. More particularly, the dielectric loss, dielectric constant, and conductivity of the poly(NOPMA) were measured by impedance analyzer.
Materials and methods
Materials
Epichlorohydrin, pyridine, methacrylic acid, potassium carbonate, α-naphthol were purchased from Merck and Sigma-Aldrich and used as received. The AIBN (2,2′-azobisizobutyronitrile) was crystallized from methanol–chloroform mixture.
Measurements
The NMR spectra (1H and 13C-NMR) were obtained using an AVANCE III 400 MHz Bruker, using CDCl3 as the solvent. The infrared and calorimetric measurements were carried out using a Perkin-Elmer Spectrum One FT-IR spectrometer and a Shimadzu DSC-50 thermal analyzer (at a heating rate of 20 °C/min), respectively. TGA measurements were carried out on approximately 4–6 mg samples at a heating rate of 10 °C/min nitrogen atmosphere (under flow of 10 cm3/min). Average-molecular weights were estimated using on an Agilent 1100 series, GPC (gel permeation chromatography), using poly(MMA) as a standard. GPC measurements were carried out at 25 °C using a silica gel column with tetrahydrofuran (1 mL/min) as a solvent and a refractive-index (RI) detector. The dielectric properties of polymers were obtained using a QuadTech 7600 precision LRC meter impedance analyzer range from 50 Hz to 2 MHz. The degradation products were analyzed by GC–MS from Agilent Technologies 6890N (Network GC System and Agilent Technologies 5973 Inert Mass Selective Detector).
Synthesis of 2-[(2-naphthyloxy)methyl] oxirane
To a 100-mL flask were added α-naphthol (3 g, 0.02 mol) and epichlorohydrin (15 mL) and then poured excess of powdered NaOH (1.38 g, 0.034 mol). The mixture was refluxed at 110 °C for 5 h and then stirred for 12 h at room temperature. After the reaction is completed, the salt formed was removed by filtration. The residue was dissolved in dichloromethane and washed with cold water. The solvent was evaporated under reduced pressure. The 2-[(2-naphthyloxy)methyl] oxirane was formed as a yellow solid and the structure of compound is illustrated in Scheme 1.

The structure of 2-[(2-naphthyloxy)methyl]oxirane
FT-IR (cm−1, the most characteristic bands): 3054 and 3010 cm−1 aromatic =C–H; 2874 and 2925 cm−1 aliphatic –C–H; 1595 and 1579 cm−1 aromatic C=C and 915 cm−1 stretch in epoxide ring.
1H-NMR (CDCI3, δ ppm): 8.39 (1H, H7), 7.87 (1H, H4); 7.56–7.41 (3H, H2, H5, H6); 7.39 (1H, H3), 6.83 (1H, H1); 4.1 and 4.4 (2H, H8); 3.51 (2H, H9); 2.85 and 2.98 (2H, H10).
Synthesis of 2-hydroxy-3-(1-naphthyloxy)propyl methacrylate, NOPMA
NOPMA was prepared from the reaction of methacrylic acid with 2-[(2-naphthyloxy)methyl] oxirane via the method given for the epoxy-carboxy reaction [22, 23]. For this purpose, methacrylic acid (1.5 g, 0.017 mol) added to the flask added was dissolved in pyridine (1 mL). A solution of 2-[(2-naphthyloxy)methyl] oxirane (3.4 g, 0.017 mol) in dry dioxane (30 mL) was slowly added to the flask over 1 h. Hydroquinone was added to prevent polymerization of the monomer to the reaction mixture, and then was stirred for 18 h at 85 °C. The formed monomer was dissolved in chloroform and washed three times with water. The 2-hydroxy-3-(1-naphthyloxy)propyl methacrylate (NOPMA) was obtained as a pure product. The structure of monomer is illustrated in Scheme 2.

