
Abstract
Activation of signal transducer and activator of transcription 3 (STAT3) has been identified as a key cardioprotective signal not only in animal studies but also in humans—in animals, STAT3 is causally involved in cardioprotection. In response to late ischemic conditioning, canonical function of STAT3 activation upregulates the expression of cardioprotective and anti-apoptotic proteins. In its non-canonical function, STAT3 is activated during ischemic conditioning and is part of the cardioprotective cytosolic survival activating factor enhancement pathway. Activated STAT3 is imported and localized to the mitochondria. Mitochondrial STAT3 stimulates the activity of mitochondrial electron transport chain complex I, reduces mitochondrial reactive oxygen species production and mitochondrial permeability transition pore opening. Finally, two novel aspects of STAT activation in cardioprotection are discussed: a genetic variance of the STAT encoding region as a potential primordial confounding variable for cardioprotection, and the cardioprotective potential of sodium–glucose cotransporter 2 inhibitors through STAT3 activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cardioprotection: strategies and relevance of STAT signaling
In addition to rapid reperfusion and current therapy there is still a need for cardioprotection to reduce morbidity and mortality in patients with acute myocardial infarction. In experimental settings, there are mechanical and pharmacological interventions that reduce myocardial infarct size. The strongest and most robust cardioprotective intervention is ischemic conditioning, which is effective in all species tested so far, including humans [45, 52]. Reduction of infarct size by ischemic conditioning can be induced by cycles of brief ischemia/reperfusion before (ischemic preconditioning, IPC) [79] or after (ischemic postconditioning, POCO) [119] sustained myocardial ischemia with reperfusion. Ischemic conditioning can also be induced remotely from the heart (remote IPC, RIC) [43, 45]. Among these cardioprotective strategies, RIC has been successfully translated from experimental studies to clinical trials. In patients undergoing elective surgical coronary revascularization, there are several single-center trials in which RIC provided perioperative myocardial protection (e.g., [15, 40, 57, 89, 100, 103]), and one of them also reported improved patient prognosis [65, 100]. However, two prospectively designed multi-center phase III trials in patients undergoing elective surgical coronary revascularization and valve surgery, i.e., ERICCA and RIPHEART, were neutral [37, 77], possibly because use of propofol rather than volatile anesthesia [49]. Similarly, in patients with acute myocardial infarction, RIC attenuated myocardial injury in single-center trials (e.g., [13, 23, 108, 115]), and again one of them also reported an improved patient prognosis [94]. However, the prospectively designed larger phase III multi-center follow-up CONDI-2/ERIC-PPCI trial was neutral on myocardial injury and clinical outcome [39]. Only the prospectively designed single-center RIC-STEMI trial truly reported an improved clinical outcome as a primary endpoint with RIC [29]. A detailed and more comprehensive review of available clinical trials on cardioprotective strategies is found in: [42, 45]. Potentially confounding factors of the cardioprotective strategies in patients [24, 61, 90] as well as errors in the planning and design of preclinical and clinical trials are discussed in detail in the other reviews [44, 47, 70]. Irrespective of all these valid considerations, one reason for the lack of success in translating cardioprotective strategies from experimental studies to the clinical situation is that the underlying signaling pathways are incompletely understood and much more basic research is needed to improve our understanding of the signaling pathways involved in cardioprotective interventions that are in principle applicable.
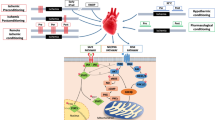
Currently, the underlying myocardial signal transduction of cardioprotection [43, 45] can be conceptually classified by their (sub-) cellular localization (extracellular molecules, cytosolic signal transduction and target organelle/structure). Extracellular molecules (e.g., autacoids, calcium, cytokines, neurohormones, nitric oxide, or reactive oxygen species) are released during conditioning cycles from cardiomyocytes, endothelial cells, neurons, etc., but the exact subcellular origin and detailed biochemical reactions of how these extracellular molecules are generated and released are unclear. Through sarcolemmal receptors or receptor-independently, these molecules then activate cytosolic signaling cascades. Within the cardiomyocyte, a variety of proteins are activated as cytosolic signal transducers. Again, conceptually, cytosolic signaling pathways are divided into three major cardioprotective pathways: the nitric oxide/protein kinase G (NO/PKG) pathway [20], the reperfusion injury salvage kinase (RISK) pathway [41], and the survival activating factor enhancement (SAFE) pathway [69]. The RISK pathway and its interaction with mitochondrial function is the subject of a detailed discussion in the current issue of “Mitochondria at the heart of cardioprotection” [116]. Key protein of the SAFE pathway is the signal transducer and activator of transcription (STAT)3 [6, 43, 45, 67, 69]. In response to ligand binding [e.g., interleukin 6-like cytokines, tumor necrosis factor alpha (TNF)] through sarcolemmal glycoprotein 130 or TNF receptors, Janus kinase (JAK) is activated and phosphorylates STAT3 [tyrosine (tyr) 701 and serine (ser) 727]. STAT3 phosphorylation is required for the protein dimerization, its subsequent translocation to the nucleus, and its function as a transcription factor; the ser727 phosphorylation seems to be the boost for transcriptional activity of STAT3 [26]. STAT3 is constitutively expressed, and under physiological conditions its expression is tightly controlled—also in myocardial cells (i.e., cardiomyocytes, endothelial cells, smooth muscle cells, and fibroblasts). STAT3 regulates the expression of genes encoding proteins mainly involved in angiogenesis, apoptosis, inflammation, and oxidative stress—the canonical function of STAT3 [6, 21, 36, 82, 117]. STATs canonical functions are too slow for acute protection [6, 59]. However, subacute cardioprotection by late IPC and late RIC (ischemic conditioning is induced 24 h before myocardial infarction) involves the canonical function of STAT3 [11, 113] and STAT5 [17]. STAT3 activation during late preconditioning upregulates the expression of anti-apoptotic and cytoprotective proteins (e.g., cyclooxygenase 2, heme oxygenase-1, manganese sodium dismutase, myeloid leukemia protein 1, apoptosis regulator protein Bcl-2 family, c-FLIP a natural homologue of caspase 8, heat shock protein 70) [11, 17, 113] (Fig. 1). In contrast to the acute non-canonical and the subacute canonical STAT3 activation which serve a protective function, chronic STAT3 activation after myocardial infarction contributes to inflammatory processes and cardiac remodeling [6, 33, 34, 36, 55]. Such chronic STAT3 activation occurs mostly in macrophages which invade from the circulating blood and are recruited from bone marrow and spleen [112]. Up to four days after myocardial infarction, macrophages initiate or maintain inflammatory processes that contribute to debris clearance. Later on, however, macrophages express anti-inflammatory cytokines which then stimulate scar formation and angiogenesis (Fig. 1). The influence of chronic STAT3 activation after myocardial infarction on the processes described above is based on studies in rodents in the absence of a cardioprotective intervention, for further details please see: [6, 33, 34, 36, 55]. Importantly, chronic systemic STAT3 activation may also promote malignant transformation, which is of concern because RIC is a systemic phenomenon that may through STAT3 activation also promote cancer [46, 53].

