
Abstract
Multiple tools have been used to determine the sensitivity of phosphates from the early Archaean Barberton greenstone belt to transformation. The assessment of the degree of transformation is crucial for verifying data about the parameters of the paleo-environment. From the obtained results, three generations of phosphates can be distinguished. Group A is observed in cherts and banded iron formation BIF early-generation fluor-hydroxyapatite that precipitated from seawater. It is characterized by flat rare earth element (REE) patterns with a positive Eu anomaly and high Y/Ho ratio in the range of 54–70. Apatites in this group lack any visible indicators of secondary alterations at the micro- and nanoscales. Fourier transform infrared spectra indicate that these apatites are relatively rich in water, and, due to cationic substitution, their OH-stretching regions exhibit complex ordering and numerous component bands. The characteristics observed in the cherts and silicified felsic volcaniclastics of group B imply advanced metasomatic alteration. They exhibit light and heavy REE depletion and an absence of water in the halogen site. Nanoscale investigations reveal cracks, pores, nanofluid inclusions and nanochannel-like structures, as well as inclusions. Group C is represented by igneous-derived apatites that partially reflect their igneous origin. The phosphates are predominantly fluorapatite with typical magmatic apatite REE distribution patterns. Imaging at the micro- and nanoscales indicates that they partially preserve the signature of igneous origin. It seems that some of the analyzed apatite partially preserved their primordial features; therefore, they might be used for the reconstruction of Archaean abiotic systems.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The calcium phosphate mineral apatite, with the general formula Ca10(PO4)6(F,Cl,OH)2, occurs in a wide range of geologic and biologic settings on Earth. Due to its ubiquity and ability to incorporate water, halogens, S, C, and trace elements including Sr, U, Th, and the rare earth elements (REEs), apatite is a powerful indicator of primary formation and transformation processes. Phosphate of sedimentary origin precipitates from seawater via organic matter sedimentation and by scavenging onto iron–manganese oxide particles (Filippelli 2008). Apatite of igneous origin crystallizes from melts (Webster and Piccoli 2015), whereas metasomatic apatite is formed due to interaction with fluids (Harlov 2015).
Many papers have studied phosphates of Precambrian age (e.g., Mojzsis et al. 1996; Lepland et al. 2002, 2013; Nishizawa et al. 2004, 2005; Blake et al. 2010; Joosu et al. 2015, 2016; Alibert 2016; Wudarska et al. 2018) with the scope to deduce their primary formation conditions. In the oldest known supracrustal rocks from the Isua Supracrustal Belt (≈ 3.7 Ga) (Moorbath et al. 1973; Nutman et al. 1997), metamorphism (e.g., Rosing et al. 1996; Rollinson 2003) caused obliteration of primordial isotopic signatures (Wudarska et al. 2018), whereas REE signatures have been partially preserved (Lepland et al. 2002; Wudarska et al. 2018). In contrast, the Barberton greenstone belt (BGB) in southern Africa (3.55–3.22 Ga), one of the oldest well-preserved volcanic and sedimentary sequences on Earth, has been metamorphosed to relatively low grades. The carbonaceous material in these rocks has been interpreted as being of biological origin and may contain the oldest recognized microfossils (e.g., Walsh 1992; Walsh and Lowe 1999; Westall et al. 2001; Javaux 2006; Hickman-Lewis et al. 2018). These unique features make the rocks of the BGB ideal candidates to investigate apatite formation conditions during the early Archean. Because apatites/phosphates show some sensitivity to transformation, we try on the basis of their composition and structure to determine the degree of their susceptibility to secondary changes.
The objective of our study is to use a set of compositional and structural data of apatite for the reconstruction of Archaean phosphate formation environments, including the behavior of volatiles. It is an attempt to analyze and describe features that are characteristic of primary phosphates of sedimentary and igneous origin not influenced by secondary processes and of phosphates that were metasomatically transformed, e.g., overprinted by interaction with fluids. Most of the samples in our research were previously analyzed for δ18Ophosphate by Blake et al. (2010). Special emphasis was placed on the volatile compositions and concentrations as their positions in the crystal structures (electron probe microanalyzer—EPMA, Fourier transform infrared spectroscopy—FTIR). The structure and its transformation were investigated by transmission electron microscopy (TEM), and an auxiliary tool was cathodoluminescence (CL). Paleoproterozoic apatite from the Zaonega formation is used as a reference material, giving insight into the relationship between the primary/secondary signatures resulting from the formation/transformation of the crystals (Joosu et al. 2015, 2016).
Geological setting
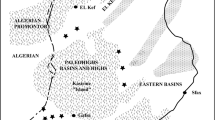
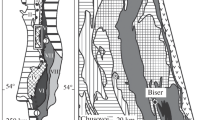
The BGB is located in the Kapvaal Craton of southern Africa (Fig. 1), one of the oldest pieces of the continental crust. The BGB consists of a NE–SW-striking succession of Archaean supracrustal rocks, termed the Swaziland Supergroup, which is surrounded and intruded by granitoids (Viljoen and Viljoen 1969; de Ronde and de Wit 1994; Lowe and Byerly 2007). The Swaziland Supergroup is subdivided into three stratigraphic units, the Onverwacht, Fig Tree and Moodies groups (Fig. 2). These rock units were tightly folded into a number of synclines that are separated by narrow anticlines or shear zones. The metamorphic grade is greenschist facies but locally reaches amphibolite facies close to contact with the surrounding granitoid domes that intruded during several episodes of magmatism at 3.45, 3.22, and 3.1 Ga (Kamo and Davis 1994; Dziggel et al. 2002). The Onverwacht Group formed between 3.55 and 3.26 Ga (Kröner et al. 2016; Byerly et al. 2018) and consists of ultramafic and mafic volcanic rocks (komatiites, komatiitic basalts, and basalts), with minor dacitic volcanic and sedimentary rocks that formed in a deep-to-shallow-marine environment (Fig. 2). The Onverwacht Group is subdivided into the Sandspruit, Theespruit, Komati, Hooggenoeg, Kromberg and Mendon formations (Fig. 1) (Viljoen and Viljoen 1969; Lowe and Byerly 2007). The Sandspruit and Theespruit Formations are separated from the overlying units by a major fault zone, but the younger units are stratigraphically conformable. Thrust faults that were active during and following the deposition of the Moodies Group are common in the uppermost sedimentary package. The Fig Tree Group, 3.26–3.23 Ga, consists of a several kilometer thick siliciclastic sedimentary sequence that is capped by felsic volcanic rocks (Hofmann 2005). In the northern part of the belt, the Fig Tree Group is characterized by turbiditic sandstone and shale interbeds that formed in a relatively deep-water environment (Hofmann 2005). In the southern part of the belt, a variety of lithofacies, including shale, graywacke, banded iron formation (BIF), conglomerate, and barite, occur that formed in a deep-to-shallow-water to alluvial environment (Fig. 2). The Moodies Group was probably deposited close to 3.227 Ga but not later than 3.11 Ga (Kamo and Davis 1994) and consists of shallow-marine to fluvial sandstone and conglomerate with minor shale and BIF.

General geological map of the southwestern part of the Barberton greenstone belt (BGB), South Africa and Swaziland, adopted form Blake et al. (2010). The red dots indicate the sample locations

Generalized stratigraphy and geochronology of the rocks in the upper part of the Onverwacht Group and Fig Tree Group in the southern part of BGB, modified after Knauth and Lowe (2003)
In this study, we analyzed some of the samples of Blake et al. (2010) from sedimentary and felsic volcaniclastic units of the Kromberg and Mendon Formations and the Fig Tree Group as well as two additional samples of metachert and dacite-derived sandstone from the Hooggenoeg Formation.
Sample description
The main idea of our sampling was to reflect the character of the sedimentary and felsic units of the BGB and their petrologic and geochemical characteristics. Therefore, the analyzed samples cover all the complexity and diversity of the upper part of the BGB (Table 1; Figs. 1, 2). We selected six samples from units within the Hooggenoeg, Kromberg and Mendon formations of the Onverwacht Group and the Fig Tree Group (Fig. 2) that were visually the most representative, free of secondary alterations and therefore able to preserve the primordial signature of the environment of their origin.
Sample AL03-28I was collected from the Footbridge Chert in the Kromberg Formation (Blake et al. 2010) and is a carbonaceous metachert. Quartz is the major phase in this sample, and the minor phases are represented mostly by Fe-oxides, i.e., hematite and magnetite (Fig. 3). Locally, in iron-enriched layers, Fe-oxides became the major phase. In addition, calcite, dolomite and pyrite are present. Apatites are very rare, highly porous and small (< 30 µm) in the analyzed sample (Table 1; Fig. 3).