The structure of 2-hydroxy-3-(1-naphthyloxy) propyl methacrylate (NOPMA)
FT-IR (KBr): 3453 cm−1 (O–H stretch); 1738 cm−1 (ester C=O stretch); 1638 cm−1 (C=C in the vinyl group) and 1600 cm−1 (C=C stretch on aromatic ring).
1H-NMR (CDCI3, δ ppm): 8.27 (1H, H12), 7.84 (1H, H9), 7.6–7.4 (4H, H7, H8, H10, H11) 6.8 (1H, H6), 6.2 and 5.6 (2H, H2), 4.1–4.6 (5H, H3, H4, H5), 2.1 (3H, H1).
13C-NMR (CDCI3, δ ppm): 18.3 (C1), 65.8 (C5), 68.8 (C6), 69.0 (C7), 104.8 (C9), 120.9 (C11), 121.8 (C15), 125.4 (C2), 125.7 (C10), 126.3 (C14), 126.5 (C13), 127.5 (C12), 134.55 (C17), 135.90 (C3), 154.0 (C8), 162.2 (C4).
Free radical polymerization of NOPMA
2-Hydroxy-3-(1-naphthyloxy)propyl methacrylate (NOPMA) was washed once with 5 % water solution of natrium hydroxide and then washed four times with pure water and dried in vacuo before polymerization. The NOPMA was polymerized at 70 °C in the presence of 1,4-dioxane using AIBN as initiator. The formed polymer was poured drop by drop within n-hexane. The polymer was dissolved in dichloromethane and reprecipitated in n-hexane and then dried at 40 °C for 24 h under vacuum.
Results and discussion
Characterization
Poly(NOPMA) having various functional side groups such as hydroxyl, carbonyl, ether and aryl was synthesized by free radical polymerization method at 70 °C in the presence of 1,4-dioxane using AIBN as initiator. The 1H-NMR spectrum of the monomer (Fig. 1a) showed important signals such as olefinic protons at 6.17 and 5.54 ppm, 8.3–6.8 (protons on aromatic ring), 2.1 ppm (CH3), 2.98 ppm (OH), 4.25 ppm (CH2 protons next to ester group), 4.5 ppm (CH proton next to OH group). The 13C-NMR spectrum of the monomer (Fig. 1b) showed important signals at 18.3 ppm (CH3), 65.8 ppm (–OCH2–), 154.0 ppm (ipso carbon on aromatic ring), 124.8–128.0 ppm (CH on aromatic ring), and 135.9 ppm C=CCH3), 166.0 ppm (C=O). The IR spectrum of poly(NOPMA) showed the most characteristic bands at 3397 cm−1 (O–H stretch), 1727 cm−1 (–C=O in ester), 1243 cm−1 [(aryl)C–O–]. The 13C-NMR spectrum of the polymer monitored the most characteristic signals at 177 ppm (C=O), 154 ppm (ipso carbon on aromatic ring), 44 ppm (quaternary carbon on the main backbone). The 1H-NMR spectrum contains important peaks at 8.6–7.0 ppm for the protons on the naphthyl group and 4.45 ppm ((–COOCH 2 CH(OH)). The number average-molecular weight of poly(NOPMA) prepared by free radical polymerization method is 7600 g/mol and its polydispersity is M w/M n = 1.61. A DSC thermogram for poly(NOPMA) was generated using a DSC-50 (at heating rate of 20 °C/min). The T g curve of poly(NOPMA) is displayed in Fig. 2. The T g values for poly(2-hydroxyethyl methacrylate) and poly(2-hydroxypropyl methacrylate), poly(phthalimido-2-hydroxypropyl methacrylate) [poly(PHPMA)] [24] bearing hydroxyl side group have been recorded as 100, 95 and 135 °C in the literature, respectively. The T g of poly(NOPMA) prepared in this study is 110 °C. Although the average-molecular weight of poly(NOPMA) is low, its T g is relatively high. The T g of the polymer reflects the movement ability of the chain or side group.

a 1H NMR and b 13C-NMR spectra of monomer (NOPMA)