Schematic overview of the time-dependent—acute, subacute, chronic—and non-canonical vs. canonical effects of STAT3 activation on myocardial ischemia/reperfusion; created with BioRender.com. ATP adenosine triphosphate, MPTP mitochondrial permeability transition pore, ROS reactive oxygen species, STAT3 signal transducer and activator of transcription 3
Outside the nucleus, the non-canonical function of STAT3 plays a unique role in acute cardioprotection: in contrast to all other cardioprotective signals, STAT3 activation is consistently not only associated with the reduction of infarct size by all ischemic conditioning procedures, but also causally involved in cardioprotection in all species tested to date [6, 43, 45, 67, 69]—also in in larger mammals, i.e., in pigs, which are more similar to humans in their cardiovascular physiology than rodents [54, 70]. In detail: Interleukin 6-like cytokines and TNF appear to be the major activators of the cardioprotective SAFE pathway [36]. Downstream of activation of sarcolemmal glycoprotein 130 or TNF receptors, the cytosolic SAFE pathway is activated [6, 43, 45, 67, 69]. In response to IPC [27, 30, 31], POCO [50], and RIC [88, 92, 98] in rodents [4, 27, 31, 88, 92, 98] and pigs [30, 50, 60, 63, 66, 71, 92, 93], STAT3 is phosphorylated at tyr705. The causal involvement of STAT3 activation in infarct size reduction by different types of ischemic conditioning was demonstrated by pharmacological blockade of STAT3 activation [6, 43, 45, 67, 69]. In STAT3 knock-out mice and aged mice (which had reduced STAT3 protein), a more potent local ischemic conditioning stimulus overcame the STAT3-associated loss of cardioprotection [5], suggesting that other cardioprotective pathways are also involved. Apart from and in addition to a potential activation of NO/PKG and RISK pathways, STAT5 may have been activated. STAT5 knock-out in mice prevented infarct size reduction by local ischemic conditioning [114]. The ser727 phosphorylation of STAT3 was described in crosstalk with key proteins of the RISK pathway [36]. Present data suggest a dynamic balance between STAT3 phosphorylation at tyr705 and ser727 for STAT3 transcriptional activity; whether this may also play a role in cardioprotection is unclear. Notably, only STAT3/5 activation induced by ischemic conditioning appears to be causal for infarct size reduction, whereas basal STAT3/5 activity is not relevant for infarct size—neither pharmacological blockade nor knock-out increases infarct size per se.
The common intracellular target of all cardioprotective pathways, including the SAFE pathway, are mitochondria [8]. As such, mitochondria are critical elements of cardiomyocyte function and viability [12, 25, 32, 78, 91], and preservation of mitochondrial function is central for the reduction of ischemia/reperfusion injury [8, 43, 45] (Fig. 1). There is a prior comprehensive review on the non-canonical function of STAT3 in cardioprotection by targeting mitochondrial function [21]. The focus of my present somewhat personal and opinionated article is on the state-of-the-art of mitochondrial STAT3 and its potential causal role in cardioprotective strategies (Table 1). Mitochondrial STAT3 has been detected across species, in myocardium from mice [7, 85, 86, 96], rats [7, 84, 99, 109, 110], and pigs [50, 85].
STAT3 phosphorylation site, import into, and localization within mitochondria
In vitro studies with isolated rat heart mitochondria and labeled STAT3 proposed an energy-dependent import of STAT3 mediated through chaperone-like activity of a complex I subunit protein—the protein of the gene associated with retinoid interferon-induced cell mortality 19 (GRIM-19, Fig. 2) [99]. In rat left ventricular protein extracts, mitochondrial import receptor subunit Tom20 co-immunoprecipitated with ser727 STAT3 and total STAT3 [7], suggesting a Tom20-dependent import. In heat shock protein 22 (HSP22) knock-out mice, STAT3 translocation into the mitochondria was reduced, and HSP22 co-immunoprecipitated with total STAT3 [86], indicating another import mechanism (Fig. 2). The initial study suggested that ser727 phosphorylation is required for STAT translocation to mitochondria, a ser727 mutation in STAT3 reduced the import and the STAT3 GRIM-19 assembly in isolated rat heart mitochondria [99]. As mentioned above, it is still unclear whether ser727 phosphorylation of STAT3 is indeed causally involved in infarct size reduction. However, while in mouse [86, 107] and rat [84, 109, 110] heart mitochondria ser727 phosphorylation was described, a tyr705 phosphorylation has been detected in rat heart mitochondria in addition to the ser727 phosphorylation [7, 84], and in pig heart mitochondria, only the tyr705 phosphorylation was reported [50]. Beside potential species-specific differences, it is also conceivable that simple methodological reasons, such as the antibodies used and their species-specificity, may have led to the reported differences. Regardless of the exact phosphorylation site, given that phosphorylation of STAT3 is a prerequisite for mitochondrial import, the cytosolic non-canonical function and, thus, phosphorylation of STAT3 seems to be a prerequisite for mitochondrial import of STAT3.

Schematic overview of STAT3 import, localization, and function in mitochondria; created with BioRender.com. I, II, II, IV indicates respiratory chain complexes; ATP adenosine triphosphate, GRIM-19 gene associated with retinoid interferon-induced cell mortality 19, HSP22 heat shock protein 22, MCU mitochondrial calcium uniporter, MPTP mitochondrial permeability transition pore, Tom20 mitochondrial import receptor subunit Tom20, ROS reactive oxygen species, STAT3 signal transducer and activator of transcription 3
Using enzymatic digestion of isolated mouse and rat heart mitochondria and Western blot technique with specific marker proteins for intra-mitochondrial localization, STAT3 was identified in the matrix of mitochondria [7, 86, 107] as well as in the inner mitochondrial membrane [86, 99]. Using immunofluorescence imaging, a colocalization of STAT3 with the mitochondrial calcium uniporter—which is localized in the inner mitochondrial membrane—was identified in rat hearts. Co-immunoprecipitation confirmed an interaction with the N-terminal domain of the mitochondrial calcium uniporter and STAT3 in isolated rat cardiomyocytes [109] (Fig. 2, Table 1).
Function of mitochondrial STAT3
Phillips et al. had questioned the functional relevance of mitochondrial STAT3 by quantitative biochemical analyses: the abundance of mitochondrial STAT3 compared with respiratory chain proteins was low, with a ratio of electron transport complex proteins to STAT3 of ~ 105, and relevance to adenosine triphosphate (ATP) production was, thus, ruled out [85]. However, studies on mitochondrial function indicate the opposite. Cardiomyocyte-specific STAT3 knock-out mice had not only less mitochondrial STAT3, but also selective defects in complex I of the electron transport chain [7]. Cardiomyocyte-specific overexpression of mitochondrial STAT3 in mice improved complex I respiration during ischemia [96] (Fig. 1). In mice that overexpress a mitochondrially targeted, transcriptionally inactive STAT3 in cardiomyocytes, a partial and persistent blockade of complex I was evident. Smaller infarct size in these mice in comparison to wild-type mice was associated with an attenuated reactive oxygen species release and attenuation of mitochondrial permeability transition pore opening at the onset of reperfusion [97]. In isolated rat heart mitochondria, pharmacological STAT3 inhibition with stattic (STAT3 inhibitory compound 6 nitrobenzol(b)thiopene 1,1-dioxide) reduced mitochondrial respiration and ATP production; opening of the mitochondrial permeability transition pore and generation of reactive oxygen species were reduced [10] (Fig. 2). There is evidence for STAT3 not only in isolated mitochondria from rodent hearts but also in those from larger mammals. In mitochondria isolated from pig myocardium, POCO induced an increase in mitochondrial STAT3 phosphorylation. This phosphorylation improved mitochondrial complex I respiration and increased calcium retention capacity [50]. The biochemical mechanisms, however, by which mitochondrial STAT3 regulates the electron transport chain complex activities and mitochondrial permeability transition pore opening are not clear. GRIM-19 enhanced in isolated rat heart mitochondria the integration of STAT3 into complex I [99], indicating a direct interaction of STAT3 with complex I activity (Fig. 2, Table 1). In a postconditioning protocol by intermittent hypobaric hypoxia, the colocalization of STAT3 and the mitochondrial calcium uniporter was associated with a reduction in mitochondrial calcium overload during reperfusion [109] (Fig. 2, Table 1).
Causal evidence for mitochondrial STAT3 in cardioprotection
A number of studies have suggested an association between mitochondrial STAT3 activation and infarct size reduction during maneuvers such as IPC [84] or intermittent hyperbaric hypoxia [109, 110]. However, there is only one study describing a causal evidence for mitochondrial STAT3 in infarct size reduction with POCO. In pigs, improved mitochondrial function after POCO was causally related to increased STAT3 tyr705 phosphorylation, but not to total STAT3. AG490 (tyrphostin B42), a JAK–STAT inhibitor, when administered in vivo before POCO, not only abolished the reduction in infarct size but also the protective effect on mitochondrial function. In vitro, stattic (a non-peptide small molecule inhibitor that inhibits STAT3 activity by binding to its SH2 region, which is essential for the tyr705 phosphorylation) abrogated better preservation of mitochondrial function when isolated after POCO. The fact that in mitochondria only STAT3 tyr705 was increased, but not total STAT3, suggests that STAT3 import into mitochondria does not play a role in acute cardioprotection [50]. In rats, POCO did not activate mitochondrial STAT3, but activated proteins of the RISK pathway which were subsequently translocated to the mitochondria [84]. In this study, however, it remained unclear whether or not the POCO maneuver that was performed actually reduced the infarct size (Table 1).