Backscattered electron images of the metachert (AL03-28I, AL03-14A, and H12), igneous-derived units (H30 and AL03-29B), and BIF (AL03-26B) samples collected from the BGB
Sample AL03-14A is carbonaceous metachert from the Buck Reef Chert in the Kromberg Formation (Blake et al. 2010). It is predominantly composed of quartz. Accessory minerals are represented by Fe-oxides (hematite and magnetite), carbonates (calcite and dolomite) and pyrite (Fig. 3). Furthermore, the sample is characterized by a relatively high concentration of organic matter and/or organic carbon (Blake et al. 2010). The apatites observed in sample AL03-14A are also very scarce and small (< 30 µm) (Table 1; Fig. 3).
Sample H12 is a black-banded metachert from the Hooggenoeg formation (Hofmann et al. 2013). Quartz is the major phase in this sample. Hematite, together with some magnetite, is the most common Fe-oxide (Fig. 3). Rare carbonates are typically represented by an Fe-rich phase (siderite) that contains minor Mg, Mn, and Ca, but dolomite, rhodochrosite, and calcite are also present. Chlorite is the secondary phase and occurs in contact with cross-cutting veins (Fig. 3). In sample H12, apatite crystals are associated with a system of veins and are relatively small (< 50 µm) (Table 1; Fig. 3).
Sample AL03-29B is silicified felsic volcaniclastics from the Mendon Formation (Blake et al. 2010) and contains mostly quartz. Chlorite is a minor compound and occurs in cross-cutting veins (Fig. 3). These veins are a clear indication of secondary hydrothermal/metamorphic overprinting in this sample. The accessory phases are represented by magnetite, pyrite, siderite, dolomite, rhodochrosite, calcite, titanite, and zircon. The apatite crystals in sample AL03-29B are relatively large and more common (up to 100 µm) and are observed in the cross-cutting chlorite veins in this sample (Table 1; Fig. 3).
Sample H30 represents dacite-derived, silicified sandstone (i.e., the erosional product of underlying dacitic volcanic rocks) from the Hooggenoeg Formation of the Onverwacht Group (Hofmann and Harris 2008). Quartz and siderite are the major phases in this sample, and hematite, dolomite, rhodochrosite, and calcite are also present (Fig. 3). In addition, some alteration minerals are observed. Sericite has developed after feldspars and is a principal Al carrier phase in sample H30. The accessory mineral assemblage includes pyrite and titanite, as well as monazite and zircon. In sample H30, the apatite crystals are randomly scattered and are up to 100 µm in size, but mainly < 50 µm (Table 1; Fig. 3).
Sample AL03-26B is a BIF from the Fig Tree Group (Blake et al. 2010). It is composed of intercalated Fe-oxide- and quartz-rich layers. Hematite, together with some magnetite, is the most common Fe-oxide, with minor goethite due to recent weathering. The minor phases are carbonates (calcite, dolomite and rhodochrosite) and pyrite. Apatite is rare in the analyzed sample but is of a size up to 50 µm and was observed in association with Fe-rich layers (Table 1; Fig. 3).
Analytical approach
Sample preparation
The apatite crystals for the Fourier transform infrared (FTIR) spectroscopy were extracted from the rock samples by multistage mineral separation: the rock fragments were crushed and sieved to obtain a fraction less than 315 µm, and the finest fraction was removed by careful and repeated washing; then, the samples underwent magnetic separation, heavy liquid (tetra-bromoethane and diiodomethane) separation and careful hand-picking under a binocular microscope. The samples were too small to orient them crystallographically, and so randomly oriented apatite crystals were polished on two opposite sides to make the two surfaces parallel. The sample thicknesses were determined using the eyepiece reticule and stage micrometer scale of an optical microscope. Based on our calibration, this procedure has an uncertainty of ± 5%. The final thickness of the doubly polished crystals varied between 20 and 40 µm. Petrographic thick sections (approx. 300 µm thick) were used for the REE measurements, whereas the EMP analyses of the major and trace elements, CL observations, and preparation using FIB-milled foils for the transmission electron microscope (TEM) analyses were performed on the same crystals in petrographic-thin sections.
Cathodoluminescence (CL)
CL measurements were performed at the Institute of Mineralogy of the TU Bergakademie Freiberg on carbon-coated, polished thin sections using a “hot cathode” CL microscope HC1-LM (cf., Neuser et al. 1995). The system was operated at a 14 kV accelerating voltage and a current of 0.2 mA (current density of approximately 10 µA/mm2). Luminescence images were captured “on-line” during CL operations using a Peltier-cooled digital video camera (OLYMPUS DP72). CL spectra in the wavelength range of 380–1000 nm were recorded with an Acton Research SP-2356 digital triple-grating spectrograph with a Princeton Spec-10 CCD detector that was attached to the CL microscope by a silica–glass fiber guide. CL spectra were measured under standardized conditions (wavelength calibration by a Hg-halogen lamp, spot width of 30 µm, and measuring time of 1 s).
Transmission electron microscope (TEM) observations
TEM analyses were conducted to determine the phosphate structure and possible presence of REE-phosphate, graphite, sulfide and/or other inclusions. Sample preparation was accomplished using focused ion beam (FIB) milling. The FIB–TEM analyses were performed at the German Research Centre for Geosciences (GFZ) in Potsdam, Germany. Areas of 20 × 20 µm were selected in each sample for analysis. First, the samples were coated with a 1.5 µm protective layer of Pt to prevent sample damage caused by FIB sputtering and Ga-ion implantation. Then, material was sputtered out from the frontal and lateral sides of the region with the FIB operating at an accelerating voltage of 30 kV and a beam current of 30 nA. The frontal surface was then “polished” with a 3-nA FIB beam current. The final size of the TEM-foils cut directly from the grains in the thin sections was approximately 20 × 10 × 0.15 µm. The TEM-ready foils were placed on a perforated carbon film on a copper grid. No carbon coating was required to prevent charging under the electron beam. A detailed description of the sample preparation was published by Wirth (2004, 2009). The TEM observations were performed using an FEI Tecnai G2 F20 X-Twin TEM with a Schottky field emitter as an electron source. The TEM was equipped with a Fishione high-angle annular dark field detector (HAADF), an EDAX X-ray analyzer and a Gatan electron energy-loss spectrometer (EELS). High-resolution lattice fringe images were used to calculate the diffraction pattern (fast Fourier transform, FFT) of the host apatite as well as solid inclusions within its structure and subsequently identify them. The observed d-spacings and angles between adjacent planes were compared with the d-spacings and angles calculated from literature data. The error in the observed d-spacings is within the third decimal place of the measurement. The angles between adjacent planes must match within 1° for positive phase identification. The error in the angular measurements from the FFT was < 0.5°.
Electron microprobe analysis (EMPA)
The quantitative chemical analysis of the apatite grains was performed using a JEOL SuperProbe JXA-8230 microprobe at the Laboratory of Critical Elements AGH-KGHM (Cracow, Poland). The samples were coated with a 20 nm thick carbon film. To minimize the known potential problems associated with the EMP analysis of F (predominantly) and Cl in apatite, i.e., crystallographic orientation relationship and apatite F and Cl X-ray intensities driven by the diffusion of halogen ions due to primary electron beam operation (Stormer et al. 1993; Pyle et al. 2002), the samples were analyzed using two protocols. The first protocol, proposed by Słaby et al. (2016) and Wudarska et al. (2018), consisted of an acceleration voltage of 15 kV, a beam current of 5 nA and a spot size of 8 µm. The following natural and synthetic standards, corresponding spectral lines, peak counting times and used crystals were applied: fluorite (F Kα, 10 s, LDE1), albite (Na Kα, 20 s, TAPH), fluorapatite (Ca Kα, 10 s, PETJ and P Kα, 20 s, PETL), YPO4 (Y Lα, 10 s, PETJ), CePO4 (Ce Lα, 20 s, LIFH), tugtupite (Cl Kα, 10 s, PETH), fayalite (Fe Kα, 10 s, LIFL) and kyanite (Al Kα, 20 s, TAPH). Tests have shown that the detection limits obtained with this protocol are relatively high, and we were therefore unable to track the concentrations of the trace elements in the apatite structure and to estimate the mean formula of the apatite. Additionally, the earlier-mentioned halogen diffusion was undetectable using this protocol. The second protocol consisted of an acceleration voltage of 15 kV, a beam current of 20 nA and a spot size of 5 µm. The natural and synthetic standards, corresponding spectral lines, peak counting times and used crystals were as follows: fluorite (F Kα, 10 s, LDE1), albite (Si Kα, 20 s, TAP and Na Kα, 20 s), SrSO4 (Sr Lα, 20 s, TAP), fluorapatite (Ca Kα, 10 s, PETH and P Kα, 20 s, PETH), anhydrite (S Kα, 20 s, PETJ), YPO4 (Y Lα, 10 s, PETJ), CePO4 (Ce Lα, 20 s, LIFH), barite (Ba Lα, 10 s, PETJ), tugtupite (Cl Kα, 10 s, PETL), fayalite (Fe Kα, 10 s, LIFL), rhodonite (Mn Kα, 10 s, LIFL), kyanite (Al Kα, 20 s, TAP), diopside (Mg Kα, 20 s, TAP), LaPO4 (La Lα, 20 s, LIF), NdPO4 (Nd Lα, 20 s, LIFL) and GdPO4 (Gd Lα, 20 s, LIFH). The apatite formulae were calculated based on the EMPA data using the method by Ketcham (2015) and were normalized based on 25 oxygen equivalents. The concentrations of all components were compared to the detection limits (DL) of each element, and only those results above the DL were taken into consideration. The OH content was calculated with the assumption that the X-site is fully occupied by F, Cl and OH (F + Cl + OH = 2).