DSC curve of poly(NOPMA)
Thermogravimetric study
The thermal decomposition temperature of poly(NOPMA) was determined by thermogravimetric analysis. The results of poly(2-hydroxypropyl methacrylate) (HPMA) and poly(2-hydroxyethyl methacrylate) (HEMA) bearing hydroxy side group are given in Table 1 as compared with that of poly(NOPMA) [25, 26]. The thermogram of poly(NOPMA) clearly indicates that it undergoes a decomposition in two stages. The first decomposition stage of the poly(NOPMA) was at 187 °C with a weight loss of 37 %, and the other is at 310 °C with a weight loss of 46 %. Although the initial decomposition temperature of poly(NOPMA) was lower than those of poly(HEMA) and poly(HPMA) its thermal stability at progressive temperatures was observed to be lower. This phenomenon means that the decomposition temperature of poly(NOPMA) starts (formation of α-naphthol) by side group elimination without breaking the chain in the structure of the polymer. The TG thermograms of poly(NOPMA) at different heating rates are displayed in Fig. 3. The E a (activation energy) for the thermal decomposition of poly(NOPMA) was determined by Flynn–Wall–Ozawa method, which is widely used. The degradation occurred in two stages up to a weight loss of 40 %. If a series of experiments are run at different heating rates, ΣE a can be obtained from the slope of the linear plot of log(heating rate) [logβ] vs. 1/T. For a study of the kinetics in the thermal degradation of polymers, TG (thermogravimetry) or ITG (isothermal thermogravimetry) at different heating rates can be selected [27]. For the thermal degradation of polymers, in which depolymerization is competing with cross-linking or cyclization because of the side groups, TG studies at different heating rates are much more suitable than ITG (isothermal thermogravimetry) studies for the research of thermal degradation kinetics. According to the Flynn–Wall–Ozawa method [28], the apparent thermal decomposition E a, can be defined from the TGA thermogram values. The TGA measurements of poly(NOPMA) were performed in the scanning state, from temperature range from 25 to 500 °C, under a nitrogen flow (10 mL/min), at different heating rates (5.0, 10.0, 20.0, 30.0 and 40.0 °C/min). The TGA curves were recorded at the end of each measurement and the weight losses were then transferred to a computer at diverse temperatures.

TGA curves of poly(NOPMA) measured at different heating rates under argon flow in 10 ml/min
The plot logβ vs. 1/T obtained from a series of experiments performed at several heating rates, should be a straight line whose slope allows evaluation of E a:
where R is a constant and β is the heating rate (°C/min). According to Eq. (1) above mentioned, the E a of degradation can be obtained from the slope of the linear relationship between logβ and the reciprocal of the temperature, as given in Fig. 4a, b; it was found that the activation energy for weight loss to 30 % was decreased as function of decomposition: hence, the E a values decreased from 207.60 to 113.78 kJ/mol for decomposition to 30 %. The average activation energies determined by the Flynn–Wall–Ozawa method for weight loss to 0–30 and 40–80 % were E a = 148.35 and 127.6 kJ/mol, respectively. It is reported that activation energies (E a) calculated from TGA curves are based on formation of all products and not on the individual products formed during degradation. The activation energies calculated for the thermal degradation of poly(NOPMA) are summarized in Table 2.

Log(β) vs temperature plots for poly(NOPMA)
Changes in FT-IR spectra during degradation of poly(NOPMA)
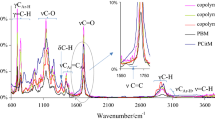
The FT-IR spectra of poly(NOPMA) partially degraded at different temperatures are shown in Fig. 5 by comparing with structural changes. For this purpose, a poly(NOPMA) film prepared on salt plates was heated to 240, 260, 320, 340, 360, 380 and 420 °C under argon atmosphere at a heating rate of 10 °C/min. The FT-IR spectra for degradation of poly(NOPMA) were recorded after some heating stages and illustrated in Fig. 5. As displayed in Fig. 5, the FT-IR spectrum of poly(NOPMA) heated to 240 °C was similar to same that of the original polymer. While the O–H band at 3418 cm−1 is decreased to 320 °C, the anhydride peaks (1801, 1759 cm−1) started to appearing and became maximum at 360 °C, and at 380 °C the anhydride band almost disappeared. The elimination of H2O and the formation of anhydride band during decomposition of poly(NOPMA) showed that depolymerization at any temperature to the corresponding monomer was not prominent in the thermal degradation of poly(NOPMA). The FT-IR spectrum of poly(NOPMA) heated to 260 °C showed a new shoulder which characterizes the carbonyl absorption at 1801 cm−1. This band became a maximum in the FT-IR spectrum of the polymer heated at 360 °C. With increasing temperature for degradation of the polymer, the intensities of bands at 1801, 1759 and 1016 cm−1 in the infrared spectra were observed. The residue after degradation at 240 °C is still soluble in organic solvents such as CHCl3, CH2Cl2, and DMSO. These spectroscopic changes suggested that considerable disappearance of the ester structures in the polymers accompanied their degradation, while cyclic anhydride structure was formed. The formation of a six-membered cyclic anhydride has been also observed for the thermal degradation of many poly(methacrylate ester)s [29–31].

