
Abstract
Faeces from cows with different milk yield and non-lactating cows were applied to soil to investigate whether soil N2O efflux is related to feeding-induced differences in faecal microbiome and abundances of nitrification and denitrification genes. Fungal 18S-rRNA gene abundance was the highest in the faeces of the non-lactating group. The 18S-rRNA/ergosterol ratio showed a strong positive correlation with the 18S-rRNA/fungal glucosamine ratio. The milk-yield groups did not affect the gene abundances of bacterial 16S rRNA, AOB amoA, nirS and nosZ clade I, or the 16S-rRNA/muramic acid (MurN) ratio. In contrast, nirK gene abundance was generally the lowest in the high-yield group. The 16S-rRNA/MurN ratio showed a strong positive correlation with the 16S-rRNA/bacterial PLFA ratio. Cow faeces application to soil increased microbial biomass and ergosterol contents as well as the gene abundances of 18S-rRNA and nosZ clade I, compared with the non-amended control soil. Cumulative ΣCO2 efflux was roughly twice as high as the control, without differences between the faeces treatments. Cumulative ΣN2O efflux showed a 16-fold increase after applying high-yield cow faeces to soil, which was above the sevenfold increase in the non-lactating faeces treatment. The ΣN2O efflux from soil was positively related to faecal MurN and total PLFA concentration but also to soil nirK at day 14. The comparison of genome markers with cell wall (glucosamine) and cell membrane components (ergosterol) showed that the fungal cells were much larger in energy-rich faeces than in C-limited soil. A cow diet reduced in protein decreased the ΣN2O efflux from faeces amended soil.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Permanent grassland ecosystems cannot be used for human food production without ruminants such as dairy cows, which act as plant biomass fermenters in many parts of the world. This is generally true for organic farming systems, where dairy farming is an important means for closing nutrient cycles and maintaining soil fertility, not only in grassland but also in arable land use systems (Mäder et al. 2002). Faeces are the cow product that enters soil as composted farmyard manure (Heinze et al. 2010) or as solid dung patches (Wachendorf and Joergensen 2011; Rashid et al. 2017). Feedstuff ration composition greatly affects the quality of faecal fertilisers for plant growth (Siegfried et al., 2013; Jost et al. 2013a; Schick et al. 2019). The threat of global warming has increasingly directed research interest toward the ruminants as a source of greenhouse trace gases, i.e., CH4 and N2O (Soussana et al. 2010; Voglmeier et al. 2019, 2020). Significant amounts of CH4 (Ali et al. 2019; Voglmeier et al. 2019, 2020; Goopy et al. 2020) and N2O are emitted from faecal dung patches after excretion (Flessa et al. 1996; Pelster et al. 2016).
The proportions of easily and hardly fermentable and digestible protein and carbohydrate fractions in feeding rations of dairy cows have been shown to modify the microbial faeces composition (van Vliet et al. 2007; McGovern et al. 2017). Less dietary protein and more fibre increased the contribution of fungi (Jost et al. 2013b; Meyer et al. 2019) and Gram-positive (Gram +) bacteria to the faecal microbiome of heifers (Jost et al. 2013b) and non-lactating cows (Meyer et al. 2019) in comparison with faeces of high-yield cows. Jost et al. (2013b) investigated the effects of feedstuff composition on the faecal microbial biomass under the fully controlled conditions of an experimental farm. Meyer et al. (2019) studied the interrelationships between chemical and microbial composition of faeces, obtained from a higher number of cows under the practical conditions of four German farms, all remote from each other. In this study, the faecal microbiome was analysed by fumigation extraction, cell wall components (amino sugars) and cell membrane components (ergosterol, archaeol and phospholipid-fatty acids (PLFA)).
In the current study, the identical faecal samples investigated by Meyer et al. (2019) were further analysed for the abundance of fungal 18S-rRNA and bacterial 16S-rRNA genes to corroborate the contribution of fungi to the faecal microbiome, using polymerase chain reaction (PCR). Nitrification, mediated by ammonia-oxidizing bacteria (AOB) and ammonia-oxidizing archaea (AOA), is an important prerequisite for N2O production and denitrification. Functional genes involved in denitrification processes are nirK (Cu-nitrite reductase) and nirS (cytochrome cd1-nitrite reductase), as well as nosZ clade I and nosZ clade II (nitrous oxide reductases clades I and II). Especially the abundance of nirK was related to N2O efflux in a variety of soils (Jones et al. 2014). In the current study, the abundance of amoA (ammonia monooxygenase subunit A) and denitrification genes was analysed not only in pure faeces but also in faeces-fertilised soil, as both microbiomes might interact in a yet unknown way (Kindler et al. 2006; Gattinger et al. 2007; Jost et al. 2013a).
In previous studies, dietary changes mainly affected the contribution of fungi and Gram + bacteria to faecal microbial biomass (Jost et al. 2013b; Meyer et al. 2019). In contrast, nitrification is dominated by autotrophic Gram-negative (Gram-) AOB activity (Xia et al. 2011), although the soil nitrifying community is usually dominated by AOA amoA gene copies (Leininger et al. 2006; Beule et al. 2019, 2020). However, less dietary protein and more fibre reduced the N2O efflux from soil, suggesting that changes in diet may not affect total gut bacteria but specifically AOA and AOB. These conflicting results might be explained by abundances of genes involved in nitrification and denitrification, giving potential indications of their relative activities.
The expanded analysis of faeces from high and low yielding, as well as non-lactating cows, makes it possible to investigate the following three hypotheses: (1) the higher N2O efflux from high-yield cows is reflected by higher abundance of denitrifying genes; (2) the comparison of genome markers with cell wall and cell membrane components in faeces and soil gives information on the cell size; (3) cells of fungi and copiotrophic faecal bacteria are larger in energy rich faeces than in C-limited soil, where the microorganisms are starving.