Critical considerations on the detection and function of mitochondrial STAT3
For methodological reasons, most studies investigated mitochondria isolated from the total myocardium; thus, mitochondrial fractions from all myocardial cell types are included. The estimated proportion of cardiomyocytes in the left ventricular myocardium in rodents is between 75% (estimated from cell volume data) and 50% (estimated from nuclei data) [73, 102]. In adult human ventricular myocardium, transcriptome analysis identified 49% cardiomyocytes, whereas studies using nuclear labeling techniques found only about 30% cardiomyocytes [74]. Since mitochondria in cardiomyocytes occupy about 30–40% of the total volume [75], and mitochondrial density in other cells (e.g., endothelial cells) is much lower at 2–5% [58], studies on mitochondria isolated from myocardial tissue mainly refer to cardiomyocyte mitochondria, but not exclusively. Considering that STAT3 activation also plays an important role in non-cardiomyocyte cells such as fibroblasts and endothelial cells, modulating there cell proliferation, differentiation, oxidative stress, cell metabolism, and survival [33], it is reasonable to assume that mitochondrial STAT3 may originate not exclusively from cardiomyocytes. Of note, the above numerical estimates may vary species-specifically, as there are significant species-specific differences in the cellular composition between mouse, rat, and human hearts [3]. In cardiomyocytes, it has been estimated that the majority of mitochondria in cardiomyocytes are interfibrillar mitochondria (IFM), a much smaller proportion are subsarcolemmal mitochondria (SSM), and the smallest proportion are perinuclear mitochondria [87]. Although many studies in rodents have documented differences between SSM and IFM not only in location, but also in function (e.g.. see [9, 56, 68, 83]), a functional difference in ischemic reperfused myocardium, however, was not confirmed in a study in pigs [16]. In rat myocardium, STAT3 was quantified in preparations of SSM and IFM [7]. Because selective isolation of perinuclear mitochondria is methodologically difficult [68], perinuclear mitochondria have not been studied in this regard.
Based on the findings discussed above, it is reasonable to assume that mitochondrial STAT3 plays a critical role in cardioprotection (Table 1). Again, however, using 3 different proteomic approaches in mouse myocardium, the abundance of mitochondrial STAT3 was estimated to be very low (10% of the total cytoplasmic STAT3 and a ratio of electron transport complex proteins to STAT3 of ~ 105) [85], so a relevance of protein–protein interaction in the mitochondria seems indeed questionable. The low abundance of STAT3 in mitochondria is indirectly confirmed by the fact that without prior immunoprecipitation of STAT3, e.g., as done in the study by Boengler et al. [7], detection in isolated mouse mitochondria failed [97]. Further, in a Percoll-purified mitochondrial preparation from mouse myocardium, STAT3 was neither detectable under baseline conditions nor after hypoxia/reoxygenation via Western blot, and confocal imaging showed no colocalization of STAT3 signal with mitochondrial proteins. In this study, only a STAT3 overexpression in a H9C2 cardiomyoblast cell line led to detectable translocation of STAT3 into mitochondria [35]. Overall, it seems most plausible that differences in mitochondrial STAT3 detection are due to the methods used (different purification, enrichment, and denaturation protocols, antibodies, etc.) and which of the available results best reflects the biological reality remains unclear. The functional studies in genetically modified mice and on mitochondria with pharmacological blockade are definitely not affected by these methodological aspects and clearly indicate a relevance of mitochondrial STAT3 for mitochondrial function. However, both the use of genetically modified animal models and the use of pharmacological blockers have different, but also fundamental limitations (e.g., for animal models, the genetic compensation of the knock-out and for pharmacological blockers side effects, non-specificity, toxicity, etc.).
In conclusion, the precise function and importance of mitochondrial STAT3 during cardioprotection is still unclear. Finally, to what extent the acute cardioprotective effect is mediated by cytosolic STAT3 or mitochondrial STAT3 activation remains open.
Evidence for STAT3/5 in humans
Indeed, there is even evidence from the human myocardium that STAT is associated with cardioprotection by ischemic conditioning. In left ventricular biopsies, taken at early reperfusion after cardioplegic ischemic arrest from patients undergoing bypass surgery [51], the activation and expression of 22 signaling proteins, key signaling proteins of the NO/PKG, RISK and SAFE pathway were analyzed using Western blot analysis. Among these 22 proteins, only the activation of STAT5 was associated with reduction of perioperative myocardial injury by RIC [51]. Confirming results in right ventricular outflow tract biopsies of children undergoing tetralogy of Fallot repair surgery, activation of STAT3 and STAT5 was also associated with perioperative myocardial protection by RIC [111]—highlighting again the potentially relevant role of STATs in cardioprotection. Even when human myocardium is investigated, cardioprotective strategies improve mitochondrial function [1, 62, 111]. Since mitochondrial STAT has not yet been detected in human myocardium, it is difficult to predict the relevance of the available data for the translation to patients. In principle, also other members of the STAT family (STAT1, STAT2, STAT5, and STAT6) are present in the mitochondria [7, 76]. Because there are several independent lines of evidence that STAT is associated with cardioprotection in human myocardium, further and more detailed analysis of (mitochondrial) STAT signaling in human myocardium is warranted.
Lack of STAT3 responsiveness: a novel confounding factor
Ossabaw minipigs, a particular strain of minipigs, are characterized by a genotype associated with a thrifty phenotype. Like humans, they develop a metabolic syndrome when fed a hypercaloric, atherogenic diet and consequently coronary atherosclerosis and occasional myocardial infarction [95, 101, 118]. The unequivocally strongest and most robust stimulus for cardioprotection, IPC, failed to reduce infarct size in a power analysis-based experimental design in these Ossabaw minipigs—even when they were lean and only predisposed to metabolic syndrome. Bioinformatic analysis of genetic differences between these Ossabaw minipigs and Göttingen minipigs, in which IPC confers robust protection, identified several clusters of protein-coding genes. One cluster was related to mitochondrial and one to JAK–STAT signaling. Indeed, the lack of infarct size reduction with IPC in the Ossabaw minipigs was associated with a lack of STAT3 activation in the myocardium [64]. RIC also failed to reduce infarct size in the Ossabaw minipigs, but RIC still induced a release of cardioprotective factors into the circulation in these Ossabaw minipigs, as evidenced by their protective effect after transfer to isolated rat hearts; thus, the lack of cardioprotection was attributed to myocardial—i.e., STAT3-dependent—non-responsiveness [72]. These studies in Ossabaw minipigs once again independently underscored the importance of STAT3 signaling for cardioprotection. However, the extent to which mitochondrial STAT3 plays here a role is unclear.
The neutral results of this prospectively designed experimental study in the Ossabaw minipigs are similar to the neutral results of several larger all-comer randomized controlled trials on RIC in patients undergoing interventional reperfusion of myocardial infarction [39] or cardiovascular surgery [37, 77]. In addition to the often discussed confounders such as comorbidities and co-medications which are typical for patients with acute myocardial infarction [61], genetic variance may be newly considered as a potential confounder for cardioprotective measures [48, 104]. In this context, it is noteworthy that STAT3 levels were reduced in aged mice and that this reduction in STAT3 levels was associated with a loss of the cardioprotective effect of POCO [5], suggesting that in addition to a genetic heterogeneity also age may act as confounding factor. A genetic heterogeneity of STATs exists also in humans. The European Lymphoma Risk Study identified human single-nucleotide polymorphisms belonging to the JAK–STAT pathway—including STAT3 and STAT5 [14]. The unique Ossabaw minipig strain may, therefore, be a suitable model to further investigate a genetically determined lack of susceptibility to cardioprotection and to develop therapeutic strategies and to possibly circumvent this blockade of cardioprotective signaling.