FT-IR spectra of residual poly(NOPMA) partially degraded under argon atmosphere on NaCl windows
Product identification studies
In a degradation ampoule having a condenser, product collection ring and a rotary pump, about 450 mg poly(NOPMA) was heated at a heating rate of 10 °C/min from room temperature to 320 °C and then its residue was again heated to 500 °C. The two CRFs, one at 320 °C (CRF1) and the other were collected at 500 °C (CRF2). The mixture of products (CRF1 and CRF2) produced from the degradation of poly(NOPMA) was characterized using FT-IR, 1H-NMR, GC–MS techniques. In addition, the condensable gas and evaporating liquid generated from ambient temperature to 320 °C were collected and characterized by FT-IR. The FT-IR spectra of fraction trapped at −196 °C (Fig. 6a), CRF1 (Fig. 6b), and CRF2 (Fig. 6c) are compared with each other in Fig. 6. As seen in Fig. 6a, the FT-IR spectrum of the fraction trapped at liquid nitrogen (−196 °C) during degradation of poly(NOPMA) heated to 320 °C contains characteristic bands 3450 cm−1 (O–H stretch), 1670 cm−1 (O–H bending vibrations). These bands showed that poly(NOPMA) heated to 320 °C considerably eliminate their water. The FT-IR spectrum (Fig. 6a) of volatile fraction trapped at −196 °C confirmed the formation of H2O. The absence of C=O band between 1700 and 1800 cm−1 for CRF1 illustrated in Fig. 6b is an important evidence for formation of α-naphthol by side group elimination during degradation of poly(NOPMA). The formation mechanism of the water and α-naphthol forming about decomposition temperature during the degradation of poly(NOPMA) is shown in Scheme 3.

FT-IR spectra of a fraction trapped at −196 °C during degradation of poly(NOPMA) heated to 320 °C, b CRF1, c CRF2

The formation mechanism of the water and α-naphthol beginning about decomposition temperature during the degradation of poly(NOPMA)
The FT-IR spectrum of the CRF2 obtained from degradation of poly(NOPMA) heated between 320 and 500 °C showed the important bands such as OH, C=O, =C–H, C–O. The most characteristic C=O bands are designed at 1801, 1787, 1762, 1723 and 1698 cm−1. The CRF2 (320–500 °C) from decomposition of poly(NOPMA) was investigated using FT-IR and GC–MS. The FT-IR spectrum of CRF2 showed characteristic bands (aliphatic C–H stretching), 1802, 1764 and 1020 cm−1 (anhydride structures) as different from that of CRF1. For the degradation of poly(NOPMA), formation of the cyclic anhydride structures begins at about 320 °C (Scheme 4).

The formation mechanism of the cyclic anhydride, epoxide and diol structures that begins at 320 °C during the degradation of poly(NOPMA)
The 1H NMR spectra of α-naphthol and CRF1 produced during degradation of poly(NOPMA) heated to 320 °C (CDCl3) are shown in Fig. 7a as compared with each other. The 1H-NMR spectrum (Fig. 7b) of CRF1 contains signals at δ 6.93 ppm (β-hydrogene), 7.35–7.65 ppm (c, d, e hydrogenes on ring), 8.1 and 8.5 ppm (f and g hydrogenes on ring, respectively). 1H-NMR spectrum illustrated for CRF1 in Fig. 7b is very similar to that of α-naphthol. So, these signals are an important evidence for formation of α-naphthol during degradation of poly(NOPMA) heated to 320 °C. GC–MS investigation of CRF1 showed that α-naphthol (retention time, r.t.: 61.01 min, m/e: 144) formed as a major product, and allyl 1-naphthyl ether (m/e: 184), 1-(1-naphthyloxy)acetone (m/e: 200), 2-[(1-naphthyloxy)methyl]oxirane (m/e: 200) as minor products, and no monomer was observed. In the case of CRF2 indicated that 3,3,5-trimethyldihydro-2H-pyran-2,6(3H)-dione (m/e:168), 2-[(1-naphthyloxy)methyl]oxirane (m/e: 200), 3-(1-naphthyloxy)propane-1,2-diol (m/e: 218), methacrylic acid (m/e: 86), naphthalene (m/e: 128). So, all the FT-IR, 1H-NMR data confirmed that depolymerization is not during degradation of poly(NOPMA) to 320 °C and then to 500 °C. So, the main product in degradation of poly(NOPMA) is α-naphthol. This may be due to the absence of β-hydrogen atom adjacent to the ester oxygen or CH next to OH in NOPMA moieties. The formation mechanisms of some products during degradation are suggested in Schemes 5 and 6.