Materials and methods
Sampling and preparation of faeces
Three feeding regime treatments were established at four different private farms according to the milk yield level of dairy cows (Bos primigenius taurus, var. German Holstein): high-yield, low-yield and non-lactating cows. Five cows were randomly selected per treatment on each of the four farms, i.e., a total of 60 faecal samples were taken for the analysis of faeces (Meyer et al. 2019). At Wolkramshausen (Thuringia) and Oederan (Saxonia), the cows were fed with a total mixed ration based on maize silage. The diets of low- and high-yield cows were supplemented with concentrate, individually allocated by transponder control. At Aurich (Lower Saxony) and Rotenburg (Schleswig–Holstein), cows were fed with a mixed ration based on grass and maize silage in the stable in addition to daily grazing during the vegetation period. Low-yield and high-yield cows received different concentrates, individually allocated by transponder control. On each farm, faecal samples were taken rectally, homogenised, frozen in liquid N2 and stored at − 18 °C to be used for the analysis of microbial biomass (MBC).
One subsample of the faeces was dried at 60 °C, ground and used for dry mass (DM) determination and chemical analyses via near-infrared spectroscopy (Althaus et al. 2013). Another subsample was freeze-dried at -32 °C (Christ Alpha l–5) for ergosterol, PLFA and amino sugar analyses (Meyer et al. 2019), who described the methods for determination of chemical and biological faeces composition (Table 1) in detail.
Design of the incubation experiment
Soil was sampled at 0–20 cm depth from the site ‘Saurasen’ (51° 22′ 34.5″ N, 9° 53′ 53.0″ E), which is in Neu-Eichenberg near Witzenhausen (Northern Hessia, Germany). The site is located at 280 m above sea level, with a mean annual precipitation of 625 mm and a mean temperature of 6.5 °C. Developed from eroded loess overlying clayey sandstone, the soil is classified as Stagnic Luvisol (IUSS 2015). Soil organic C and total N contents were 12.8 mg g−1 and 1.19 mg g−1 soil, respectively. The soil had a water holding capacity of 57.4% and a pH-H2O of 6.6. After sampling, soil was sieved (< 2 mm) and stored in polyethylene bags at room temperature until the experiment started.
The incubation experiment had the following four treatments with five replicates: (1) non-amended control soil, (2) soil + faeces from high milk-yield level cows, (3) soil + faeces from low milk-yield and (4) soil + faeces from non-lactating dairy cows. Faeces treatments comprised individual faecal samples from five cows. The experiment was carried out in 1-l incubation vessels, containing four 50-ml beakers with 70 g soil each at 50% water holding capacity. The bottom of the 1-l vessel was covered with 20 ml water to prevent drying. Freshly thawed faeces were mixed into the soil at a rate of 170 kg N ha−1. Then, the vessels were incubated for 82 days in the dark at 22 °C. One beaker was removed after 14, 28, 56 and 82 days from the 1-l incubation vessel and analysed for inorganic N, ergosterol, microbial biomass and (de)nitrifying community. One additional beaker was analysed immediately after the incubation started at day 0. CO2 and N2O emissions were measured at day 0, 1, 2, 7, 14, 28, 56 and 82. A 10-ml gas sample was taken out of the jar through a 3-layer silicone septum (Hamilton, Nevada, USA) with a vacutainer. The samples were analysed immediately using a gas chromatograph GC-14B (Shimadzu, Kyoto, Japan), attached to an automated sample changer (Loftfields Analytical Solutions, Neu Eichenberg, Germany).
Soil microbial biomass indices
Soil microbial biomass C (MBC) and N (MBN) were estimated by fumigation extraction (Brookes et al. 1985; Vance et al. 1987). Fumigated (24 h with ethanol-free CHCl3 at 25 °C) and non-fumigated soil samples equivalent to 10 g dry mass were extracted with 40 ml 0.5 M K2SO4 by 30 min horizontal shaking at 200 rev min−1 and filtered. Organic C and total N in the extracts were determined, using a Multi N/C 2100S analyser (Jena Analytik, Germany). MBC was calculated as EC/kEC, where EC = (organic C extracted from fumigated soil) – (organic C extracted from non-fumigated soil) and kEC = 0.45 (Wu et al. 1990). MBN was calculated as EN/kEN, where EN = (total N extracted from fumigated soil) – (total N extracted from non-fumigated soil) and kEN = 0.54 (Brookes et al. 1985).
Ergosterol was extracted and measured according to Djajakirana et al. (1996). Soil samples of 2 g were extracted with 100 ml ethanol for 30 min by horizontal shaking at 200 rev min−1 and measured by HPLC analysis.
Fungal 18S-rRNA, bacterial 16S-rRNA, nitrification and denitrification genes
All samples were freeze-dried for 72 h and finely ground using a swing mill (MM 400, Retsch, Haan, Germany) at 25 Hz for 1 min. DNA was extracted from 50 mg of ground faeces and soil material according to a cetyl trimethyl ammonium bromide (CTAB) based protocol (Brandfass and Karlovsky 2008), modified according to Beule et al. (2017). DNA quality was assessed by electrophoresis in 0.8% (v/w) agarose gels. Prior to real-time PCR, samples were tested for the presence of inhibitors of amplification as described by Guerra et al. (2020). Faecal samples of the four farms were diluted 1:50 (v/v); soil samples of the incubation experiment were diluted 1:20 (v/v) with double-distilled H2O. Fungal 18S-rRNA, bacterial 16S-rRNA and ammonia-oxidizing genes for nitrification, i.e., archaeal (AOA) and bacterial (AOB) amoA genes, as well as genes for denitrification, i.e., nirK, nirS, nosZ clade I and nosZ clade II genes, were quantified as described by Beule et al. (2019). Real-time PCRs were performed in 4-µL reaction volumes in a CFX384 Thermocycler (BioRad, Rüdigheim, Germany). Composition of reaction volumes and thermocycling programs is also described in detail by Beule et al. (2019).
Statistical analysis
The results presented in table and figures are expressed on an oven-dry basis. Normality was tested by the Shapiro–Wilk test and equal variance by the Levene test. If data did not fulfil these two requirements, they were ln-transformed. In soil samples, this was true for MBC, K2SO4 extractable C and abundances of 18S-rRNA, 16S-rRNA, nirK, nirS, nosZ clades I and II, as well as the ratio 18S-rRNA/ergosterol. The significance of treatment effects was tested by one-way ANOVA, using sampling time as repeated measure, followed by the Holm-Sidak post-hoc test.