Cardioprotective effects of SGLT2 inhibitors through STAT3 activation
Sodium–glucose cotransporter 2 (SGLT2) inhibitors—also known as gliflozins—are a class of drugs originally developed to treat type 2 diabetes via inhibition of the sodium–glucose transport protein 2 in the kidney [2]. However, clinical trials have impressively shown that in addition to lowering blood glucose levels, gliflozins also significantly improve cardiovascular outcomes in patients with and without type 2 diabetes, indicating multifaceted cardioprotective effects that are beyond inhibition of the sodium–glucose transport protein 2 in the kidneys [2]. The larger clinical trials did not identify a reduction in the incidence of acute coronary syndromes, however, dapagliflozin and empagliflozin reduced the incidence of recurrent myocardial infarction, possibly reflecting attenuated ischemia/reperfusion injury [2]. Indeed, a recent study in SGLT2 knock-out mice demonstrated that the infarct size reduction by empagliflozin was completely independent of its initial target, sodium–glucose transport protein 2 inhibition [18]. Among all potential mediators discussed in the context of gliflozin-induced but SGLT2-independent cardioprotection [2], myocardial STAT3 activation could be a common denominator. In fact, one-week of treatment with dapagliflozin and empagliflozin before induction of myocardial infarction in mice reduced infarct size, and STAT3 activation was causally involved. In association with cardioprotection by empaliflozin, mitochondrial complex I and II respiration was preserved after myocardial infarction [81]. A more acute administration of empaliflozin (4 or 24 h before myocardial infarction in mice), however, failed to reduce myocardial infarct size, and STAT3 was not activated [80]. However, 24 h pretreatment of empaliflozin increased STAT3-dependently the survival of cultured human endothelial cells after hypoxia/reoxygenation [80]. Coronary endothelial cells are relative resistant to ischemia, but with endothelial swelling obstructing capillary blood flow, they contribute to microvascular damage in response to myocardial ischemia/reperfusion injury, and microvascular obstruction has a strong impact on patient prognosis [38]. Notably, knock-out of endothelial STAT3 in mice resulted in reduced recovery of left ventricular function during reperfusion after myocardial infarction [106]. In the EMMY trial [105] in patients with recent myocardial infarction, daily administration of SGLT2 inhibitors (with onset no later than 72 h after interventional reperfusion) preserved left ventricular function, possibly through anti-inflammatory effects [28]. However, a number of issues on the role of STAT3 in the gliflozin action remain to be resolved: Is, in response to gliflozins, STAT also activated in the human heart? In cardiomyocytes or endothelial cells, possibly also in inflammatory cells? Does STAT3 serve a canonical or non-canonical function? Are mitochondria involved?
Recently, also other compounds have been described that activate STAT3 and are causally involved in cardioprotection under experimental conditions. The anabolic steroid nandrolone decanoate [22] and dexmedetomidine—a selective alpha 2 adrenoceptor agonist [19] reduced infarct size/biomarker release reflecting myocardial injury in rodents via STAT3 activation. Again, improved mitochondrial function was also associated with cardioprotection. Thus, these agents may also be of interest for use in patients. For these agents, it is even more important to conduct further studies to determine the extent to which the results of the initial experimental studies can be transferred to patients.
Conclusion
STAT3 plays an important role in cardioprotection, also in the human heart. In animal models, there is evidence of mitochondrial STAT3 involved in mitochondrial function. The assumed causal relationship between mitochondrial STAT3 and cardioprotection is based on only one study in pigs. Data from human myocardium on mitochondrial STAT are lacking. There is a primordial non-responsiveness to cardioprotection in pigs which involves STAT3. Combination therapies that acutely activate STAT3—possibly mitochondrial STAT3—through different pathways could therefore be particularly effective, as they could bypass possible blockades upstream of STAT3. The timing of STAT activation seems to be relevant for therapeutic approaches. Acute activation and the non-canonical function of STAT3 are cardioprotective, subacute activation of canonical STAT3 function in late preconditioning is also cardioprotective. However, chronic activation of canonical STAT3 function may be more detrimental than beneficial.
References
Ale-Agha N, Jakobs P, Goy C, Zurek M, Rosen J, Dyballa-Rukes N, Metzger S, Greulich J, von Ameln F, Eckermann O, Unfried K, Brack F, Grandoch M, Thielmann M, Kamler M, Gedik N, Kleinbongard P, Heinen A, Heusch G, Gödecke A, Altschmied J, Haendeler J (2021) Mitochondrial telomerase reverse transcriptase protects from myocardial ischemia/reperfusion injury by improving complex I composition and function. Circulation 144:1876–1890. https://doi.org/10.1161/CIRCULATIONAHA.120.051923
Andreadou I, Bell RM, Bøtker HE, Zuurbier CJ (2020) SGLT2 inhibitors reduce infarct size in reperfused ischemic heart and improve cardiac function during ischemic episodes in preclinical models. Biochim Biophys Acta Mol Basis Dis. https://doi.org/10.1016/j.bbadis.2020.165770,16577010.1016/j.bbadis.2020.165770
Anto Michel N, Ljubojevic-Holzer S, Bugger H, Zirlik A (2022) Cellular heterogeneity of the heart. Front Cardiovasc Med 9: 868466 doi:https://doi.org/10.3389/fcvm.2022.868466
Billah M, Ridiandries A, Rayner BS, Allahwala UK, Dona A, Khachigian LM, Bhindi R (2019) Egr-1 functions as a master switch regulator of remote ischemic preconditioning-induced cardioprotection. Basic Res Cardiol 115:3. https://doi.org/10.1007/s00395-019-0763-9
Boengler K, Buechert A, Heinen Y, Roeskes C, Hilfiker-Kleiner D, Heusch G, Schulz R (2008) Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res, 102: 131-135. doi: https://doi.org/10.1161/CIRCRESAHA.107.164699
Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R (2008) The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther 120:172–185. https://doi.org/10.1016/j.pharmthera.2008.08.002
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R (2010) Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105:771–785. https://doi.org/10.1007/s00395-010-0124-1
Boengler K, Lochnit G, Schulz R (2018) Mitochondria “THE” target of myocardial conditioning. Am J Physiol Heart Circ Physiol 315:H1215–H1231. https://doi.org/10.1152/ajpheart.00124.2018
Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R (2009) Presence of connexin 43 in subsarcolemmal but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol 104:141–147. https://doi.org/10.1007/s00395-009-0007-5
Boengler K, Ungefug E, Heusch G, Schulz R (2013) The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Curr Pharm Des 19:6890–6895
Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y, Xuan YT (2011) A murine model of inducible, cardiac-specific deletion of STAT3: Its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol 50:589–597. https://doi.org/10.1016/j.yjmcc.2011.01.002
Bonora M, Wieckowski MR, Sinclair DA, Kroemer G, Pinton P, Galluzzi L (2019) Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat Rev Cardiol 16:33–55. https://doi.org/10.1038/s41569-018-0074-0
Bøtker HE, Kharbanda R, Schmidt MR, Bøttcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sorensen HT, Redington AN, Nielsen TT (2010) Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375:727–734. https://doi.org/10.1016/S0140-6736(09)62001-8
Butterbach K, Beckmann L, de Sanjose S, Benavente Y, Becker N, Foretova L, Maynadie M, Cocco P, Staines A, Boffetta P, Brennan P, Nieters A (2011) Association of JAK-STAT pathway related genes with lymphoma risk: results of a European case-control study (EpiLymph). Br J Haematol 153:318–333. https://doi.org/10.1111/j.1365-2141.2011.08632.x
Candilio L, Malik A, Ariti C, Barnard M, Di Salvo C, Lawrence D, Hayward M, Yap J, Roberts N, Sheikh A, Kolvekar S, Hausenloy DJ, Yellon DM (2015) Effect of remote ischaemic preconditioning on clinical outcomes in patients undergoing cardiac bypass surgery: a randomised controlled clinical trial. Heart 10:185–192. https://doi.org/10.1136/heartjnl-2014-306178
Chandra Shekar K, Yannopoulos D, Kosmopoulos M, Riess ML (2022) Differential effects of reperfusion on cardiac mitochondrial subpopulations in a preclinical porcine model of acute myocardial infarction. Front Cell Dev Biol 10:843733. https://doi.org/10.3389/fcell.2022.843733
Chen H, Jing XY, Shen YJ, Wang TL, Ou C, Lu SF, Cai Y, Li Q, Chen X, Ding YJ, Yu XC, Zhu BM (2018) Stat5-dependent cardioprotection in late remote ischaemia preconditioning. Cardiovasc Res 114:679–689. https://doi.org/10.1093/cvr/cvy014
Chen S, Wang Q, Christodoulou A, Mylonas N, Bakker D, Nederlof R, Hollmann MW, Weber NC, Coronel R, Wakker V, Christoffels VM, Andreadou I, Zuurbier CJ (2023) Sodium glucose cotransporter-2 inhibitor empagliflozin reduces infarct size independently of sodium glucose cotransporter-2. Circulation 147:276–279. https://doi.org/10.1161/CIRCULATIONAHA.122.061688
Chen ZR, Hong Y, Wen SH, Zhan YQ, Huang WQ (2023) Dexmedetomidine pretreatment protects against myocardial ischemia/reperfusion injury by activating STAT3 signaling. Anesth Analg 137:426–439. https://doi.org/10.1213/ANE.0000000000006487
Cohen MV, Downey JM (2007) Cardioprotection: spotlight on PKG. Br J Pharmacol 152:833–834. https://doi.org/10.1038/sj.bjp.0707453
Comita S, Femmino S, Thairi C, Alloatti G, Boengler K, Pagliaro P, Penna C (2021) Regulation of STAT3 and its role in cardioprotection by conditioning: focus on non-genomic roles targeting mitochondrial function. Basic Res Cardiol 116:56. https://doi.org/10.1007/s00395-021-00898-0
Domingos AE, Seara FAC, Oliveira DF, Maciel L, Barbosa RAQ, Barcellos LC, Pinto VS, Fortunato RS, Nascimento JHM (2023) Mitochondrial dysfunction and cardiac ischemia/reperfusion injury are attenuated by nandrolone: role of JAK-STAT3 pathway. Steroids 197:109247. https://doi.org/10.1016/j.steroids.2023.109247
Eitel I, Stiermaier T, Rommel KP, Fuernau G, Sandri M, Mangner N, Linke A, Erbs S, Lurz P, Boudriot E, Mende M, Desch S, Schuler G, Thiele H (2015) Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: the randomized LIPSIA CONDITIONING trial. Eur Heart J 36:3049–3057. https://doi.org/10.1093/eurheartj/ehv463
Ferdinandy P, Andreadou I, Baxter GF, Bøtker HE, Davidson SM, Dobrev D, Gersh BJ, Heusch G, Lecour S, Ruiz-Meana M, Zuurbier CJ, Hausenloy DJ, Schulz R (2023) Interaction of cardiovascular nonmodifiable risk factors, comorbidities and comedications with ischemia/reperfusion injury and cardioprotection by pharmacological treatments and ischemic conditioning. Pharmacol Rev 75:159–216. https://doi.org/10.1124/pharmrev.121.000348
Ferrari R, Pedersini P, Bongrazio M, Gaia G, Bernocchi P, Di Lisa F, Visioli O (1993) Mitochondrial energy production and cation control in myocardial ischaemia and reperfusion. Basic Res Cardiol 88:495–512. https://doi.org/10.1007/BF00795415
Fischer P, Hilfiker-Kleiner D (2007) Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res Cardiol 102:393–411. https://doi.org/10.1007/s00395-007-0658-z
Fuglesteg BN, Suleman N, Tiron C, Kanhema T, Lacerda L, Andreasen TV, Sack MN, Jonassen AK, Mjos OD, Opie LH, Lecour S (2008) Signal transducer and activator of transcription 3 is involved in the cardioprotective signalling pathway activated by insulin therapy at reperfusion. Basic Res Cardiol 103:444–453. https://doi.org/10.1007/s00395-008-0728-x
Garcia-Ropero A, Santos-Gallego CG, Badimon JJ (2019) The anti-inflammatory effects of SGLT inhibitors. Aging (Albany NY) 11:5866–5867. https://doi.org/10.18632/aging.102175
Gaspar A, Lourenco AP, Pereira MA, Azevedo P, Roncon-Albuquerque R Jr, Marques J, Leite-Moreira AF (2018) Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Basic Res Cardiol 113:14. https://doi.org/10.1007/s00395-018-0672-3
Gent S, Skyschally A, Kleinbongard P, Heusch G (2017) lschemic preconditioning in pigs: a causal role for signal transducer and activator of transcription 3. Am J Physiol Heart Circ Physiol 312:H478–H484. https://doi.org/10.1152/ajpheart.00749.2016
Goodman MD, Koch SE, Afzal MR, Butler KL (2011) STAT subtype specificity and ischemic preconditioning in mice: is STAT-3 enough? Am J Physiol Heart Circ Physiol 300:H522–H526. https://doi.org/10.1152/ajpheart.00231.2010
Gustafsson AB, Gottlieb RA (2008) Heart mitochondria: gates of life and death. Cardiovasc Res 77:334–343. https://doi.org/10.1093/cvr/cvm005
Haghikia A, Ricke-Hoch M, Stapel B, Gorst I, Hilfiker-Kleiner D (2014) STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res 102:281–289. https://doi.org/10.1093/cvr/cvu034
Haghikia A, Stapel B, Hoch M, Hilfiker-Kleiner D (2011) STAT3 and cardiac remodeling. Heart Fail Rev 16:35–47. https://doi.org/10.1007/s10741-010-9170-x
Harhous Z, Badawi S, Bona NG, Pillot B, Augeul L, Paillard M, Booz GW, Canet-Soulas E, Ovize M, Kurdi M, Bidaux G (2019) Critical appraisal of STAT3 pattern in adult cardiomyocytes. J Mol Cell Cardiol 131:91–100. https://doi.org/10.1016/j.yjmcc.2019.04.021
Harhous Z, Booz GW, Ovize M, Bidaux G, Kurdi M (2019) An update on the multifaceted roles of STAT3 in the heart. Front Cardiovasc Med 6:150. https://doi.org/10.3389/fcvm.2019.00150
Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, Knight R, Kunst G, Laing C, Nicholas J, Pepper J, Robertson S, Xenou M, Clayton T, Yellon DM, Investigators ET (2015) Remote ischemic preconditioning and outcomes of cardiac surgery. N Engl J Med 373:1408–1417. https://doi.org/10.1056/NEJMoa1413534
Hausenloy DJ, Chilian W, Crea F, Davidson SM, Ferdinandy P, Garcia-Dorado D, van Royen N, Schulz R, Heusch G (2019) The coronary circulation in acute myocardial ischaemia/reperfusion injury - a target for cardioprotection. Cardiovasc Res 115:1143–1155. https://doi.org/10.1093/cvr/cvy286