1H NMR spectra of a α-naphthol and b CRF1 during degradation of poly(NOPMA) heated to 320 °C (CDCl3)

The formation mechanism of the epoxide and diol structures that begins at about 300 °C during the degradation of poly(NOPMA)

The formation mechanism of the naphthalene derivatives during the degradation of poly(NOPMA)
The dielectric properties
The surfaces of the poly(NOPMA) were painted with silver paste to make better contact between two the electrodes. It was dried at 45 °C under vacuum and then kept in a dried environment, for the elimination of any moisture effects. The capacitance (C p) and the loss factor, the AC conductivity of the poly(NOPMA) were investigated at frequency ranging from 50 Hz to 2 MHz, and for different temperatures at 1 kHz. The dielectric constant (ε′) is a measure of the influence of a particular dielectric on the capacitance of a condenser. The dielectric constant value says how much bigger or smaller the area becomes. The dielectric constant was calculated from C p (capacitance) using Eq. (2):
where C is the capacitance; d, the thickness; A, surface area of the sample and ε o, is 8.85 × 10−12 F/m. The loss factor, ε″ = ε′·tanδ [32], d is thickness of the polymer sample, and A is surface area of the sample. Figure 8a, b shows dependence of the dielectric constant and dielectric loss on frequency (50 Hz–2 kHz) for poly(NOPMA). The dielectric constant and the loss factor for poly(NOPMA) are ε′ = 5.56 and ε″ = 0.0956 at 1 kHz, respectively. The dielectric constant was decreased with increasing frequency. This trend in dielectric constant has been reported in many studies observed at frequency ranges of a few MHz [33, 34]. A somewhat more rapid decrease in ε′ can be seen over the frequency range 1–300 kHz. The hydroxyl (OH) and C=O groups of poly(NOPMA) are comprehensively responsible for the permittivity. The dielectric permittivity of a material, ε′, represents the amount of dipole alignment and the loss factor, ε″, measures the energy required to align dipoles or move ions [35]. Also, it arises due to the polarization of molecules and the permittivity increases with polarization. This may be due to the tendency of induced dipoles in the macromolecules to orient themselves in the direction of the applied field in this frequency range. The dielectric constant calculated for poly(NOPMA) (ε′ = 5.56) is much better than that of most of commercial polymers such as polystyrene (PS) (ε′ = 2.2), poly(methylmethacrylate (PMMA) (ε′ = 3.5), polytetrafluoroethylene (PTFE) (ε′ = 2.1). The conductivity depending on frequency and temperature is due to hopping of the charge carriers in the localized states and a increase in the dielectric constant with temperature indicates that thermal energy converts the bound charges to the charge carriers [36]. Figure 9 shows the plot of lnσac versus 103/T from which activation energy was calculated using the Arrhenius equation. The temperature dependence of the AC conductivity can be written as follows [37]:
where σo is the pre-exponential factor, k is the Boltzmann constant and ∆E is the apparent activation energy. Figure 9 shows temperature (T) dependence of AC conductivity σac measured at different frequencies in the temperature range of 303–473 K. Figure 8 shows the variation of lnσac versus 103/T from which the activation energy was found to be 0.837 eV using Eq. (3).