Multiple linear relationships were calculated between chemical and biological faecal properties as independent variables, selected by stepwise forward regression analysis, and 18S-rRNA18S-rRNA/ergosterol, 16S-rRNA, nirK, nosZ clade I, 16S-rRNA/MurN as dependent variables for faeces from all four farms, as well as soil ΣCO2 efflux and soil ΣN2O efflux from the incubation experiment with faeces from Oederan. All regression models were tested for normality (Shapiro–Wilk), constancy of variance, the absence of correlation between the residuals (Durbin–Watson statistics) and the absence of multi-collinearity, calculating the variance inflation factor (VIF). Variables were removed from the model if the VIF value exceeded 4.0. All statistical analyses were performed using SigmaPlot 13.0 (Systat Inc., San José, USA).
Results
Composition of cow faeces
Faecal pH was the highest in the non-lactating group and lowest in the high-yield group (Table 1). However, the differences between the three groups were not always significant, leading to significant farm × milk yield interactions. Total N concentration was in most cases significantly the lowest in the faeces of the non-lactating group, except Rotenburg, where the three milk yield groups did not differ. Faecal lignin concentration was again always highest in the non-lactating group but did not significantly differ at Rotenburg and Wolkramshausen.
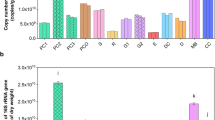
Fungal 18S-rRNA gene abundance was always significantly the highest in the faeces of the non-lactating group, with a mean 6.1 × 108 g−1 DM (Table 1), i.e., more than five times higher than the average of the lactating groups. However, the differences in fungal gene abundance between these two groups varied in a farm-specific way. Fungal gene abundance was positively related to faecal lignin, cellulose and MBN but negatively to total N concentrations according to multiple linear regression analysis (Table 2). The 18S-rRNA/ergosterol ratio was also always the highest in the faeces of the non-lactating group but showed non-significant differences to the other yield groups at Rotenburg and Wolkramshausen (Table 1). The 18S-rRNA/ergosterol ratio was negatively related to the undigested dietary N (UDN) concentration and the fungal/bacterial PLFA ratio (Table 2). The 18S-rRNA/ergosterol ratio showed a strong positive correlation with the 18S-rRNA/fungal GlcN ratio (Fig. 1a).

taken from Meyer et al. (2019)
a Linear relationships between a the 18S-rRNA/ergosterol and the 18S-rRNA/fungal GlcN ratio (y = − 4.11 × 1.10x, r = 0.93, n = 60, P < 0.01) as well as b 16S-rRNA/bacterial PLFA and the 16S-rRNA/MurN ratios of cow faeces obtained from three milk yield groups taken at four farms (y = 1.84 × 0.82x, r = 0.72, n = 60, P < 0.01); ergosterol, fungal GlcN (glucosamine), bacterial PLFA (phospholipid fatty acids) and MurN (muramic acid) data were
Differences in feed ingredients of dairy cows did not affect the abundances of bacterial 16S rRNA, AOB amoA, nirS and nosZ clade I genes, or the 16S-rRNA/MurN ratio (Table 1). In contrast, the abundance of nirK genes was generally the lowest in the faeces from the high-yield group. In contrast to the milk yield groups, all gene abundances showed strong farm-specific differences in 16S-rRNA, nirK and nosZ clade I genes as well as the 16S-rRNA/MurN ratio. They were significantly the lowest at Rotenburg, but the highest at Wolkramshausen. AOB genes were not detected at these two farms, whereas almost no nirS genes were identified at Wolkramshausen. AOA amoA and nosZ clade II genes were generally not detected in the current cow faeces.
Bacterial gene abundance was positively related to faecal pH as well as UDN and hemicellulose concentrations but negatively to cellulose (Table 2). The 16S-rRNA/MurN ratio was positively related to faecal MBN, WSN and hemicellulose concentrations as well as the Gram + /Gram- PLFA ratio (Table 2). The 16S-rRNA/MurN ratio showed a strong positive correlation with the 16S-rRNA/bacterial PLFA ratio (Fig. 1b). The abundances of nirK and nosZ clade I genes were both positively related to the faecal MBC and ergosterol concentrations as well as the Gram + /Gram- PLFA ratio (Table 2).
Cow faeces effects on microbial biomass and gene abundances in soil
The application of cow faeces to soil increased MBC and ergosterol contents as well as the abundances of 18S-rRNA and nosZ clade I genes in comparison with the non-amended control soil (Table 3). The same was true for MBN (Fig. 2a) and nirK gene abundance (Fig. 2b). The highest ergosterol content and ergosterol/MBC ratio, as well as the highest 18S-rRNA and nosZ clade I gene abundances, occurred in the non-lactating faeces treatment (Table 3). During incubation, the majority of soil microbiome and gene abundance indices significantly increased initially and decreased later, i.e., MBC, MBN (Fig. 2a), ergosterol, 18S-rRNA, AOA, AOB, nosZ clade I (Table 3) and nirK (Fig. 2b), as well as the 18S-rRNA/ergosterol ratio (mean 71 × 107 µg−1). Only highly variable 16S-rRNA (mean 86 × 108) and nirS (mean 6 × 106) gene abundances did not significantly respond to faeces treatment or to incubation time (Table 3).

a MBN (microbial biomass N; probability values: treatment P = 0.05, day P < 0.01, treatment × day P < 0.01) and b nirK gene abundance (probability values: treatment P = 0.05, day P < 0.01, treatment × day P < 0.01) during an 82-day incubation at 22 °C from a non-amended control soil and from soils amended with faeces of high milk yield, low milk yield and non-lactating cows from the farm Oederan; bars show one standard error of mean; different letters on top of a bar indicate a significant difference between incubation days (P < 0.05 Holm-Sidak test)
Cow faeces effects on microbial activity indices
The application of cow faeces increased the CO2 efflux rates during the whole 82-day incubation period (Fig. 3a) and the N2O efflux rates until day 14 (Fig. 3b), although the majority of N2O was produced immediately after application. The cumulative ΣCO2 efflux was roughly twice as high as the control, without differences between the faeces treatments (Fig. 4). The cumulative ΣCO2 efflux from soil was positively related to faecal MBN and negatively to the Gram + /Gram- PLFA ratio (Table 2). In contrast, the cumulative ΣN2O efflux showed a 16-fold increase after applying faeces from the high-yield cows to soil, which was significantly above the sevenfold increase in the treatment with faeces from non-lactating cows (Fig. 4). The ΣN2O efflux from soil was positively related to faecal MurN and total PLFA but also to soil nirK at day 14 (Table 2).