Laser ablation inductively coupled plasma mass spectrometry (LA–ICP–MS)
For trace element analyses, LA–ICP–MS was carried out at the Geochemisches Institute, University of Göttingen, using an Element 2 (Thermo-Fischer) instrument combined with a RESOlution Laser (193 nm ArF Excimer) by Resonetics equipped with the Laurin S-155 sample cell. Single measurements with a spot size of 23 µm were conducted on the apatite crystals in low mass resolution mode, and ablation was performed in a helium atmosphere. The total measurement time was typically 100 s: the signal was collected for 35 s from the target mineral, and the measurements started after 40 s of prewarming and ended after 20 s of delay. This protocol was intended to avoid peaks that do not reveal the actual concentrations. The laser was operated at a pulse energy of 80 mJ, a fluence of 2–3 J/cm2 and a repetition rate of 7 Hz. The measured intensities were transformed into concentrations by applying an external calibration with NIST 610 and 612 glass reference materials. The calcium concentration was used as an internal standard. The mass spectra were visualized with Iolite v2.1528, a noncommercial freeware solution implemented as a self-contained package running in the Igor Pro v6.34A (Wavemetrics Incorporated, Portland, Oregon, USA) environment. The concentration values were obtained by integrating the results over defined time intervals. The rare earth element (REE) distribution profiles were normalized to chondritic abundances using values given by Evensen et al. (1978).
Fourier transform infrared (FTIR) spectroscopy
Unpolarized FTIR spectra across the OH-stretching range were collected using a Bruker Hyperion 2000 IR-microscope attached to a Vertex 80v FTIR-spectrometer at the German Research Centre for Geosciences (GFZ) in Potsdam, Germany. We used a CaF2 beamsplitter and an InSb detector and placed the crystals on KBr pellets. The reference spectra were taken next to the crystals through the KBr. The spectra were averaged over 512 scans with a spectral resolution of 2/cm. The peak separation and peak area Ai (/cm) were fitted using PeakFit software, and the bands were assigned according to Tacker (2004). Using unpolarized spectra, we can only estimate the water content, as the quantification of the water content by FTIR spectroscopy requires the measurement of polarized spectra on two orthogonal sections oriented parallel to each of two axes of the indicatrix ellipsoid (Libowitzky and Rossman 1996). We averaged the thickness-normalized integrated absorbance values measured on several crystals of each sample and multiplied the average by 2 to get the total integrated absorbance values. This procedure is based on the fact that the OH-vector in apatite is aligned parallel the crystallographic c axis, and the OH-bands yield maximum absorbance in the spectra parallel to the c axis and zero absorbance perpendicular. Multiplying the absorbance values of the unpolarized spectra by 3 as done for biaxial crystals (see discussion in Qiu et al. 2018) would overestimate the water content. To estimate the hydrogen content, we applied the Beer–Lambert law (with a density for apatite of 3.1 g cm3) and used the absorption coefficient as determined by Wang et al. (2011). Based on our experience with polarized and unpolarized measurements, this procedure gives the water content with an uncertainty of ± 30%.
Results
Metacherts (samples AL03-28I, AL03-14A and H12)
CL data
The apatite from the metacherts show a wide range of luminescence values. For sample AL03-28I, the apatite shows weak, patchy greenish luminescence in the core and very strong orange luminescence in the narrow rim (Fig. 4f). The luminescence is activated by manganese (Fig. 4c). The inhomogeneity is not visible under the polarizing microscope and is most likely related to secondary alterations. Sample AL03-14A shows weak luminescence (Fig. 5g), yet it is violet and activated by REE (Dy3+, Tb3+, Sm3+, Nd3+, and Eu3+) substitution. The collected CL spectrum shows very strong and sharp REE3+ emission bands (Fig. 5c). The CL images of sample H12 reveal that the apatite crystals are generally patchy, with orange-brownish luminescence (Fig. 6f) related to the presence of REEs (Dy3+, Tb3+, Sm3+, Nd3+, and Eu3+) and dominant Mn2+ emission (Fig. 6c).

Data collected for sample AL03-28I. a Results of electron microprobe measurements of apatite plotted in F-OH-Cl ternary diagram representing mole fraction occupancy of the X-site. b chondrite-normalized (Evensen et al. 1978) rare earth elements distribution patterns. Red line corresponds to REE distribution pattern of Archaean sedimentary apatite (Lepland et al. 2002). c CL spectrum of apatite in Fig. 4f. d HAADF image of apatite. Highly elongated and preferentially oriented pyrrhotite inclusions are marked (black arrow). e HAADF image of a rutile (Rt), carbonate minerals (Cb) and pyrrhotite (Po) inclusions in apatite. Furthermore, nano-size pores are visible (black arrow). f CL image of apatite. g FTIR spectrum

Data collected for sample AL03-14A. a Results of electron microprobe measurements of apatite plotted in F–OH–Cl ternary diagram representing mole fraction occupancy of the X-site. b chondrite-normalized (Evensen et al. 1978) rare earth elements distribution patterns. Red line corresponds to REE distribution pattern of Archaean sedimentary apatite (Lepland et al. 2002). c CL spectrum of apatite in g. d HAADF image of highly elongated and preferentially oriented pyrrhotite inclusions (black arrow) in host apatite. e EDX analysis of pyrrhotite inclusions. Interference from host apatite is visible as Ca and P peaks. f HREM image of pyrrhotite (Po) inclusions in host apatite (Ap). g CL image of apatite in sample AL03-14A. h FTIR spectrum

Data collected for sample H12. a Results of electron microprobe measurements of apatite plotted in F–OH–Cl ternary diagram representing mole fraction occupancy of the X-site. b chondrite-normalized (Evensen et al. 1978) rare earth elements distribution patterns. Red line corresponds to REE distribution pattern of Archaean sedimentary apatite (Lepland et al. 2002). c CL spectrum of apatite in f. d HAADF image of porosity in apatite. Low-angle grain boundaries are marked with black arrows. e Bright field image of nano-size inclusion rich zone. f CL image of apatite in sample H12. g FTIR spectrum
TEM analysis
Three foils (no. 4332, sample AL03-28I; no. 4803, sample AL03-14A; and no. 4831, sample H12) were cut for the TEM analysis. Zones with abundant nanopores were observed in all three samples (Figs. 4d, e, 5d, 6d, e). Moreover, highly elongated iron sulfide inclusions were observed in samples AL03-28I and AL03-14A (Fig. 4d, e and 5d–f). The diffraction patterns (FFT) from the high-resolution lattice fringe image of sample AL03-28I were used to determine the structural parameters (dhkl) of the inclusions and to identify the iron sulfide phase. The structural parameters indicate that the iron sulfide phase is pyrrhotite (Figs. 4e, 5d, f). Furthermore, silica, rutile and carbonate mineral inclusions have been identified (Fig. 4e).
In sample H12, the apatite is composed of slightly misoriented subgrains that show individual diffraction contrast patterns when tilting the foil in the TEM. The subgrains are separated by arrays of dislocations that form low-angle grain boundaries (Fig. 6d). Some crystal parts are porous (Fig. 6d), whereas others are rich in nanosize inclusions (Fig. 6e). The presence of highly elongated CaYP7O20 inclusions, with lengths of up to 300 nm and widths of approximately 15 nm, was identified. A more detailed description can be found in Birski et al. (2018).