Plots of a dielectric constant (ε′) and b dielectric loss (ε″) versus frequency

Plot of ln of ac conductivity (lnσac) versus 103/T
Conclusion
NOPMA was polymerized by free radical polymerization method in the presence of AIBN at 60 °C. The glass transition temperature of poly(NOPMA) was measured as 110 °C by differential scanning calorimetry (DSC). Poly(NOPMA) was heated from room temperature to 320 °C, and then 500 °C, respectively. The cold ring fractions (CRFs) were collected at two different temperatures, initially fraction-1 (CRF1) is from room temperature to 320 °C and the other fraction-2 (CRF2) is from 320 to 500 °C. All fractions were investigated by FT-IR, 1H and 13C-NMR spectroscopic techniques and the cold ring fractions (CRFs) were also characterized by GC–MS. For the degradation of polymer, the major products of CRFs are α-naphthol. The GC–MS, IR and NMR data showed that depolymerization below 320 °C to the corresponding monomer was not prominent in the thermal degradation of poly(NOPMA). The mode of thermal degradation including formation of the major products was suggested. The dielectric permittivity (ε′ = 5.56), the loss factor (ε″ = 0.0956) and conductivity (σac) were measured using an impedance analyzer in the frequency range of 50 Hz to 20 kHz. Both dielectric loss and dielectric constant seem to decrease with increase in the applied field frequency and increase on increasing temperature.
References
Lewis E, Manring LE (1988) Thermal degradation of saturated poly(methyl methacrylate). Macromolecules 2(2):528–530
Hirata T, Kashiwagi T, Brown JE (1985) Thermal and oxidative degradation of poly(methyl methacrylate): weight loss. Macromolecules 18(7):1410–1418
Holland BJ, Hay JN (2001) The kinetics and mechanisms of the thermal degradation of poly(methyl methacrylate) studied by thermal analysis-Fourier transform infrared spectroscopy. Polymer 42:4825–4835
Coşkun M, Demirelli K, Erol I, Ahmedzade M (1998) Thermal degradation of poly[2-(3-aryl-3- methylcyclobutyl)-2-hydroxyethyl methacrylate]. Polym Degrad Stab 61(3):493–497
Cauich-Rodríguez JM, Vázquez-Torres JV, Vázquez-Torres H, Licea-Claveríe A (2006) TGA/FTIR study on thermal degradation of polymethacrylates containing carboxylic groups. Polym Degrad Stab 91:3312–3321
McNeill IC (1997) Thermal degradation mechanisms of some addition polymers and copolymers. J Anal Appl Pyrol 40–41:21–41
McNeill IC, Mahmood T (1994) Thermal degradation studies of methacrylonitrile polymers and copolymers 1. Polymethacrylonitrile prepared using normal free-radical initiators. Polym Degrad Stab 45:285–291
Zulfigar S, Zulfiqar M, Kausar T, McNeill IC (1987) Thermal degradation of phenyl methacrylate methyl methacrylate copolymers. Polym Degrad Stab 17:327–339
Czech Z, Kowalczyk A, Kabatc J, Ṧwiderska J (2013) Thermal stability of poly(2-ethylhexyl acrylates) used as plasticizers for medical application. Polym Bull 70:1911–1918
Peniche C, Zaldivar D, Bulay A, Roman JS (1993) Study of the thermal degradation of poly(furfuryl methacrylate) by thermogravimetry. Polym Degrad Stab 40(3):287–295
Manring LE (1991) Thermal-degradation of poly(methyl methacrylate). 4. Random side-group scission. Macromolecules 24(11):3304–3309
Coskun M, Demirelli K (1996) Thermal degradation of sulphonylated polystyrene. Polym Degrad Stab 51(2):173–178
Tsuchiya Y, Sumi K (1969) Thermal decomposition products of poly(vinyl alcohol). J Polym Sci A 7:3151–3158
McNeill IC, Mahmood T (1994) Thermal degradation studies of methacrylonitrile polymers and copolymers. 1. Polymethacrylonitrile prepared using normal free-radical initiators. Polym Degrad Stab 45(3):285–291
Demirelli K, Kaya E, Coşkun M, Bağci E (2013) Thermal degradation of two different polymers bearing amide pendant groups prepared by ATRP method. J Therm Anal Calorim 114(2):917–926
Jobish J, Charoen N, Praveen P (2012) Dielectric properties and AC conductivity studies of novel NR/PVA full-interpenetrating polymer networks. J Non-Cryst Solids 358(8):1113–1119