a CO2 production rates and b N2O evolution rates during an 82-day incubation at 22 °C from a non-amended control soil and from soils amended with faeces of high milk yield, low milk yield and non-lactating cows from the farm Oederan; bars show one standard error of mean

Cumulative (Σ) CO2 efflux and ΣN2O efflux at the end of an 82-day incubation at 22 °C from a non-amended control soil and from soils amended with faeces of high milk yield, low milk yield and non-lactating cows from the farm Oederan; bars show one standard error of mean; different letters on top of a bar indicate a significant difference between incubation days (P < 0.05 Holm-Sidak test)
Faeces application to soil led to a marked 50% increase in the K2SO4 extractable C content in comparison with the non-amended control (Fig. 5a). This increase rapidly declined within 14 days (high-yield and non-lactating faeces treatments) or 28 days (low-yield faeces treatment) and reached a level of approximately 10% above the control. In the non-amended control soil, the inorganic N content continuously increased during the incubation (Fig. 5b). The application of cow faeces led to an immediate net-N immobilisation, which increased in the order non-lactating < low-yield < high-yield group during the incubation. At day 28, all yield groups reached a similar net-N immobilisation level, followed by re-mineralisation. This led to a small net-N mobilisation in the high-yield faeces treatment at the end of the incubation, whereas the strongest net N-immobilisation was now observed in the non-lactating faeces treatment.

a 0.5 M K2SO4 extractable C (probability values: treatment P < 0.01, day P < 0.01, treatment × day P = 0.04) and b inorganic N (NO3−-N + NH4+-N) in the non-amended control soil and net N mineralisation rate in soil amended with faeces of high milk yield, low milk yield and non-lactating cows from the farm Oederan during an 82-day incubation at 22 °C; bars show one standard error of mean; different letters on top of a bar indicate a significant difference between incubation days (P < 0.05 Holm-Sidak test)
Discussion
Faeces-induced gas fluxes from soil
The ΣN2O efflux from soil was the microbial property showing the clearest effects of feeding-induced response to cow faeces composition. This means that a diet for cows, reduced in protein and increased in NDF and ADF, will reduce N2O emission after application of their faeces to soil, even for feeding under practical farming conditions. The results are in line with the observations of Jost et al. (2013a).
The ΣN2O efflux from soil was most closely related to faecal MurN, indicating bacterial biomass in faeces (Jost et al. 2011). The predictive capacity of this cell-wall biomarker exceeded that of all other indices derived from cell-membrane components, such as specific PLFA or ergosterol, or from total molecular genetic tools, such as 16S-rRNA or specific gene abundances. Faecal concentration of total PLFA and bacterial nirK gene abundance also explained some variance of the ΣN2O efflux from soil, according to multiple linear regression analysis. Similarly to MurN and nirK, total PLFA also have a strong focus on bacteria, as the concentration and number of indicator PLFA are much higher in bacteria than in fungi (Joergensen and Wichern 2008; Frostegård et al. 2011). This specific role of bacterial MurN in faeces for ΣN2O efflux from soil is remarkable, as the fungal biomarkers ergosterol and 18S-rRNA in faeces revealed a much stronger response to feeding changes than all faecal biomarkers of bacteria.
In the feeding experiment of Jost et al. (2013a), the ΣN2O efflux was most closely related to faecal MBN, 80% of which is accounted for by bacteria (Meyer et al. 2019). It is an interesting feature of the current experiment that the ΣCO2 efflux from soil was now most closely related to faecal MBN. This suggests that feeding-induced changes in the mineralisation of faeces in soil are more closely related to small shifts in bacterial biomass indices than to the strong response of fungal biomass indices.
Extractable inorganic N and organic C
Direct application of cow faeces to soil resulted in a net-N immobilisation for at least 56 days, which has been observed repeatedly (Griffin et al. 2005; Peters and Jensen 2011). Immediate net-N immobilisation was the highest after application of faeces from high-yield cows but the lowest after faeces application from the non-lactating group. However, it is interesting that the reverse was true for the subsequent re-mineralisation rate, so that a small net-N mineralisation was ultimately observed after faeces application from high-yield cows. The high-yield faeces from Oederan exhibited the highest hemicellulose and the lowest lignin concentrations (Meyer et al. 2019), leading to a microbiome with high MBC and MurN concentrations but a low Gram + /Gram- PLFA ratio. After application to soil, these features strongly increased the soil N turnover, leading to an immediate growth-induced uptake of inorganic N from the soil solution.
Microbial growth also reduced the increased K2SO4 extractable C contents in the faeces amended soil, accompanied by a strong increase in CO2 efflux. A similar increase followed by a decrease in K2SO4 extractable C was observed after straw incorporation (Joergensen et al. 1994). The K2SO4 extractable C contents in the faeces amended soil remained approximately 25% above the non-amended control, presumably due to the presence of hardly available heterocyclic purines and pyrimidines. This might explain the generally increased ratio of K2SO4 extractable C to SOC and it indicates long-term manure fertilization, due to the accumulation of non-mineralizable but mobilizable organic matter (Sradnick et al. 2018). Usually, the content of K2SO4 extractable C remains roughly constant in incubation experiments (Wolters and Joergensen 1991; Barduca et al., 2021) and does not indicate microbially available SOC (Poeplau et al. 2018).
Fungal abundance and biomass
The number of fungal 18S-rRNA gene copies is in the range observed for soil (Spohn et al. 2016; Grüning et al. 2018; Beule et al. 2019, 2020), whereas no quantitative information exists for faeces. Fungal abundance was increased in the faeces of the non-lactating group according to the current 18S-rRNA data. The higher NDF and ADF concentrations in the feedstuff of the non-lactating in comparison with the lactating cows resulted in higher faecal cellulose and lignin concentrations (Meyer et al. 2019). This led consequently to a higher biomass of saprotrophic fungi according to faecal ergosterol and fungal GlcN concentrations (Meyer et al. 2019). Not only the abundance of 18S-rRNA was increased in the faeces of the non-lactating group but also the 18S-rRNA/ergosterol ratio. The close correlations of the 18S-rRNA/ergosterol and 18S-rRNA/fungal GlcN ratios indicate that all three fungal indices give valid information on the contribution of fungi to the faecal microbiome of cows.