Hausenloy DJ, Kharbanda RK, Møller UK, Ramlall M, Aarøe J, Butler R, Bulluck H, Clayton T, Dana A, Dodd M, Engstrom T, Evans R, Flensted Lassen J, Frischknecht Christensen E, Garcia-Ruiz JM, Gorog DA, Hjort J, Houghton RF, Ibanez B, Knight R, Lippert FK, Lønborg JT, Maeng M, Milasinovic D, More R, Nicholas JM, Okkels Jensen L, Perkins A, Radovanovic N, Rakhit RD, Ravkilde J, Ryding AD, Schmidt MR, SkogstadRiddervold I, Toft Sørensen H, Stankovic G, Varma M, Webb I, JuhlTerkelsen C, Greenwood JP, Yellon DM, Bøtker HE (2019) Effect of remote ischemic conditioning on clinical outcomes at 12 months in acute myocardial infarction patients: the CONDI-2/ERIC-PPCI trial. Lancet 394:1415–1424. https://doi.org/10.1016/S0140-6736(19)32039-2
Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, Ashley E, Vichare S, Di Salvo C, Kolvekar S, Hayward M, Keogh B, MacAllister RJ, Yellon DM (2007) Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomized controlled trial. Lancet 370:575–579. https://doi.org/10.1016/S0140-6736(07)61296-3
Hausenloy DJ, Yellon DM (2004) New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc Res 61:448–460. https://doi.org/10.1016/j.cardiores.2003.09.024
Heusch G (2013) Cardioprotection: chances and challenges of its translation to the clinic. Lancet 381:166–175. https://doi.org/10.1016/S0140-6736(12)60916-7
Heusch G (2015) Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 116:674–699. https://doi.org/10.1161/CIRCRESAHA.116.305348
Heusch G (2018) Cardioprotection research must leave its comfort zone. Eur Heart J 39:3393–3395. https://doi.org/10.1093/eurheartj/ehy253
Heusch G (2020) Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Heusch G (2023) Cardioprotection in cardio-oncology – a case for concern? Cardiovas Res cvad111. https://doi.org/10.1093/cvr/cvad111
Heusch G (2023) Cardioprotection and its translation: a need for new paradigms? Or for new pragmatism? An opinionated retro- and perspective. J Cardiovasc Pharmacol Ther. https://doi.org/10.1177/10742484231179613
Heusch G, Bøtker EH, Ferdinandy P, Schulz R (2023) Primordial non-responsiveness – a neglected obstacle to cardioprotection. Eur Heart J 44:1687–1689. https://doi.org/10.1093/eurheartj/ehad160
Heusch G, Gersh BJ (2016) ERICCA and RIPHeart: two nails in the coffin for cardioprotection by remote ischemic conditioning? Probably not! Eur Heart J 37:200–202. https://doi.org/10.1093/eurheartj/ehv606
Heusch G, Musiolik J, Gedik N, Skyschally A (2011) Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 109:1302–1308. https://doi.org/10.1161/CIRCRESAHA.111.255604
Heusch G, Musiolik J, Kottenberg E, Peters J, Jakob H, Thielmann M (2012) STAT5 activation and cardioprotection by remote ischemic preconditioning in humans. Circ Res 110:111–115. https://doi.org/10.1161/CIRCRESAHA.111.259556
Heusch G, Rassaf T (2016) Time to give up on cardioprotection? A critical appraisal of clinical studies on ischemic pre-, post-, and remote conditioning. Circ Res 119:676–695. https://doi.org/10.1161/CIRCRESAHA.116.308736
Heusch G, Rassaf T (2021) Protection from cardiotoxicity of cancer chemotherapy - a novel target for remote ischaemic conditioning? Cardiovasc Res 117:985–986. https://doi.org/10.1093/cvr/cvaa199
Heusch G, Skyschally A, Schulz R (2011) The in-situ pig heart with regional ischemia/reperfusion - ready for translation. J Mol Cell Cardiol 50:951–963. https://doi.org/10.1016/j.yjmcc.2011.02.016
Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, Forster O, Quint A, Landmesser U, Doerries C, Luchtefeld M, Poli V, Schneider MD, Balligand JL, Desjardins F, Ansari A, Struman I, Nguyen NQ, Zschemisch NH, Klein G, Heusch G, Schulz R, Hilfiker A, Drexler H (2007) A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 128:589–600. https://doi.org/10.1016/j.cell.2006.12.036
Holmuhamedov EL, Oberlin A, Short K, Terzic A, Jahangir A (2012) Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PLoS One 7:e44667. https://doi.org/10.1371/journal.pone.0044667
Hong DM, Jeon Y, Lee CS, Kim HJ, Lee JM, Bahk JH, Kim KB, Hwang HY (2012) Effects of remote ischemic preconditioning with postconditioning in patients undergoing off-pump coronary artery bypass surgery. Circ J 76:884–890. https://doi.org/10.1253/circj.CJ-11-1068
Kirkman DL, Robinson AT, Rossman MJ, Seals DR, Edwards DG (2021) Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am J Physiol Heart Circ Physiol 320:H2080–H2100. https://doi.org/10.1152/ajpheart.00917.2020
Kishore R, Verma SK (2012) Roles of STATs signaling in cardiovascular diseases. JAKSTAT 1:118–124. https://doi.org/10.4161/jkst.20115
Kleinbongard P, Amanakis G, Skyschally A, Heusch G (2018) Reflection of cardioprotection by remote ischemic perconditioning in attenuated ST-segment elevation during ongoing coronary occlusion in pigs: evidence for cardioprotection from ischemic injury. Circ Res 122:1102–1108. https://doi.org/10.1161/CIRCRESAHA.118.312784
Kleinbongard P, Bøtker HE, Ovize M, Hausenloy DJ, Heusch G (2020) Co-morbidities and co-medications as confounders of cardioprotection - does it matter in the clinical setting? Br J Pharmacol 177:5252–5269. https://doi.org/10.1111/bph.14839
Kleinbongard P, Gedik N, Kirca M, Stoian L, Frey U, Zandi A, Thielmann M, Jakob H, Peters J, Kamler M, Heusch G (2018) Mitochondrial and contractile function of human right atrial tissue in response to remote ischemic conditioning. J Am Heart Assoc 7:e009540. https://doi.org/10.1161/JAHA.118.009540
Kleinbongard P, Lieder H, Skyschally A, Heusch G (2023) No sex-related differences in infarct size, no-reflow and protection by ischaemic preconditioning in Göttingen minipigs. Cardiovasc Res 119:561–570. https://doi.org/10.1093/cvr/cvac062
Kleinbongard P, Lieder HR, Skyschally A, Alloosh M, Gödecke A, Rahmann S, Sturek M, Heusch G (2022) Non-responsiveness to cardioprotection by ischaemic preconditioning in Ossabaw minipigs with genetic predisposition to, but without the phenotype of the metabolic syndrome. Basic Res Cardiol 117:58. https://doi.org/10.1007/s00395-022-00965-0
Kleinbongard P, Peters J, Jakob H, Heusch G, Thielmann M (2018) Persistent survival benefit from remote ischemic preconditioning in patients undergoing coronary artery bypass surgery. J Am Coll Cardiol 71:251–262. https://doi.org/10.1016/j.jacc.2017.10.083
Kleinbongard P, Skyschally A, Gent S, Pesch M, Heusch G (2018) STAT3 as a common signal of ischemic conditioning: a lesson on “rigor and reproducibility” in preclinical studies on cardioprotection. Basic Res Cardiol, 113: 3. doi:https://doi.org/10.1007/s00395-017-0660-z
Kleinbongard P, Skyschally A, Heusch G (2017) Cardioprotection by remote ischemic conditioning and its signal transduction. Pflügers Arch - Eur J Physiol 469:159–181. https://doi.org/10.1007/s00424-016-1922-6
Koncsos G, Varga ZV, Baranyai T, Ferdinandy P, Schulz R, Giricz Z, Boengler K (2018) Nagarse treatment of cardiac subsarcolemmal and interfibrillar mitochondria leads to artefacts in mitochondrial protein quantification. J Pharmacol Toxicol Methods 91:50–58. https://doi.org/10.1016/j.vascn.2018.01.004
Lecour S (2009) Activation of the protective survivor activating factor enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK path? J Mol Cell Cardiol 47:32–40. https://doi.org/10.1016/j.yjmcc.2009.03.019