Kamath A, Devendrappa H (2015) Concentration-dependent ionic conductivity and dielectric relaxation of methyl blue-dyed polyethylene oxide films. Polym Bull 72:2705–2724
Figueiro SD, Macedo AAM, Melo MRS, Freitas ALP, Moreira RA, de Oliveira RS, Goes JC, Sombra ASB (2006) On the dielectric behaviour of collagen-algal sulfated polysaccharide blends: effect of glutaraldehyde crosslinking. Biophys Chem 120(2):154–159
Beers KL, Boo Sohyun, Gaynor Scott G, Krzysztof M (1999) Atom transfer radical polymerization of 2-hydroxyethyl methacrylate. Macromolecules 32:5772–5776
Das N (2013) Preparation methods and properties of hydrogel: a review. Int J Pharm Pharm Sci 5(3):112–117
Eswaramma S, Krishna Rao KSV, Madhusudana Rao K (2016) Diffusion and controlled release characteristics of pH-sensitive poly(2-(dimethyl amino)ethyl methacrylate-co-2-hydroxyethylacrylate) hydrogels. Int J Polym Mater Polym Biomater 65(3):134–142
Madec PJ, Marechal E (1983) Study of epoxy-carboxy reaction on models in solvents of low or medium dielectric constants. Macromol Chem 184:323–334
Boutevin B, Pietrasanta Y, Parisi JP (1987) Synthesis of photocrosslinkable telomers-8-grafting of acrylic acid onto glycidyl methacrylate telomers with thiols. Makromol Chem 188:1621–1629
Rao Y, Wong CP (2004) Material characterization of a high-dielectric-constant polymer-ceramic composite for embedded capacitor for RF applications. J Appl Polym Sci 92(4):2228–2231
Coskun MF, Demirelli K, Güzel D, Coskun M (2002) Free-Radical Copolymerization of 3-phthalimido-2-hydroxypropyl methacrylate with styrene: the determination of monomer reactivity ratios and thermal analysis studies. J Polym Sci Part A Polym Chem 40:650–658
Wendlandt WW (1986) Thermal analysis. Anal Chem 58(5):R1–R6 (review)
Flynn JH, Wall LA, Quick A (1966) Direct method for the determination of activation energy from thermogravimetric data. Polym Lett 4:323–328
Demirelli K, Coskun MF, Kaya E, Coskun M (2002) Investigation of the thermal decomposition of poly(2-hydroxypropyl methacrylate). Polym Degrad Stab 78(2):333–339
Demirelli K, Coşkun M, Kaya E (2001) A detailed study of thermal degradation of poly(2-hydroxyethyl methacrylate). Polym Degrad Stab 72(1):75–80
Grant DH, Grassie N (1960) The thermal decomposition of polymethacrylic acid. Polymer 1:125–134
Coskun MF, Demirelli K, Coskun M, Doğru M (2001) Thermal decomposition of poly[3-Pht halimido-2-hydroxypropyl methacrylate]. Polym Degrad Stab 76(1):145–154
Coskun M, Barım G, Demirelli K (2006) Thermal stabilities of poly(N-acryloyl-N′-methylpiperazine), its blends with poly(methyl methacrylate), and poly(N-acryloyl-N′-methylpiperazine-co-methyl methacrylate). J Macromol Sci Pure Appl Chem A 43(1):83–93
Seven P, Coskun M, Demirelli K (2008) Synthesis and characterization of two-armed graft copolymers prepared with acrylate and methacrylate using atom transfer radical polymerization. React Funct Polym 68(5):922–930
Stefanec D, Krajnc P (2005) 4-vinylbenzyl chloride based porous spherical polymer support derived from water-in-oil-water emulsion. React Funct Polym 65(1–2):37–45
Mohamed K, Moussy F, Harmon JP (2006) Dielectric analyses of a series of poly(2-hydroxyethyl methacrylate-co-2,3-dihydroxypropyl methacrylate) copolymers. Polymer 47:3856–3865
Ku CC, Liepins R (1987) Electrical properties of polymers. Hanser, Munich
Raja V, Sharma AK, Rao VVR (2004) Impedance spectroscopic and dielectric analysis of PMMA-co-P4VPNO polymer films. Mater Lett 58(26):3242–3247
Acknowledgments
The authors thank the Fırat University Research Fund for financial support to this Project (FUBAP FF.12.13).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Biryan, F., Demirelli, K., Torğut, G. et al. Synthesis, thermal degradation and dielectric properties of poly[2-hydroxy,3-(1-naphthyloxy)propyl methacrylate]. Polym. Bull. 74, 583–602 (2017). https://doi.org/10.1007/s00289-016-1731-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-016-1731-2