An increased 18S-rRNA/ergosterol ratio has been observed after application of rice straw to soil (Wichern et al. 2020). Also, the application of faeces increased the ratio of genome to cell membrane components, indicating larger fungal cells after growth. This is even clearer when comparing the roughly constant 18S-rRNA/ergosterol ratio in soil, which was approximately 15 times wider than that in faeces. This indicates that the fungal cells were much larger in energy-rich faeces than in C-limited soil.
In the current faeces, fungi contributed only 0.1% to the total gene copies, i.e., the sum of fungal 18S-rRNA and bacterial 16S-rRNA. In contrast, a considerable mean percentage of roughly 20% to the biomass of the microbiome, according to PLFA and amino sugar data (Meyer et al. 2019). In the current soil, fungi contributed even 3.3% to the sum of bacterial and fungal gene copies, but 75% to microbial tissue, according to amino sugar data (Khan et al. 2016). These differences in fungal and bacterial gene copies have been observed repeatedly (Spohn et al. 2016; Grüning et al. 2018; Beule et al. 2019, 2020), but the causes are still not fully understood.
Apart from the difference in cell size between fungi and bacteria, other problems might have been created by the obstructive thicker fungal cell walls and the embedding of eukaryotic DNA into the protein structure. Methodological differences in DNA extraction, DNA yield, the choice of primer sets (Xue et al. 2019) and PCR efficiencies bias fungal community studies (Kohout et al. 2014; Grüning et al. 2018). Hartmann et al. (2015) obtained a bacterial/fungal OTU ratio of 1.5 in the arable soils of the DOK long-term fertilisation trial, using a qualitative amplicon sequencing approach, which is more in line with amino sugar-based calculations of fungal to bacterial biomass in soil (Joergensen and Wichern 2008; Joergensen 2018).
Bacterial abundance
The number of bacterial 16S-rRNA gene copies is within the range for soil (Spohn et al. 2016; Grüning et al. 2018; Beule et al. 2019, 2020), whereas no quantitative information exists for faeces. The ratio of 16S-rRNA to MBC in faeces was approximately 0.6 × 107, whereas the respective ratio was 3 × 107 in soil. These two ratios would decline to 0.5 × 107 and 0.8 × 107, respectively, considering that bacteria contributed 80% to the faecal microbial biomass (Meyer et al. 2019) and 25% to the soil microbial biomass (Khan et al. 2016). These data suggest that the cell size of copiotrophic faecal bacteria is also larger than that of starving soil bacteria. However, the difference in cell size between faeces and soil is smaller for bacteria than for fungi.
The higher NDF and ADF concentrations in the feeding ration of the non-lactating cows not only promotes fungi but also increases the ratio of Gram + /Gram- bacterial PLFA (Meyer et al. 2019). As Gram + bacteria contain 3.8 times more MurN (Appuhn and Joergensen 2006), the 16S-rRNA/MurN should be lower in the non-lactating group. However, the reverse is true, suggesting again a change in cell volume, especially of Gram + bacteria, i.e., the larger the cell volume, the larger the 16S-rRNA/MurN ratio. Another possibility is that not all 16S-rRNA was extracted from bacterial phyla with thick cell walls. This would explain why PLFA data indicate the dominance of Gram + bacteria in faeces, whereas DNA sequencing approaches usually find markedly more Gram- bacteria (Pitta et al. 2016; Jose et al. 2017; Kala et al. 2017; Thomas et al. 2017). However, Kim et al. (2014) observed a dominant contribution of Gram + Firmicutes, ranging between 50 and 77% of total sequences caused by differences in diet, which is in line with recent results of others (Faulkner et al. 2017; Cendron et al. 2020; Huang et al. 2020).
Abundance of nitrification and denitrification genes
No AOA amoA genes were detected in cow faeces at any farm, while AOB amoA genes were found in only 50% of the farms. The low abundance of AOA amoA genes is surprising, considering the large concentration of archaeal PLEL (16% of total PLFA) in cow faeces (Gattinger et al. 2007). Qualitative amplicon sequencing approaches suggested that archaea often contribute less than 1.3% to the faecal microbiome (Pitta et al. 2016; Jose et al. 2017; Kala et al. 2017), whereas Cendron et al. (2020) observed an average of even 6%, indicating the strong demand for methodological reconciliation.
In contrast to faeces, large numbers of AOA and AOB amoA genes could be detected in soil, where the abundance of genes related to nitrification and denitrification responded more dynamically to faeces application than the biomass indices MBC, MBN and ergosterol. However, the close relationship between bacterial MurN in faeces and the ΣN2O efflux from soil indicates that mainly bacteria contributed nirK, nirS and nosZ clade I genes and, thus, to N2O production. The pseudo-murein of archaea does not contain MurN (Kandler and König 1998). Sadly, limited information exists on the cell-wall composition of archaea (Joergensen 2018). The current relationship between MurN and the ΣN2O efflux is in line with several studies suggesting that AOB amoA have a higher nitrification capacity than AOA amoA (Xia et al. 2011; Sterngren et al. 2015; Ouyang et al. 2016).
The positive effects of faecal MBC on nirK and nosZ clade I gene abundance indicate their general relationship with the gut microbiome. Those of the Gram + /Gram- PLFA ratio suggest the stronger importance of Gram + bacteria for denitrifying microorganisms, whereas the positive effects of fungal ergosterol on nirK and nosZ clade I gene abundance point to the possibility of fungal denitrification and N2O production (Ma et al. 2017; Zhong et al. 2018; Xu et al. 2019).
Gas efflux from soil and faecal gene abundance were not directly quantitatively related, an exception being the significant effect of soil nirK gene abundance at day 14 on the final ΣN2O efflux. This suggests that a mismatch between N2O production rate and denitrifying gene abundance often occurs (Gschwendtner et al. 2014), as genes have a longer persistence in soil than the rapid release of gaseous N2O (Grüning et al. 2018). Relationships between nirK gene abundance and ΣN2O efflux have been observed repeatedly (Morales et al. 2010; Rasche et al. 2011; Levy-Booth et al. 2014; Wu et al. 2017). NirK-containing organisms mostly lack nosZ genes (Graf et al. 2014), which explains why soil ΣN2O efflux is predominantly determined by nirK rather than by nirS-gene containing microorganisms (Grüning et al. 2018). Recently, new primer sets have been reported for nosZ clade I (Zhang et al. 2021) and clade II (Chee-Sanford et al. 2020), which will provide more comprehensive assessment of denitrifier gene hosts in combination with their ecological patterns. As the detection of genes only gives potential indications of the relative activities, it is necessary to determine the expression of genes, e.g., by mRNA analysis, to obtain true indications of their contribution to N2O production. However, this would require more sampling points immediately after faeces application, due the rapid changes of gene expression.