Electron microprobe analysis
Apatite crystals were analyzed in three metachert samples (AL03-28I, AL03-14A and H12). The EMP data indicate that the apatite from metacherts can be divided into two groups with different volatile compositions. The first group (samples AL03-28I and AL03-14A; Figs. 4a, 5a) is fluorapatite with a significant but variable amount of OH, and the second group (sample H12) is fluorapatite (Fig. 6a). In general, both groups are Cl-poor varieties. The OH-rich fluorapatite contains substantial concentrations of S (0.28–1.11 wt%), Si (up to 1.02 wt%), Y and REEs (up to 1.72 wt%), whereas the fluorapatite is generally depleted in all trace elements (Supplementary Tables S1 and S2). The calculated OH content (assuming that the X-site is completely filled, i.e., F + Cl + OH = 2) is in the range of 30.4–40.6 mol% for sample AL03-28I, 13.9–28.0 mol% for sample AL03-14A and up to 5.7 mol% for sample H12. This is equal to 0.53–0.71, 0.24–0.49 and up to 0.10 wt% H2O, respectively (Table 2).
Trace element composition
The REE patterns of the apatite from sample AL03-28I are flat, and europium shows a distinct positive anomaly (Fig. 4b). In addition, the AL03-14A REE pattern is relatively flat and characterized by a positive Eu, yet the anomaly is not as distinctive as that in sample AL03-28I (Fig. 5b). Furthermore, in contrast to AL03-28I, some crystals show low LREE values. The REE patterns of both samples do not differ from those considered as sedimentary (Lepland et al. 2002; Wudarska et al. 2018).
Based on the abundance of REEs, two populations (A and B) of apatite can be distinguished in H12 (Fig. 6b). Both populations of apatite are LREE-depleted and are characterized by similar LREE concentrations. They show increasing amounts of MREEs without any distinct anomaly. One of the patterns from population B displays a weak, positive Sm anomaly. Furthermore, the apatite grains from population B are more HREE-depleted than the apatite grains from population A. Depletion in both LREEs and HREEs is typical for metasomatic apatite (e.g., Lepland et al. 2002; Wudarska et al. 2018). The Y-Ho ratios for samples AL03-28I and AL03-14A have ranges of 54–58 and 64–66, respectively, whereas sample H12 has a low Y-Ho ratio in the range of 25–52. No differences in the Y-Ho ratio have been observed between the A and B populations. The ratio values for samples AL03-28I and AL03-14A are characteristic of modern seawater (Bau and Dulski 1994), whereas the values for H12 have a broad range between the chondrite and modern seawater values (Bau and Möller 1992; Bau and Dulski 1994). A full data set for samples AL03-28I, AL03-14A and H12 is included in Supplementary Table S3.
FTIR spectroscopy
The FTIR spectra collected from the metachert samples (Figs. 4g, 5g, 6g) show prominent OHF bands (Tacker 2004) at 3538/cm, 3538/cm and 3545/cm in samples AL03-28I, AL03-14A and H12, respectively (Table 3). Additionally, in all three samples, bands indexed as OHOH peaks were observed, and their positions are 3572/cm, 3572/cm and 3576/cm, respectively (Table 3). No additional peaks were observed in sample H12 (Fig. 6g), whereas in samples AL03-28I and AL03-14A, additional weak bands were marked, namely, four bands (Mn–OHF, Sr–OHF, Mn–OHOH and Sr–OHOH) in sample AL03-28I (Fig. 4g) and two bands (Mn–OHF and an additional band indexable as Mn–OHOH or Sr–OHF) in sample AL03-14A (Fig. 5g). These peaks correlate with small amounts of Mn and Sr, which have been confirmed by EMP analyses (Supplementary Table S2). With the Aint,tot values, we estimated the wt% H2O in the apatite using the calibration given by Wang et al. (2011). The values of Aint of 11.62/cm, 11.03/cm and 5.71/cm and thicknesses of 15.6 µm, 25.3 µm and 47.4 µm correspond to 0.56 wt% H2O, 0.33 wt% H2O and 0.09 wt% H2O in samples AL03-28I, AL03-14A and H12, respectively (Table 3).
Igneous-derived units (H30 and AL03-29B)
CL data
The apatite in sample AL03-29B shows strong and patchy orange luminescence (Fig. 7f) related to the presence of Mn2+ and REE3+ (Dy3+, Tb3+, Sm3+, Nd3+, Eu3+) emission centers that are confirmed in the CL spectra (Fig. 7c). In sample H30, the luminescence is weak, with blue-violet luminescence in the core and very strong orange luminescence in the narrow rim (Fig. 8c). This inhomogeneity is related to secondary alterations or it could be a new growth related to metamorphic events. The CL spectra indicate the presence of REE3+ and Mn2+ emission centers in variable amounts. The manganese emission center is dominant over bands characteristic of REE emission centers in areas that are closer to the rims (Fig. 8f).

Data collected for sample AL03-29B. a Results of electron microprobe measurements of apatite plotted in F–OH–Cl ternary diagram representing mole fraction occupancy of the X-site. b chondrite-normalized (Evensen et al. 1978) rare earth elements distribution patterns. c CL spectrum of apatite in f. d HAADF image of metasomatically altered apatite in sample AL03-29B. Nano-size inclusion rich zone (white dots), porosity (black arrow) and Ca3Y(PO4)3 inclusion (white arrow; Birski et al. 2018) are visible. e Bright field image of nano-size inclusion rich zone (white dots), low-angle grain boundaries (white arrow) and single dislocations (black arrows). f CL image of apatite in sample AL03-29B. The spectrum was collected from area marked by blue circle. g FTIR spectrum

Data collected for sample H30. a Results of electron microprobe measurements of apatite plotted in F–OH–Cl ternary diagram representing mole fraction occupancy of the X-site. b chondrite-normalized (Evensen et al. 1978) rare earth elements distribution patterns. c CL image of apatite in sample H30. d HREM image of xenotime inclusion in host apatite. e HREM image of monazite inclusion in host apatite. f CL spectrum of apatite in c. Color of spectrum agrees with the area marked in c. g FTIR spectrum. h HAADF image of the area being the border between reacted and unreacted fluorapatite (black dashed lines). REE-phosphate inclusions are marked by black arrows
TEM analysis
Four foils, two (nos. 4332 and 4334) from AL03-29B and two (nos. 4441 and 4482) from H30, were cut. In sample AL03-29B, the apatite is composed of slightly misoriented subgrains that show individual diffraction contrast patterns (Fig. 7e). The micrometer-sized pores are partially filled with Ca3Y(PO4)3 inclusions up to 200 nm in size (Fig. 7d). Furthermore, cracks, pores and nano channel-like structures have been reported by Birski et al. (2018).
Both foils from sample H30 reveal the absence of dislocations or low-angle grain boundaries in apatite, but (REE + Y)-phosphate inclusions were observed. The diffraction patterns (FFT) from the high-resolution lattice fringe image provide information about the lengths of the individual diffraction vectors and the angles between them, allowing mineral identification. Therefore, FFTs were collected from the (REE + Y)-phosphate inclusions in both foils. In foil no. 4332, the angles between the vectors were measured in three inclusions. The first inclusion featured observed angles of 37.3° and 50.6° (Supplementary Table S4 a), the second featured observed angles of 34.0° and 66.7° (Supplementary Table S4 b), and the third featured observed angles of 28.0°, 21.0°, 81.3° and 48.7° (Supplementary Table S4 c). All three inclusions were indexed as xenotime (Supplementary Table S4, Fig. 8d). In foil no. 4334, the angles between the vectors were measured in three inclusions as well. All three inclusions featured observed angles of approximately 40.0° and 53.0° (Supplementary Table S4) and were recognized as monazite (Supplementary Table S4, Fig. 8e). Furthermore, an orientation relationship between the monazite inclusion and host apatite was observed (Supplementary Table S4). The (010) direction of monazite is parallel to the (0001) direction of the apatite for all indexed monazite inclusions ([010]Mnz || [0001]Ap). The presence of hexagonal symmetry in the monazite inclusions is also a characteristic feature of the interfaces observed in high-resolution images of sample H30 (Fig. 8e). In foil no. 4482, two zones could be discriminated (Fig. 8h). The first zone has no inclusions and is visibly brighter than the second zone, which commonly contains solid inclusions. The differences in brightness in the high-angle annular dark-field (HAADF) Z-contrast images are most likely due to changes in the relative concentration of REEs of apatite caused by the removal of REEs and Y from the host apatite structure and the later nucleation of (REE + Y)-phosphate inclusions within the visibly darker zone. The boundary between the two zones is very clear and sharp (Fig. 8h).