Lecour S, Andreadou I, Bøtker HE, Davidson SM, Heusch G, Ruiz-Meana M, Schulz R, Zuurbier CJ, Ferdinandy P, Hausenloy DJ, CA16225 obotEU-CCA (2021) IMproving Preclinical Assessment of Cardioprotective Therapies (IMPACT) criteria: guidelines of the EU-CARDIOPROTECTION COST action. Basic Res Cardiol, 116: 52, https://doi.org/10.1007/s00395-021-00893-5
Lieder HR, Kleinbongard P, Skyschally A, Hagelschuer H, Chilian WM, Heusch G (2018) Vago-splenic axis in signal transduction of remote ischemic preconditioning in pigs and rats. Circ Res 123:1152–1163. https://doi.org/10.1161/CIRCRESAHA.118.313859
Lieder HR, Skyschally A, Sturek M, Heusch G, Kleinbongard P (2022) Remote ischemic conditioning in Ossabaw minipigs induces the release of humoral cardioprotective triggers, but the myocardium does not respond with reduced infarct size. Am J Physiol Heart Circ Physiol 323:H1365–H1375. https://doi.org/10.1152/ajpheart.00580.2022
Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, Nadelmann ER, Roberts K, Tuck L, Fasouli ES, DeLaughter DM, McDonough B, Wakimoto H, Gorham JM, Samari S, Mahbubani KT, Saeb-Parsy K, Patone G, Boyle JJ, Zhang H, Zhang H, Viveiros A, Oudit GY, Bayraktar O, Seidman JG, Seidman CE, Noseda M, Hubner N, Teichmann SA (2020) Cells of the adult human heart. Nature 588:466–472. https://doi.org/10.1038/s41586-020-2797-4
Lother A, Kohl P (2023) The heterocellular heart: identities, interactions, and implications for cardiology. Basic Res Cardiol 118:30. https://doi.org/10.1007/s00395-023-01000-6
Lukyanenko V, Chikando A, Lederer WJ (2009) Mitochondria in cardiomyocyte Ca2+ signaling. Int J Biochem Cell Biol 41:1957–1971. https://doi.org/10.1016/j.biocel.2009.03.011
Meier JA, Larner AC (2014) Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 26:20–28. https://doi.org/10.1016/j.smim.2013.12.005
Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C, Coburn M, Schaelte G, Boning A, Niemann B, Roesner J, Kletzin F, Strouhal U, Reyher C, Laufenberg-Feldmann R, Ferner M, Brandes IF, Bauer M, Stehr SN, Kortgen A, Wittmann M, Baumgarten G, Meyer-Treschan T, Kienbaum P, Heringlake M, Schon J, Sander M, Treskatsch S, Smul T, Wolwender E, Schilling T, Fuernau G, Hasenclever D, Zacharowski K, Collaborators RIS (2015) A multicenter trial of remote ischemic preconditioning for heart surgery. N Engl J Med 373:1397–1407. https://doi.org/10.1056/NEJMoa1413579
Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D, Youle RJ, American Heart Association Council on Basic Cardiovascular Sciences CoCC, Council on Functional G, Translational B (2016) Mitochondrial function, biology, and role in disease: a scientific statement from the American heart association. Circ Res 118:1960–1991. https://doi.org/10.1161/RES.0000000000000104
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136. https://doi.org/10.1161/01.CIR.74.5.1124
Nikolaou PE, Efentakis P, Qourah FA, Femmino S, Makridakis M, Kanaki Z, Varela A, Tsoumani M, Davos CH, Dimitriou CA, Tasouli A, Dimitriadis G, Kostomitsopoulos N, Zuurbier CJ, Vlahou A, Klinakis A, Brizzi MF, Iliodromitis EK, Andreadou I (2021) Chronic empaglifozin treatment reduces myocardial infarct size in non-diabetic mice through STAT-3 mediated protection on microvascular endothelial cells and reduction of oxidative stress. Antioxid Redox Signal 34:551–571. https://doi.org/10.1089/ars.2019.7923
Nikolaou PE, Mylonas N, Makridakis M, Makrecka-Kuka M, Iliou A, Zerikiotis S, Efentakis P, Kampoukos S, Kostomitsopoulos N, Vilskersts R, Ikonomidis I, Lambadiari V, Zuurbier CJ, Latosinska A, Vlahou A, Dimitriadis G, Iliodromitis EK, Andreadou I (2022) Cardioprotection by selective SGLT-2 inhibitors in a non-diabetic mouse model of myocardial ischemia/reperfusion injury: a class or a drug effect? Basic Res Cardiol 117:27. https://doi.org/10.1007/s00395-022-00934-7
O’Sullivan KE, Breen EP, Gallagher HC, Buggy DJ, Hurley JP (2016) Understanding STAT3 signaling in cardiac ischemia. Basic Res Cardiol 111:27. https://doi.org/10.1007/s00395-016-0543-8
Palmer JW, Tandler B, Hoppel CL (1977) Biochemical properties of subsarcolemmal and inter-fibrillar mitochondria isolated from rat cardiac-muscle. J Biol Chem 252:8731–8739. https://doi.org/10.1016/S0021-9258(19)75283-1
Penna C, Perrelli MG, Tullio F, Angotti C, Camporeale A, Poli V, Pagliaro P (2013) Diazoxide postconditioning induces mitochondrial protein S-Nitrosylation and a redox-sensitive mitochondrial phosphorylation/translocation of RISK elements: no role for SAFE. Basic Res Cardiol 108:371. https://doi.org/10.1007/s00395-013-0371-z
Phillips D, Reilley MJ, Aponte AM, Wang G, Boja E, Gucek M, Balaban RS (2010) Stoichiometry of STAT3 and mitochondrial proteins: implications for the regulation of oxidative phosphorylation by protein-protein interactions. J Biol Chem 285:23532–23536. https://doi.org/10.1074/jbc.C110.152652
Qiu H, Lizano P, Laure L, Sui X, Rashed E, Park JY, Hong C, Gao S, Holle E, Morin D, Dhar SK, Wagner T, Berdeaux A, Tian B, Vatner SF, Depre C (2011) H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 124:406–415. https://doi.org/10.1161/CIRCULATIONAHA.110.013847
Rajab BS, Kassab S, Stonall CD, Daghistani H, Gibbons S, Mamas M, Smith D, Mironov A, AlBalawi Z, Zhang YH, Baudoin F, Zi M, Prehar S, Cartwright EJ, Kitmitto A (2022) Differential remodelling of mitochondrial subpopulations and mitochondrial dysfunction are a feature of early stage diabetes. Sci Rep 12:978. https://doi.org/10.1038/s41598-022-04929-1
Sawashita Y, Hirata N, Yoshikawa Y, Terada H, Tokinaga Y, Yamakage M (2020) Remote ischemic preconditioning reduces myocardial ischemia-reperfusion injury through unacylated ghrelin-induced activation of the JAK/STAT pathway. Basic Res Cardiol 115:50. https://doi.org/10.1007/s00395-020-0809-z
Saxena P, Aggarwal S, Misso NL, Passage J, Newman MA, Thompson PJ, d’Udekem Y, Praporski S, Konstantinov IE (2013) Remote ischaemic preconditioning down-regulates kinin receptor expression in neutrophils of patients undergoing heart surgery. Interact Cardiovasc Thorac Surg 17:653–658. https://doi.org/10.1093/icvts/ivt279
Schulz R, Andreadou I, Hausenloy DJ, Ferdinandy P (2020) Risk factors, co-morbidities, and co-medications in cardioprotection: importance for translation. Br J Pharmacol 177:5249–5251. https://doi.org/10.1111/bph.15294
Schulz R, Schlüter KD (2023) Importance of mitochondria in cardiac pathologies: focus on uncoupling proteins and monoamine oxidases. Int J Mol Sci 24:6459. https://doi.org/10.3390/ijms24076459
Skyschally A, Gent S, Amanakis G, Schulte C, Kleinbongard P, Heusch G (2015) Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by reperfusion injury salvage kinase and survival activating factor enhancement pathways. Circ Res 117:279–288. https://doi.org/10.1161/CIRCRESAHA.117.306878
Skyschally A, Kleinbongard P, Lieder HR, Gedik N, Stoian L, Amanakis G, Elbers E, Heusch G (2018) Humoral transfer and intra-myocardial signal transduction of protection by remote ischemic perconditioning in pigs, rats, and mice. Am J Physiol Heart Circ Physiol 315:H159–H172. https://doi.org/10.1152/ajpheart.00152.2018