Differences in the dietary composition of dairy cows had no significant effects on 16S-rRNA, AOA, AOB, nirK and nirS, in contrast to the farm environment: the highest faecal pH led to the highest 16S-rRNA gene abundance at Wolkramshausen, in comparison with the other three farms. The lowest faecal total N and the highest lignin concentrations resulted in the lowest 16S-rRNA gene abundance at Rotenburg. This suggests that the specific farm microbiome of the feeding environment has a stronger impact on the bacterial faeces microbiome than the chemical composition of the feedstuff, contrasting the results of the ΣN2O efflux and the ΣCO2 efflux.
Conclusions
A cow diet reduced in protein but increased in NDF and ADF decreased the ΣN2O efflux from faeces amended soil. This was also reflected by the lower abundance of denitrifying nirK genes. A large percentage of this ΣN2O efflux could be explained by faecal MurN, indicating that AOB amoA have a higher nitrification capacity in soil than AOA amoA. The comparison of genome markers with cell wall (GlcN) and cell membrane components (ergosterol) indicates that the fungal cells were much larger in energy-rich faeces than in C-limited soil. This difference in cell size was also calculated for bacteria but was less pronounced. The growth of copiotrophic faecal bacteria led to a strong initial net-N immobilisation immediately after incorporation of faeces into soil. Initial net-N immobilisation but also final net-N mineralization was the highest after application of faeces from high-yield dairy cows and the lowest after application of faeces from the non-lactating group. Future studies should evaluate the relationship of faecal MurN and ΣN2O efflux from soil on a larger number of farms, to establish the relationship between farming environment and cow diet. Gene expression analysis and sequencing of marker genes will further elucidate the interactions between faeces quality and ΣN2O efflux from soil.
References
Ali AIM, Wassie SE, Korir D, Merbold L, Goopy JP, Butterbach-Bahl K, Dickhoefer U, Schlecht E (2019) Supplementing tropical cattle for improved nutrient utilization and reduced enteric methane emissions. Animals 9:210
Althaus B, Papke G, Sundrum A (2013) Use of near infrared reflectance spectroscopy to assess nitrogen and carbon fractions in dairy cow feces. Anim Feed Sci Tech 185:53–59
Appuhn A, Joergensen RG (2006) Microbial colonisation of roots as a function of plant species. Soil Biol Biochem 38:1040–1051
Barduca L, Wentzel S, Schmidt R, Malagoli M, Joergensen RG (2021) Mineralisation of distinct biogas digestate qualities directly after application to soil. Biol Fertil Soils 57:235–243
Beule L, Grüning MM, Karlovsky P, Arnold AIM (2017) Changes of Scots pine phyllosphere and soil fungal communities during outbreaks of defoliating insects. Forests 8:316
Beule L, Corre MD, Schmidt M, Göbel L, Veldkamp E, Karlovsky P (2019) Conversion of monoculture cropland and open grassland to agroforestry alters the abundance of soil bacteria, fungi and soil-N-cycling genes. PLoS ONE 14:e0218779
Beule L, Lehtsaar E, Corre MD, Schmidt M, Veldkamp E, Karlovsky P (2020) Poplar rows in temperate agroforestry croplands promote bacteria, fungi, and denitrification genes in soils. Front Microbiol 10:3108
Brandfass C, Karlovsky P (2008) Upscaled CTAB-based DNA extraction and real-time PCR assays for Fusarium culmorum and F. graminearum DNA in plant material with reduced sampling error. Int J Mol Sci 9:2306–2321
Brookes PC, Landman A, Pruden G, Jenkinson DS (1985) Chloroform fumigation and the release of soil nitrogen: a rapid direct extraction method for measuring microbial biomass nitrogen in soil. Soil Biol Biochem 17:837–842
Cendron F, Niero G, Carlino G, Penasa M, Cassandro M (2020) Characterizing the fecal bacteria and archaea community of heifers and lactating cows through 16S rRNA next-generation sequencing. J Appl Gen 61:593–605
Chee-Sanford JC, Connor L, Krichels A, Yang WH, Sanford RA (2020) Hierarchical detection of diverse Clade II (atypical) nosZ genes using new primer sets for classical- and multiplex PCR array applications. J Microbiol Methods 172:105908
Djajakirana G, Joergensen RG, Meyer B (1996) Ergosterol and microbial biomass relationship in soil. Biol Fert Soils 22:299–304
Faulkner MJ, Wenner BA, Solden LM, Weiss WP (2017) Source of supplemental dietary copper, zinc, and manganese affects fecal microbial relative abundance in lactating dairy cows. J Dairy Sci 100:1037–1044
Flessa H, Dörsch P, Beese F, König H, Bouwman AF (1996) Influence of cattle wastes on nitrous oxide and methane fluxes in pasture land. J Environ Qual 25:1366–1370
Frostegård Å, Tunlid A, Bååth E (2011) Use and misuse of PLFA measurements in soils. Soil Biol Biochem 43:1621–1625
Gattinger A, Höfle MG, Schloter M, Embacher A, Böhme F, Munch JC, Labrenz M (2007) Traditional cattle manure application determines abundance, diversity and activity of methanogenic Archaea in arable European soil. Environ Microbiol 9:612–624
Gschwendtner S, Tejedor J, Bimueller C, Dannenmann M, Knabner IK, Schloter M (2014) Climate change induces shifts in abundance and activity pattern of bacteria and archaea catalyzing major transformation steps in nitrogen turnover in a soil from a mid-European beech forest. PLoS ONE 9:e114278
Goopy JP, Korir D, Pelster D, Ali AIM, Wassie SE, Schlecht E, Dickhoefer U, Merbold L, Butterbach-Bahl K (2020) Severe below-maintenance feed intake increases methane yield from enteric fermentation in cattle. Brit J Nutr 123:1239–1246
Graf DRH, Jones CM, Hallin S (2014) Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PLoS ONE 9:e114118