Electron microprobe analysis
The apatite grains from the igneous-derived units are predominantly fluorapatite (Figs. 7a, 8a). The Cl and trace element concentrations are below the detection limit (Supplementary Tables S1 and S2). The calculated OH content (assuming that the X-site is completely filled, i.e., F + Cl + OH = 2) is up to 7.54 mol%, which equals up to 0.13 wt% H2O (Table 2).
Trace element composition
The REE patterns of sample H30 indicate the presence of two populations of apatite crystals (Fig. 8b). Population A is slightly enriched in LREEs and slightly depleted in HREEs and has a distinct negative Eu anomaly. This REE pattern is characteristic of magmatic apatite crystallization after plagioclase fractionation. Population B is characterized by an enrichment in MREEs and a strong depletion in LREEs and HREEs compared to population A. Population B does not demonstrate any distinct anomalies. The apatite crystals from sample AL03-29B are characterized by an enrichment in MREEs (Fig. 7b). The REE distribution patterns of population B in samples H30 and AL03-29B are typical of metasomatic apatite crystals (e.g., Lepland et al. 2002; Wudarska et al. 2018), and the Y-Ho ratio ranges between 21 and 28. A full data set is included in Supplementary Table S3.
FTIR spectroscopy
The water content was also determined in apatite from the igneous-derived samples using FTIR spectrometry. The spectra of sample AL03-29B show the presence of two bands, a prominent OHF band at 3539/cm and a much weaker OHOH band at 3545/cm, whereas in the spectra of sample H30, we observed four OH-bands, i.e., a dominant OHF band at 3538/cm and three additional small bands (OHOH, Mn–OHF and a band indexable as Mn–OHOH or Sr–OHF) (Table 3; Figs. 7g, 8g). These spectra have integrated absorbance values of 5.29/cm and 5.91/cm for H30 and AL03-29B, respectively. By applying the equation by Wang et al. (2011) and using measured thicknesses of 52.6 µm (H30) and 54.2 µm (AL03-29B), a hydroxyl content equivalent to approximately 0.08 wt% H2O can be calculated for the apatite in both samples (Table 3).
Banded iron formation (sample AL03-26B)
CL data
The apatite shows very weak yellow luminescence (Fig. 9f). The collected CL spectrum indicates that this luminescence is related to Mn2+ substitution (Fig. 9c). The inhomogeneity is easily visible. The core is dull and almost completely dark, which is most likely caused by Fe substitution (or a limited Mn content), whereas the very narrow rim is much brighter. This heterogeneity is not visible under the polarizing microscope.

Data collected for sample AL03-26B. a Results of electron microprobe measurements of apatite plotted in F–OH–Cl ternary diagram representing mole fraction occupancy of the X-site. b chondrite-normalized (Evensen et al. 1978) rare earth elements distribution patterns. Red line corresponds to REE distribution pattern of Archaean sedimentary apatite (Lepland et al. 2002). c CL image of apatite in sample AL03-26B. d Bright field image of apatite. Iron oxide inclusions are marked with black arrows, whereas low-angle grain boundaries with white arrow. e HAADF image of the iron oxide inclusions in host apatite. f CL spectrum of apatite in Fig. 9c showing a broad emission band due to Mn2+. g FTIR spectrum
TEM analysis
One foil (no. 4829) was lifted out from the sample for TEM study. The studied apatite was relatively rich in micro- and nanosized iron oxide inclusions and pores (with sizes up to 200 nm) (Fig. 9e). Furthermore, deformation and annealing features such as parallel dislocations and low-angle grain boundaries were observed (Fig. 9d).
Electron microprobe analysis
In sample AL03-26B, the apatite is associated with iron oxide layers and it is fluorapatite (Fig. 9a), with Cl concentrations below the detection limit (Supplementary Table S1). Furthermore, the sample contains appreciable concentrations of Sr (approx. 0.27 wt%) (Supplementary Table S2). The OH content, calculated assuming that the X-site is completely filled, i.e., F + Cl + OH = 2, is in the range of 36.7–45.8 mol%, which equals 0.64–0.81 wt% H2O (Table 2).
Trace element composition
The apatite crystals from sample AL03-26B are relatively enriched in REEs (Fig. 9b). Moreover, they are characterized by flat REE distribution patterns with slightly positive Nd, negative Sm, and distinct positive Eu anomalies. The Y–Ho ratio varies in the range of 55–70. This REE pattern is characteristic of sedimentary apatite (e.g., Lepland et al. 2002; Wudarska et al. 2018), whereas the Y–Ho ratio is characteristic of modern seawater values (Bau and Dulski 1994). A full data set is included in Supplementary Table S3.
FTIR spectroscopy
The FTIR spectrum collected from the sample shows a multiband pattern (Fig. 9g). A prominent OH band occurs at 3539/cm (Table 3). According to Tacker (2004), the OH band corresponds to a high F content within the apatite and should be indexed as an OHF band. Additional peaks were observed at 3572/cm (Table 3) and should be correlated with the position of the OHOH band. Four additional weak bands can be marked as Mn–OHF, Sr–OHF, Mn–OHOH and Sr–OHOH (Fig. 9g). Their presence may be correlated with the presence of small amounts of Mn and Sr, as confirmed by EMPA (Supplementary Table S2). The integrated absorbance, Aint,tot, was determined to be 12.84/cm. By applying the calibration of Wang et al. (2011) and using a measured thickness of 22.7 µm and a density of 3.1 g cm3, a hydroxyl content equivalent to 0.43 wt% H2O was calculated for this sample (Table 3).
Discussion
Apatite formation/transformation pathways
The studied samples show different paths of formation and also dissimilar transformation paths. The question arises as to whether the obtained characteristics allow the confirmation of the expected environment of emergence if all samples are transformed. What part of the characteristics can be considered as an indicator for the environment of the initial formation of phosphates? Finally, given that the studied phosphates originated from the period before the Great Oxidation Event, it would be interesting to know if the turn from a prebiotic to a biotic environment somehow influenced their transformation paths.
Modern ocean waters are characterized by a positive Y anomaly (mass ratio of Y to Ho) and a negative Ce anomaly, and their REEs show a pattern of slight enrichment from light to heavy REEs. However, modern hydrothermal vent waters are characterized by a positive Eu anomaly (Bau and Dulski 1999). The larger Y anomalies (40–80) in modern ocean waters occur in open marine settings, while the smaller Y anomalies (33–40) occur in near-shore or restricted settings (De Baar et al. 1985a, b; Bau and Dulski 1994; Zhang and Nozaki 1996; Bau et al. 1997). The Y/Ho ratio also varies with the salinity, phosphate mineral solubility, and fractionation during chemical weathering, biological processes and redox cycling (Liu and Byrne 1997; Hill et al. 2000). Our understanding of the Y anomalies is somewhat limited, as many earlier studies did not include Y measurements alongside REEs (Tostevin et al. 2016).
Measurements of the REEs and Y in 2.5–3.7 Ga-old BIFs and metacherts (e.g., Bolhar et al. 2004, 2005; Nishizawa et al. 2005; Hofmann et al. 2013) as well as in apatite (Lepland et al. 2002; Nishizawa et al. 2004; Joosu et al. 2015, 2016; Wudarska et al. 2018) show that they contain “sea water fingerprints” with a Y anomaly and flat REE distribution patterns, identical to the pattern found in modern sea water (Elderfield and Greaves 1982). The Y anomaly seems to be present in geological records throughout the entire Precambrian, both after and prior to the great oxidation event (GOE) and up to the Eoarchean (Joosu et al. 2015, 2016; Wudarska et al. 2018). However, unlike modern seawater, the sedimentary rocks prior to the GOE are characterized by a lack of a negative Ce anomaly, which implies anoxic conditions (e.g., Lepland et al. 2002; Joosu et al. 2015, 2016; Wudarska et al. 2018), whereas a positive Eu anomaly indicates significant hydrothermal input into the Archaean oceans (Veizer et al. 1989). Due to the mafic melt differentiation rules, apatite crystallizes as one of the latest phases, and therefore they exhibit a negative Eu anomaly (Puchelt and Emmermann 1976). During late diagenesis and hydrothermal recrystallization, apatite typically becomes enriched in the middle REEs (MREEs) because of their preferential substitution for Ca in the apatite crystal lattice due to their similar ionic radii (Morad and Felitsyn 2001), whereas light REEs (LREEs) and heavy REEs (HREEs) remain in the fluid. Such a crystal structure-controlled MREE enrichment mechanism is supported by the modeling studies of Reynard et al. (1999), who showed that MREE enrichment is expected during the late diagenetic recrystallization.