Sloth AD, Schmidt MR, Munk K, Kharbanda RK, Redington AN, Schmidt M, Pedersen L, Sorensen HT, Bøtker HE (2014) Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J 35:168–175. https://doi.org/10.1093/eurheartj/eht369
Sturek M, Alloosh M, Sellke FW (2020) Swine disease models for optimal vascular engineering. Annu Rev Biomed Eng 22:25–49. https://doi.org/10.1146/annurev-bioeng-082919-053009
Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC (2011) Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem 286:29610–29620. https://doi.org/10.1074/jbc.M111.226209
Szczepanek K, Xu A, Hu Y, Thompson J, He J, Larner AC, Salloum FN, Chen Q, Lesnefsky EJ (2015) Cardioprotective function of mitochondrial-targeted and transcriptionally inactive STAT3 against ischemia and reperfusion injury. Basic Res Cardiol 110:53. https://doi.org/10.1007/s00395-015-0509-2
Tamareille S, Mateus V, Ghaboura N, Jeanneteau J, Croue A, Henrion D, Furber A, Prunier F (2011) RISK and SAFE signaling pathway interactions in remote limb ischemic perconditioning in combination with local ischemic postconditioning. Basic Res Cardiol 106:1329–1339. https://doi.org/10.1007/s00395-011-0210-z
Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NB (2013) The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem 288:4723–4732. https://doi.org/10.1074/jbc.M112.378984
Thielmann M, Kottenberg E, Kleinbongard P, Wendt D, Gedik N, Pasa S, Price V, Tsagakis K, Neuhäuser M, Peters J, Jakob H, Heusch G (2013) Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-centre randomised, double-blind, controlled trial. Lancet 382:597–604. https://doi.org/10.1016/S0140-6736(13)61450-6
Trask AJ, Katz PS, Kelly AP, Galantowicz ML, Cismowski MJ, West TA, Neeb ZP, Berwick ZC, Goodwill AG, Alloosh M, Tune JD, Sturek M, Lucchesi PA (2012) Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J Appl Physiol 113:1128–1140. https://doi.org/10.1152/japplphysiol.00604.2012
Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr., Akkad AD, Herndon CN, Arduini A, Papangeli I, Roselli C, Aguet F, Choi SH, Ardlie KG, Babadi M, Margulies KB, Stegmann CM, Ellinor PT (2020) Transcriptional and cellular diversity of the human heart. Circulation 142:466–482. https://doi.org/10.1161/CIRCULATIONAHA.119.045401
Venugopal V, Hausenloy DJ, Ludman A, Di Salvo C, Kolvekar S, Yap J, Lawrence D, Bognolo G, Yellon DM (2009) Remote ischaemic preconditioning reduces myocardial injury in patients undergoing cardiac surgery with cold blood cardioplegia: a randomised controlled trial. Heart 95:1567–1571. https://doi.org/10.1136/hrt.2008.155770
Vilahur G (2023) A primordial obstacle in cardioprotection: the answer resides in our genes. Cardiovasc Res 119:e122–e124. https://doi.org/10.1093/cvr/cvad034
von Lewinski D, Kolesnik E, Tripolt NJ, Pferschy PN, Benedikt M, Wallner M, Alber H, Berger R, Lichtenauer M, Saely CH, Moertl D, Auersperg P, Reiter C, Rieder T, Siller-Matula JM, Gager GM, Hasun M, Weidinger F, Pieber TR, Zechner PM, Herrmann M, Zirlik A, Holman RR, Oulhaj A, Sourij H (2022) Empagliflozin in acute myocardial infarction: the EMMY trial. Eur Heart J 43:4421–4432. https://doi.org/10.1093/eurheartj/ehac494
Wang M, Zhang W, Crisostomo P, Markel T, Meldrum KK, Fu XY, Meldrum DR (2007) Endothelial STAT3 plays a critical role in generalized myocardial proinflammatory and proapoptotic signaling. Am J Physiol Heart Circ Physiol 293:H2101-2108. https://doi.org/10.1152/ajpheart.00125.2007
Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323:793–797. https://doi.org/10.1126/science.1164551
White SK, Froehlich GM, Sado DM, Maestrini V, Fontana M, Treibel TA, Tehrani S, Flett AS, Meier P, Ariti C, Davies JR, Moon JC, Yellon DM, Hausenloy DJ (2015) Remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol Cardiovasc Interv 8:178–188. https://doi.org/10.1016/j.jcin.2014.05.015
Wu L, Tan JL, Chen ZY, Huang G (2019) Cardioprotection of post-ischemic moderate ROS against ischemia/reperfusion via STAT3-induced the inhibition of MCU opening. Basic Res Cardiol 114:39. https://doi.org/10.1007/s00395-019-0747-9
Wu L, Tan JL, Wang ZH, Chen YX, Gao L, Liu JL, Shi YH, Endoh M, Yang HT (2015) ROS generated during early reperfusion contribute to intermittent hypobaric hypoxia-afforded cardioprotection against postischemia-induced Ca(2+) overload and contractile dysfunction via the JAK2/STAT3 pathway. J Mol Cell Cardiol 81:150–161. https://doi.org/10.1016/j.yjmcc.2015.02.015
Wu Q, Wang T, Chen S, Zhou Q, Li H, Hu N, Feng Y, Dong N, Yao S, Xia Z (2018) Cardiac protective effects of remote ischaemic preconditioning in children undergoing tetralogy of fallot repair surgery: a randomized controlled trial. Eur Heart J 39:1028–1037. https://doi.org/10.1093/eurheart/ehx030
Xia T, Zhang M, Lei W, Yang R, Fu S, Fan Z, Yang Y, Zhang T (2023) Advances in the role of STAT3 in macrophage polarization. Front Immunol 14:1160719. https://doi.org/10.3389/fimmu.2023.1160719
Xuan Y-T, Guo Y, Zhu Y, Wang O-L, Rokosh G, Messing RO, Bolli R (2005) Role of the protein kinase C-e-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation 112:1971–1978. https://doi.org/10.1161/CIRCULATIONAHA.105.561522
Yamaura G, Turoczi T, Yamamoto F, Siddqui MA, Maulik N, Das DK (2003) STAT signaling in ischemic heart: a role of STAT5A in ischemic preconditioning. Am J Physiol Heart Circ Physiol 285:H476–H482. https://doi.org/10.1152/ajpheart.00079.2003
Yellon DM, Ackbarkhan AK, Balgobin V, Bulluck H, Deelchand A, Dhuny MR, Domah N, Gaoneadry D, Jagessur RK, Joonas N, Kowlessur S, Lutchoo J, Nicholas JM, Pauvaday K, Shamloll O, Walker JM, Hausenloy DJ (2015) Remote ischemic conditioning reduces myocardial infarct size in STEMI patients treated by thrombolysis. J Am Coll Cardiol 65:2764–2765. https://doi.org/10.1016/j.jacc.2015.02.082
Yellon DM, BeikoghliKalkhoran S, Davidson SM (2023) The RISK pathway leading to mitochondria and cardioprotection: how everything started. Basic Res Cardiol 118:22. https://doi.org/10.1007/s00395-023-00992-5
Zgheib C, Zouein FA, Kurdi M, Booz GW (2012) Differential STAT3 signaling in the heart: impact of concurrent signals and oxidative stress. JAKSTAT 1:101–110. https://doi.org/10.4161/jkst.19776
Zhang Y, Fan G, Liu X, Skovgaard K, Sturek M, Heegaard PMH (2021) The genome of the naturally evolved obesity-prone Ossabaw miniature pig. iScience 24:103081. https://doi.org/10.1016/j.isci.2021.103081
Zhao Z-Q, Corvera JS, Halkos ME, Kerendi F, Wang N-P, Guyton RA, Vinten-Johansen J (2003) Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 285:H579–H588. https://doi.org/10.1152/ajpheart.01069.2002
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the German Research Foundation [SFB 1116 B8] and the European Union COST ACTION [IG16225].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Additional information
This article is part of the topical collection “Mitochondria at the heart of cardioprotection”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kleinbongard, P. Perspective: mitochondrial STAT3 in cardioprotection. Basic Res Cardiol 118, 32 (2023). https://doi.org/10.1007/s00395-023-01003-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-023-01003-3