Griffin TS, He Z, Honeycutt CW (2005) Manure composition affects net transformation of nitrogen from dairy manures. Plant Soil 273:29–38
Grüning MM, Beule L, Meyer S, Karlovsky P, Arnold AIM (2018) The abundance of fungi, bacteria and denitrification genes during insect outbreaks in Scots pine forests. Forests 9:497
Guerra V, Beule L, Lehtsaar E, Liao HL, Karlovsky P (2020) Improved protocol for DNA extraction from subsoils using phosphate lysis buffer. Microorganisms 8:532
Hartmann M, Frey B, Mayer J, Mäder P, Widmer F (2015) Distinct soil microbial diversity under long-term organic and conventional farming. ISME J 9:1177–1194
Heinze S, Raupp J, Joergensen RG (2010) Effects of fertilizer and spatial heterogeneity in soil pH on microbial biomass indices in a long-term field trial of organic agriculture. Plant Soil 328:203–215
Huang S, Ji S, Wang F, Huang J, Alugongo GM, Li S (2020) Dynamic changes of the fecal bacterial community in dairy cows during early lactation. AMB Expr 10:167
Joergensen RG (2018) Amino sugars as specific indices for fungal and bacterial residues in soil. Biol Fertil Soils 54:559–568
Joergensen RG, Wichern F (2008) Quantitative assessment of the fungal contribution to microbial tissue in soil. Soil Biol Biochem 40:2977–2991
Joergensen RG, Meyer B, Mueller T (1994) Time-course of the soil microbial biomass under wheat. - a one year field study. Soil Biol Biochem 26:987–994
Jones CM, Spor A, Brennan FP, Breuil MC, Bru D, Lemanceau P, Griffiths B, Hallin S, Philippot L (2014) Recently identified microbial guild mediates soil N2O sink capacity. Nat Clim Change 4:801–805
Jose VL, Appoothy T, More RP, Arun AS (2017) Metagenomic insights into the rumen microbial fibrolytic enzymes in Indian crossbred cattle fed finger millet straw. AMB Expr 7:13
Jost DI, Indorf C, Joergensen RG, Sundrum A (2011) Determination of microbial biomass and fungal and bacterial distribution in cattle faeces. Soil Biol Biochem 43:1237–1244
Jost DI, Joergensen RG, Sundrum A (2013a) Effect of cattle faeces with different microbial biomass content on soil properties, gaseous emissions and plant growth. Biol Fertil Soils 49:61–70
Jost DI, Aschemann M, Lebzien P, Joergensen RG, Sundrum A (2013b) Microbial biomass in faeces of dairy cows affected by a nitrogen deficient diet. Arch Anim Nutr 67:104–118
Kala A, Kamra DN, Kumar A, Agarwal N, Chaudhary LC, Joshi CG (2017) Impact of levels of total digestible nutrients on microbiome, enzyme profile and degradation of feeds in buffalo rumen. PLoS ONE 12:e0172051
Kandler O, König H (1998) Cell wall polymers in Archaea (Archaebacteria). Cell Mol Life Sci 54:305–308
Khan KS, Mack R, Castillo X, Kaiser M, Joergensen RG (2016) Microbial biomass, fungal and bacterial residues, and their relationships to the soil organic matter C/N/P/S ratios. Geoderma 271:115–123
Kim M, Kim J, Kuehn LA, Bono JL, Berry ED, Kalchayanand N, Freetly HC, Benson AK, Wells JE (2014) Investigation of bacterial diversity in the feces of cattle fed different diets. J Anim Sci 92:683–694
Kindler R, Miltner A, Richnow HH, Kästner M (2006) Fate of gram-negative bacterial biomass in soil – mineralization and contribution to SOM. Soil Biol Biochem 38:2860–2870
Kohout P, Sudová R, Janoušková M, Čtvrtlíková M, Hejda M, Pánková H, Slavíková R, Štajerová K, Vosátka M, Sýkorová Z (2014) Comparison of commonly used primer sets for evaluating arbuscular mycorrhizal fungal communities: is there a universal solution? Soil Biol Biochem 68:482–493
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW, Prosser JI, Schuster SC, Schleper C (2006) Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442:806–809
Levy-Booth DJ, Prescott CE, Grayston SJ (2014) Microbial functional genes involved in nitrogen fixation, nitrification and denitrification in forest ecosystems. Soil Biol Biochem 75:11–25
Ma S, Shan J, Yan X (2017) N2O emissions dominated by fungi in an intensively managed vegetable field converted from wheat–rice rotation. Appl Soil Ecol 116:23–29
Mäder P, Fließbach A, Dubois D, Gunst L, Fried P, Niggli U (2002) Soil fertility and biodiversity in organic farming. Science 296:1694–1697
McGovern E, McCabe MS, Cormican P, Popova M, Keogh K, Kelly AK, Kenny DA, Waters SM (2017) Plane of nutrition affects the phylogenetic diversity and relative abundance of transcriptionally active methanogens in the bovine rumen. Sci Reports 7:13047
Meyer S, Thiel V, Joergensen RG, Sundrum A (2019) Relationships between feeding and microbial faeces indices in dairy cows at different milk yield levels. PLoS ONE 14:e0221266
Morales SE, Cosart T, HolbenWE, (2010) Bacterial gene abundances as indicators of greenhouse gas emission in soils. ISME J 4:799–808
Ouyang Y, Norton JM, Stark JM, Reeve JR, Habteselassie MY (2016) Ammonia-oxidizing bacteria are more responsive than archaea to nitrogen source in an agricultural soil. Soil Biol Biochem 96:4–15
Pelster DE, Gisore B, Koske JK, Goopy J, Korir D, Rufino MC, Butterbach-Bahl K (2016) Methane and nitrous oxide emissions from cattle excreta on an East African grassland. J Environ Qual 45:1531–1539
Peters K, Jensen LS (2011) Biochemical characteristics of solid fractions from animal slurry separation and their effects on C and N mineralisation in soil. Biol Fertil Soils 47:447–455
Pitta DW, Indugu N, Kumar S, Vecchiarelli B, Sinha R, Baker LD, Bhukya B, Ferguson JD (2016) Metagenomic assessment of the functional potential of the rumen microbiome in Holstein dairy cows. Anaerobe 38:50–60