Apatite of sedimentary origin
The first group of apatite (AL03-28I, AL03-14A and AL03-26B) has the signature of an Archaean sedimentary environment. Phosphate oxygen data (δ18O = 19.9‰ and 18.1‰ in AL03-28I and AL03-26B, respectively) provided the basis for Blake et al. (2010) to infer the influence of microbial activity during phosphate precipitation. The REE distributions in the Archaean sediments have been shown to typically exhibit positive Eu anomalies (Derry and Jacobsen 1990; Lepland et al. 2002; Nishizawa et al. 2004; Wudarska et al. 2018), suggesting that hydrothermal venting was the major contributor to the REE budget of the Archaean ocean (Danielson et al. 1992; Lepland et al. 2002; Wudarska et al. 2018). These data are consistent with the higher geothermal gradient in the early Earth (Ronde et al. 1997). Since the crustal differentiation was probably less evolved during the Archaean and less continental crust was available for weathering, maintaining the hydrothermal REE signature of the ocean against the contribution from the continental source was easier. Furthermore, the Eu anomalies recorded in authigenic apatite are considered stable tracers that do not change during diagenesis (Shields and Stille 2001). Therefore, the nucleation of apatite in equilibrium with the Archaean seawater in AL03-28I, AL03-14A and AL03-26B is indicated by both the Y–Ho ratio (Fig. 10) and REE distribution patterns with a positive Eu anomaly (Figs. 4b, 5b, 9b). Furthermore, the apatite in all three samples is characterized by a relatively high water concentration, as shown by EMP analyses (Table 2) and FTIR (Table 3). The obtained EMPA values are in good agreement with FTIR measurements (Tables 2, 3). All the values plotted in the ternary diagram indicate that the samples have a wide range of F- and OH-members (Figs. 4a, 5a, 9a). The TEM observations of these samples show no indications of secondary alteration, which allows treating a number of observed features as primary. The presence of zones rich in nanopores (Figs. 4e, 5f) is most likely associated with the precipitation process and is in agreement with the presence of highly elongated and preferentially oriented pyrrhotite inclusions (Figs. 4e–f, 5e–f). Similar features were previously observed by Gottesmann and Wirth (1997) in apatites of magmatic plutons from Kirchberg and Bergen in the Vogtland region of Saxony, Germany. The presence of epitactically intergrown apatite and pyrrhotite inclusions can be caused by their similar lattice spacing. This similarity may result in the observed orientation relationship. The nuclei of pyrrhotite formed due to the oversaturation of Fe and S ions at the interface of the seawater/phosphate precursors using the apatite lattice as a template for nucleation. Subsequently, they grew simultaneously with the apatite until the concentrations of Fe and S at the interface decreased enough to prevent the further growth of pyrrhotite. During the growth of apatite, the moving interface experienced increases in the Fe and S concentrations, thus inducing the formation of new pyrrhotite nuclei at the interface of the seawater/phosphate precursors. The epitactic intergrowths might also explain the lack of an amorphous rim at the phase boundary interface between the pyrrhotite inclusion and the host apatite. The CL data (Fig. 5c, d) indicate that the luminescence of the apatite crystals is activated by REE substitution, which may point to the original composition of the formation environment. Archaean ocean pH value is still a matter of debate and controversy. Proposed values are scant and conflicting. Studies have concluded that the early ocean was alkaline (Kempe and Degens 1985; Blättler et al. 2017), acidic (Sleep and Zahnle 2001; Halevy and Bachan 2017; Krissansen-Totton et al. 2018), or approximately neutral (Macleod et al. 1994; Friend et al. 2008). Nevertheless, most proposed values is in the range of 6.5–8.6 pH. This lilac/violet CL color seems to be related to alkaline conditions that favor the incorporation of REEs (Kempe and Götze 2002). CL data are in the support of circumneutral to weakly alkaline Archean ocean pH based on rare Earth element anomalies proposed by Friend et al. (2008). These assumptions correlate with determined trace element abundances (Supplementary Table S3). On the other hand, the presence of orange luminescence (activated by Mn2+) in the narrow rim of sample AL03-28I (Fig. 4c) may indicate that this sample experienced secondary transformation processes and Mn substitution to a limited extent. No evidence of metamorphic growth of apatite have been observed in CL. The lack of CL luminescence (Fig. 9f) in AL03-26B is most likely caused by Fe2+ substitution (Kempe and Götze 2002), which is confirmed by the presence of relatively high concentrations of iron in the EMP analyses (Supplementary Table S2). Moreover, the iron concentrations in apatite correlate with the presence of iron oxide inclusions (Fig. 9e) and are associated with Fe-rich layers in the BIFs (Fig. 3). The characteristics of the samples of the first group allow them to be placed in the environment before the GOE as samples that at least partially preserve their original structures and compositions.

Metasomatic/secondary apatite
The samples from the igneous-derived rocks (AL03-29B and population B in H30) and metachert (H12) show very distinct features that are characteristic of metasomatic apatite. Blake et al. (2010) reported a δ18O value of 9.3‰ for apatite crystals from the same location as sample AL03-29B. This relatively low δ18O value, together with evidence for secondary veining, indicates that these apatites formed from secondary fluids (Blake et al. 2010). The result obtained in this paper provides additional evidence for this. Secondary overprinting is indicated by the metasomatic-type REE distribution pattern and Y–Ho ratio values that are much lower than the characteristic values for modern seawater (Fig. 10) (Bau and Möller 1992; Bau and Dulski 1994). Due to the removal of LREEs and HREEs from the structure of the apatite, MREE enrichment is considered the most important characteristic to evaluate the effects of the late diagenetic alteration of apatite. This feature is clearly visible in the apatite within this group (Figs. 6b, 7b, 8b). Previous studies (Harlov et al. 2002; Harlov and Förster 2003; Wudarska et al. 2018) have shown that during metasomatic transformations, apatite evolves towards the F-rich end-member, and therefore the secondary apatite will be relatively depleted in water in comparison to the sedimentary apatite (Tables 2, 3). Furthermore, relative to the sedimentary apatite, the metasomatic apatite also seems to be depleted in trace elements due to their removal by fluids, consistent with the results for H12 from EMP analyses (Supplementary Table S2) and FTIR spectra (Fig. 6g). The features of the metasomatic influence in samples H12, H30 and AL03-29B are also very distinct under the TEM. Traces of secondary alterations have already been described (Birski et al. 2018), and they consist of slightly misoriented subgrains that are separated by arrays of dislocations forming low-angle grain boundaries, the presence of porosity, and solid inclusions that are associated with the system of subgrain boundaries (Figs. 6d, 7d, e). The presence of numerous inclusions (Ca3Y(PO4)3 and CaYP7O20) in metasomatic apatite has also been reported (Birski et al. 2018), whereas in sample H30, inclusions of monazite and xenotime have been observed. The occurrence of monazite [(Ce,LREE)(PO4)] and/or xenotime [(Y,HREE)(PO4)] as inclusions in fluorapatite [Ca10(PO8)6(F,Cl,OH)2] has been widely documented (Harlov and Förster 2003; Harlov et al. 2005; Harlov 2011, 2015; Jonsson et al. 2016). Monazite and xenotime inclusion formation has been directly linked to metasomatism by either external or internal H2O-bearing fluids (Harlov 2015). The fluid-induced formation of monazite and xenotime inclusions in apatite is supported by experiments, which indicate that their formation and growth are the result of coupled dissolution–reprecipitation processes during metasomatic alteration (Harlov et al. 2002, 2005; Harlov and Förster 2003; Harlov 2011, 2015). In the apatite structure, (REE + Y) substitutions are charge-balanced through coupled substitutions: Si4+ + (REE + Y)3+ = P5+ + Ca2+ and Na+ + (REE + Y)3+ = 2Ca2+ (Pan and Fleet 2002). The nucleation of the REE-phosphate inclusions in the host apatite structure is caused by fluids that preferentially remove Na and/or Si from the apatite-fluid system without removing REEs. Consequently, when supersaturated conditions are reached, the REEs can react with phosphate and possibly cause the nucleation and growth of (REE + Y)-phosphate phases. This process occurs via the two following mass transfer reactions: [Ca5– 2x, Nax, REEx]P3O12F + x [2 Ca2+ + P5+] (from the fluid) = Ca5P3O12F + x REEPO4 + x[Na+] (into the fluid) and [Ca5–y, REEy][P3–y, Siy]O12F + y [Ca2++ 2 P5+] (from the fluid) = Ca5P3O12F + y REEPO4 + y [Si4+] (into the fluid) (Harlov 2015). The final composition of the (REE + Y)-phosphate inclusions depends on the chemistry of the altering fluid, the P–T conditions of the alteration and the host apatite chemistry (Harlov and Förster 2003). Therefore, the presence of different (REE + Y)-phosphate inclusions in different zones of the host apatite can be explained by differences in the Y and REE compositions. Furthermore, Birski et al. (2018) concluded that the REE-F complexes in an H2O solution and the specific budgets of REEs and Y in the apatite were responsible for the nucleation and formation of the (Ca–Y)-phosphate phases in the host apatites in the rocks of the BGB. Finally, in both samples, an orientation relationship between the solid inclusions and the host apatite is observed. In AL03-29B, an epitaxial relationship between the Ca3Y(PO4)3 inclusions and the host apatite has been shown (Birski et al. 2018), whereas in sample H30, an orientation relationship has been observed between the monazite inclusions and the apatite. Previous researchers (Pan et al. 1993; Pan and Fleet 2002; Harlov 2015) have noted that REE-phosphate inclusions, e.g., monazite, can be highly elongated. This elongation often occurs along the b axis and parallel to the c axis of the host apatite (Harlov 2015). This feature has been observed in the analyzed monazite inclusions (Fig. 8e), where the [010] direction of the monazite is always parallel to the [0001] direction of the apatite. According to Harlov et al. (2007), this phenomenon can be explained by a topotactic reaction, i.e., a chemical solid-state reaction in which the orientations of the product crystals are determined by the orientation of the initial crystal. In this case, the formation of monazite does not involve exsolution from a solid solution; instead, it involves the fluid-aided growth of a separate phase that utilizes the available chemical components. This growth can be referred to as topotactic because the formation and growth of one phase (i.e., monoclinic monazite) within a second phase (i.e., apatite) can be required to accommodate the differences in the lattice parameters and d-spacings. An orientation relationship is not visible in the xenotime inclusions. This may be explained by the differences in the crystal lattice systems among the Ca3Y(PO4)3, CaYP7O20 (Birski et al. 2018), monazite and xenotime inclusions and the similarities in the lattice spacings between the host apatite and the monazite and Ca3Y(PO4)3 inclusions. All these observations together suggest that the REE + Y-phosphate inclusions originated via dissolution–reprecipitation processes. The formation is not dated, and consequently there is no evidence as to whether these changes occurred within the Archaean hydrothermal or a later system.
Igneous apatite
The REE profile of population A in sample H30 indicates that, at least to a limited extent, a signature of magmatic origin could be potentially preserved. According to previous studies (Lepland et al. 2002; Higashi et al. 2017; Wudarska et al. 2018), REEs are more resistant to metasomatic alteration than the volatile elements that substitute into the apatite structure. Therefore, the REE distribution patterns should more clearly demonstrate a magmatic origin. A very distinguished negative Eu anomaly together with a slight enrichment in LREEs seems to confirm late crystallization from a differentiated melt. The yttrium behavior in apatite is similar to that of REEs, especially holmium, which has an identical valence and similar ionic radii. Therefore, Y and Ho behave similarly in many geochemical processes, resulting in a constant chondritic weight ratio Y/Ho = 28 in igneous rocks (Bau et al. 1996) and in hydrothermal fluids (Bau and Dulski 1999). Hence, the Y-Ho ratios do not indicate whether the apatite is of magmatic or metasomatic origin (Fig. 10). The features observed in CL suggest a complex history. A blue–violet core is characteristic of REE3+ emission centers. Alkaline rocks have been proven to tend towards blue, violet or lilac CL colors (Kempe and Götze 2002). In contrast, Mn2+ emission centers become dominant over bands characteristic of REEs in areas that are closer to the rims. This observation confirms the progressive alteration and transformation of the initially magmatic apatite. Since orange luminescence (activated by Mn2+) is typical for acidic environments (Kempe and Götze 2002), the fluids that metasomatically altered apatite crystals might be related to the granitic magmatism at 3.45, 3.22, and 3.1 Ga (Kamo and Davis 1994; Dziggel et al. 2002). The acidic metasomatism of the volcani-sedimentary rocks deposited in the BGB has been confirmed (Rouchon and Orberger 2008). Moreover, differences in the Y and REE compositions are clearly visible in CL images (Fig. 8c) in the form of more blue and violet zones that might reflect variations in the REE and Y budgets of the magma during the crystallization of apatite. TEM observations seems to confirm the partial preservation of the igneous signature (Fig. 8h). The obtained results allow the assignment of the apatite from this group to the Archaean environment with the preservation of many of its original features.
Conclusions
The growth/regrowth morphology patterns, compositional variations, and alterations of the studied Archaean apatite provide valuable information on the environmental conditions during their formation and transformation. Based on that, three generations of apatite occurring in the BGB can be designated as follows:
-
A.
Early-generation apatite that precipitated from Archaean seawater. This group clearly preserved the features that can be expected from apatite precipitating before the GOE. This phosphate is Cl-depleted fluorapatite with a significant amount of OH. The REE distribution patterns are flat with positive Eu anomalies and without a negative Ce anomaly, and the preserved Y–Ho ratios of 54–70 are comparable to those of modern seawater (Bau and Dulski 1994). The CL data indicate precipitation under alkaline conditions. A characteristic of the apatite in this group is the presence of epitactically intergrown pyrrhotite inclusions. TEM investigation does not reveal any indicators of secondary alteration processes in this apatite, and the FTIR spectra indicate that the sedimentary apatites are relatively rich in water. Mn and Sr substitutions cause a stretching mode in the hydrogen bonding between OH and F and the consequent presence of additional component bands.
-
B.
Late-generation fluorapatite that experienced metasomatism/metamorphism. Their secondary origin is confirmed by REE distribution patterns depleted in both LREEs and HREEs. CL data suggest an origin in acidic metasomatism, most likely associated with granitic magmatism at 3.45–3.1 Ga (Kamo and Davis 1994; Dziggel et al. 2002). TEM observations reveal the presence of many structures such as cracks, pores and nanochannel-like structures as well as the occurrence of (REE + Y)-phosphate inclusions. FTIR measurements confirm that the metasomatic apatites are OH-group depleted. Moreover, the removal of trace elements that substitute in the Ca2 position from the structure of apatite causes a lack of any additional band present in the early-generation apatites of the first group.
-
C.
Igneous-derived apatite that partially preserves its igneous origin. These grains are predominantly fluorapatite such as those in the second group; however, their REE distribution patterns are slightly enriched in LREEs and slightly depleted in HREEs and have distinct negative Eu anomalies. The CL data indicate that the apatites in this group preserve the signature of igneous origin within their cores, whereas the rims are metasomatically altered. Due to the predominant F substitution in the X-site, the OH-stretching mode is the strongest for the OHF band. Moreover, the occupancy by Mn and Sr of the Ca2 position is visible in the FTIR spectrum as additional Mn-OHF and Sr-OHF bands.
It seems that some of the apatite partly preserved its primordial features and therefore might be used for the reconstruction of Archaean abiotic systems. In this manner, some kind of comparison can be made between the periods before and after the GOE.