Poeplau C, Don A, Six J, Kaiser M, Benbie D, Chenu C, Cotrufo MF, Derrien D, Gioacchini P, Grand S, Gregorich E, Griepentrog M, Gunina A, Haddix M, Kuzyakov Y, Kühnel A, Macdonald LM, Soong J, Trigalet S, Vermeire ML, Rovira P, van Wesemael B, Wiesmeier M, Yeasmin S, Yevdokimov I, Nieder R (2018) Isolating organic carbon fractions with varying turnover rates in temperate agricultural soils – a comprehensive method comparison. Soil Biol Biochem 125:10–26
Rasche F, Knapp D, Kaiser C, Koranda M, Kitzler B, Zechmeister-Boltenstern S, Richter A, Sessitsch A (2011) Seasonality and resource availability control bacterial and archaeal communities in soils of a temperate beech forest. ISME J 5:389–402
Rashid MI, Lantinga EA, Brussaard L, de Goede RGM (2017) The chemical convergence and decomposer control hypotheses explain solid cattle manure decomposition in production grasslands. Appl Soil Ecol 113:107–116
Schick BD, Guretzky JA, Schacht WH, Mamo M (2019) Dietary nutritive value, dung quality, decomposition, and nutrient movement into soil in smooth bromegrass pastures. Crop Sci 59:1294–1308
Siegfried K, Dietz H, Amthauer Gallardo D, Schlecht E, Buerkert A (2013) Effects of manure with different C/N ratios on yields, yield components and matter balances of organically grown vegetables on a sandy soil of northern Oman. Org Agric 3:9–22
Soussana JF, Tallec T, Blanfort V (2010) Mitigating the greenhouse gas balance of ruminant production systems through carbon sequestration in grasslands. Animal 4:3
Spohn M, Pötsch EM, Eichorst SA, Woebken D, Wanek W, Richter A (2016) Soil microbial carbon use efficiency and biomass turnover in a long-term fertilization experiment in a temperate grassland. Soil Biol Biochem 97:168–175
Sradnick A, Oltmanns M, Raupp J, Joergensen RG (2018) Microbial biomass and activity down the soil profile after long-term addition of farmyard manure to a sandy soil. Org Agric 8:29–38
Sterngren AE, Hallin S, Bengtson P (2015) Archaeal ammonia oxidizers dominate in numbers, but bacteria drive gross nitrification in N-amended grassland soil. Front Microbiol 6:1350
Thomas M, Webb M, Ghimire S, Blair A, Olson K, Fenske GJ, Fonder AT, Christopher-Hennings J, Brake D, Scaria J (2017) Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci Reports 7:12257
van Vliet PCJ, Reijs JW, Bloem J, Dijkstra J, de Goede RGM (2007) Effects of cow diet on the microbial community and organic matter and nitrogen content of feces. J Dairy Sci 90:5146–5158
Vance ED, Brookes PC, Jenkinson DS (1987) An extraction method for measuring soil microbial biomass C. Soil Biol Biochem 19:703–707
Voglmeier K, Six J, Jocher M, Ammann C (2019) Grazing-related nitrous oxide emissions: from patch scale to field scale. Biogeosciences 16:1685–1703
Voglmeier K, Six J, Jocher M, Ammann C (2020) Soil greenhouse gas budget of two intensively managed grazing systems. Agric Forest Meteorol 287:107960
Wachendorf C, Joergensen RG (2011) Mid-term tracing of 15N derived from urine and dung in soil microbial biomass. Biol Fertil Soils 47:147–155
Wichern F, Islam R, Hemkemeyer M, Watson C, Joergensen RG (2020) Organic amendments alleviate salinity effects on soil microorganisms and mineralisation processes in aerobic and anaerobic paddy rice soils. Front Sust Food Syst 4:30
Wolters V, Joergensen RG (1991) Microbial carbon turnover in beech forest soils at different stages of acidification. Soil Biol Biochem 23:897–902
Wu J, Joergensen RG, Pommerening B, Chaussod R, Brookes PC (1990) Measurement of microbial biomass C by fumigation extraction - an automated procedure. Soil Biol Biochem 22:1167–1169
Wu X, Liu H, Fu B, Wang Q, Xu M, Wang H, Yang F, Liu G (2017) Effects of land-use change and fertilization on N2O and NO fluxes, the abundance of nitrifying and denitrifying microbial communities in a hilly red soil region of southern China. Appl Soil Ecol 120:111–120
Xia W, Zhang C, Zeng X, Feng Y, Weng J, Lin X, Zhu J, Xiong Z, Xu J, Cai Z, Jia Z (2011) Autotrophic growth of nitrifying community in an agricultural soil. ISME J 5:1226–1236
Xu H, Sheng R, Xing X, Zhang W, Hou H, Liu Y, Qin H, Chen C, Wei W (2019) Characterization of fungal nirK-containing communities and N2O emission from fungal denitrification in arable soils. Front Microbiol 10:117
Xue C, Hao Y, Pu X, Penton CR, Wang Q, Zhao M, Zhang B, Ran W, Huang Q, Shen Q, Tiedje JM (2019) Effect of LSU and ITS genetic markers and reference databases on analyses of fungal communities. Biol Fertil Soils 55:79–88
Zhang B, Penton CR, Yu Z, Xue C, Chen Q, Chen Z, Yan C, Zhang Q, Zhao M, Quensen JF, Tiedje JM (2021) A new primer set for Clade I nosZ that recovers genes from a broader range of taxa. Biol Fertil Soils 57:523–531
Zhong L, Bowatte S, Newton PCD, Hoogendoorn CJ, Luo D (2018) An increased ratio of fungi to bacteria indicates greater potential for N2O production in a grazed grassland exposed to elevated CO2. Agric Ecosyst Environ 254:111–116
Acknowledgements
The technical assistance of Gabriele Dormann and Christiane Jatsch is highly appreciated.
Funding
Open Access funding enabled and organized by Projekt DEAL. This project was supported by the Research Training Group 1397 ‘Regulation of soil organic matter and nutrient turnover in organic agriculture’ of the German Research Foundation (DFG).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meyer, S., Grüning, M.M., Beule, L. et al. Soil N2O flux and nitrification and denitrification gene responses to feed-induced differences in the composition of dairy cow faeces. Biol Fertil Soils 57, 767–779 (2021). https://doi.org/10.1007/s00374-021-01566-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-021-01566-0