References
Alibert C (2016) Rare earth elements in Hamersley BIF minerals. Geochim Cosmochim Acta 184:311–328. https://doi.org/10.1016/j.gca.2016.03.026
Bau M, Dulski P (1994) Evolution of the yttrium-holmium systematics of seawater through time. Miner Mag 58A:61–62
Bau M, Dulski P (1999) Comparing yttrium and rare earths in hydrothermal fluids from the Mid-Atlantic Ridge: implications for Y and REE behaviour during near-vent mixing and for the Y/Ho ratio of Proterozoic seawater. Chem Geol 155:77–90. https://doi.org/10.1016/S0009-2541(98)00142-9
Bau M, Möller P (1992) Rare earth element fractionation in metamorphogenic hydrothermal calcite, magnesite and siderite. Mineral Petrol 45:231–246. https://doi.org/10.1007/BF01163114
Bau M, Koschinsky A, Dulski P, Hein JR (1996) Comparison of the partitioning behaviours of yttrium, rare earth elements, and titanium between hydrogenetic marine ferromanganese crusts and seawater. Geochim Cosmochim Acta 60:1709–1725. https://doi.org/10.1016/0016-7037(96)00063-4
Bau M, Mdler P, Dulski P (1997) Yttrium and lanthanides in eastern Mediterranean seawater and their fractionation during redox-cycling. Mar Chem 56:23–36
Birski Ł, Wirth R, Słaby E et al (2018) (Ca-Y)-phosphate inclusions in apatite crystals from Archean rocks from the Barberton Greenstone Belt and Pilbara Craton: first report of natural occurrence. Am Mineral 103:307–313. https://doi.org/10.2138/am-2018-6150
Blake RE, Chang SJ, Lepland A (2010) Phosphate oxygen isotopic evidence for a temperate and biologically active Archaean ocean. Nature 464:1029–1032. https://doi.org/10.1038/nature08952
Blättler CL, Kump LR, Fischer WW et al (2017) Constraints on ocean carbonate chemistry and pCO2 in the Archaean and Palaeoproterozoic. Nat Geosci 10:41–45. https://doi.org/10.1038/ngeo2844
Bolhar R, Kamber BS, Moorbath S et al (2004) Characterisation of early Archaean chemical sediments by trace element signatures. Earth Planet Sci Lett 222:43–60. https://doi.org/10.1016/j.epsl.2004.02.016
Bolhar R, Van Kranendonk MJ, Kamber BS (2005) A trace element study of siderite-jasper banded iron formation in the 3.45 Ga Warrawoona Group, Pilbara Craton—formation from hydrothermal fluids and shallow seawater. Precambrian Res 137:93–114. https://doi.org/10.1016/j.precamres.2005.02.001
Byerly GR, Lowe DR, Heubeck C (2018) Geologic evolution of the Barberton Greenstone belt—a unique record of crustal development, surface processes, and early life 3.55 to 3.20 Ga. In: Van Kranendonk MJ, Bennett VC, Hoffmann E (eds) Earth’s oldest rocks, 2nd edn. Elsevier, Berlin
Danielson A, Möller P, Dulski P (1992) The europium anomalies in banded iron formations and the thermal history of the oceanic crust. Chem Geol 97:89–100. https://doi.org/10.1016/0009-2541(92)90137-T
de Ronde CEJ, de Wit MJ (1994) Tectonic history of the Barberton greenstone belt, South Africa: 490 million years of Archean crustal evolution. Tectonics 13:983–1005. https://doi.org/10.1029/94TC00353
De Baar HJW, Bacon MP, Brewer PG, Bruland KW (1985a) Rare earth elements in the Pacific and Atlantic Oceans. Geochim Cosmochim Acta 49:1943–1959. https://doi.org/10.1016/0016-7037(85)90089-4
De Baar HJW, Brewer PG, Bacon MP (1985b) Anomalies in rare earth distributions in seawater: Gd and Tb. Geochim Cosmochim Acta 49:1961–1969. https://doi.org/10.1016/0016-7037(85)90090-0
Derry LA, Jacobsen SB (1990) The chemical evolution of Precambrian seawater: evidence from REEs in banded iron formations. Geochim Cosmochim Acta 54:2965–2977. https://doi.org/10.1016/0016-7037(90)90114-Z
Dziggel A, Stevens G, Poujol M et al (2002) Metamorphism of the granite–greenstone terrane south of the Barberton greenstone belt, South Africa: an insight into the tectono-thermal evolution of the ‘lower’ portions of the Onverwacht Group. Precambrian Res 114:221–247. https://doi.org/10.1016/S0301-9268(01)00225-X
Elderfield H, Greaves MJ (1982) The rare earth elements in seawater. Nature 296:214–219. https://doi.org/10.1038/296214a0
Evensen NM, Hamilton PJ, O’Nions RK (1978) Rare-earth abundances in chondritic meteorites. Geochim Cosmochim Acta 42:1199–1212. https://doi.org/10.1016/0016-7037(78)90114-X
Filippelli GM (2008) The global phosphorus cycle: past, present, and future. Elements 4:89–95. https://doi.org/10.2113/GSELEMENTS.4.2.89
Friend CRL, Nutman AP, Bennett VC, Norman MD (2008) Seawater-like trace element signatures (REE + Y) of Eoarchaean chemical sedimentary rocks from southern West Greenland, and their corruption during high-grade metamorphism. Contrib Mineral Petrol 155:229–246. https://doi.org/10.1007/s00410-007-0239-z
Gottesmann B, Wirth R (1997) Pyrrhotite inclusions in dark pigmented apatite from granitic rocks. Eur J Mineral 9:491–500. https://doi.org/10.1127/ejm/9/3/0491
Halevy I, Bachan A (2017) The geologic history of seawater pH. Science 355:1069–1071. https://doi.org/10.1126/science.aal4151
Harlov DE (2011) Formation of monazite and xenotime inclusions in fluorapatite megacrysts, Gloserheia Granite Pegmatite, Froland, Bamble Sector, southern Norway. Mineral Petrol 102:77–86. https://doi.org/10.1007/s00710-011-0166-6
Harlov DE (2015) Apatite: a fingerprint for metasomatic processes. Elements 11:171–176. https://doi.org/10.2113/gselements.11.3.171
Harlov DE, Förster HJ (2003) Fluid-induced nucleation of (Y + REE)-phosphate minerals within apatite: nature and experiment. Part II. Fluorapatite. Am Mineral 88:1209–1229
Harlov DE, Foerster H-J, Nijland TG (2002) Fluid-induced nucleation of (Y + REE) -phosphate minerals within apatite: nature and experiment. Part I. Chlorapatite Am Mineral 87:245–261
Harlov DE, Wirth R, Förster HJ (2005) An experimental study of dissolution-reprecipitation in fluorapatite: fluid infiltration and the formation of monazite. Contrib Mineral Petrol 150:268–286. https://doi.org/10.1007/s00410-005-0017-8
Harlov DE, Marschall HR, Hanel M (2007) Fluorapatite-monazite relationships in granulite-facies metapelites, Schwarzwald, southwest Germany. Mineral Mag 71:223–234. https://doi.org/10.1180/minmag.2007.071.2.223
Hickman-Lewis K, Cavalazzi B, Foucher F, Westall F (2018) Most ancient evidence for life in the Barberton greenstone belt: Microbial mats and biofabrics of the ∼ 3.47 Ga Middle Marker horizon. Precambrian Res 312:45–67. https://doi.org/10.1016/J.PRECAMRES.2018.04.007
Higashi Y, Itoh S, Hashiguchi M et al (2017) Hydrogen diffusion in the apatite-water system: fluorapatite parallel to the c-axis. Geochem J 51:115–122. https://doi.org/10.2343/geochemj.2.0460
Hill IG, Worden RH, Meighan IG (2000) Yttrium: the immobility-mobility transition during basaltic weathering. Geology 28:923. https://doi.org/10.1130/0091-7613(2000)28%3C923:YTITDB%3E2.0.CO;2
Hofmann A (2005) The geochemistry of sedimentary rocks from the Fig Tree Group, Barberton greenstone belt: implications for tectonic, hydrothermal and surface processes during mid-Archaean times. Precambrian Res 143:23–49. https://doi.org/10.1016/j.precamres.2005.09.005
Hofmann A, Harris C (2008) Silica alteration zones in the Barberton greenstone belt: A window into subseafloor processes 3.5–3.3 Ga ago. Chem Geol 257:221–239. https://doi.org/10.1016/j.chemgeo.2008.09.015
Hofmann A, Bolhar R, Orberger B, Foucher F (2013) Cherts of the Barberton Greenstone Belt, South Africa: petrology and trace-tlement geochemistry of 3.5 to 3.3 Ga old silicified volcaniclastic sediments. South African J Geol 116:297–322. https://doi.org/10.2113/gssajg.116.2.297
Javaux EJ (2006) Extreme life on Earth—past, present and possibly beyond. Res Microbiol 157:37–48. https://doi.org/10.1016/J.RESMIC.2005.07.008
Jonsson E, Harlov DE, Majka J et al (2016) Fluorapatite-monazite-allanite relations in the Grängesberg apatite-iron oxide ore district, Bergslagen, Sweden. Am Mineral. https://doi.org/10.2138/am-2016-5655
Joosu L, Lepland A, Kirsimäe K et al (2015) The REE-composition and petrography of apatite in 2 Ga Zaonega Formation, Russia: The environmental setting for phosphogenesis. Chem Geol 395:88–107. https://doi.org/10.1016/j.chemgeo.2014.11.013
Joosu L, Lepland A, Kreitsmann T et al (2016) Petrography and the REE-composition of apatite in the Paleoproterozoic Pilgujärvi Sedimentary Formation, Pechenga Greenstone Belt, Russia. Geochim Cosmochim Acta 186:135–153. https://doi.org/10.1016/j.gca.2016.04.043
Kamo SL, Davis DW (1994) Reassessment of Archean crustal development in the Barberton Mountain Land, South Africa, based on U-Pb dating. Tectonics 13:167–192. https://doi.org/10.1029/93TC02254
Kempe S, Degens ET (1985) An early soda ocean? Chem Geol 53:95–108. https://doi.org/10.1016/0009-2541(85)90023-3
Kempe U, Götze J (2002) Cathodoluminescence (CL) behaviour and crystal chemistry of apatite from rare-metal deposits. Mineral Mag 66:151–172. https://doi.org/10.1180/0026461026610019
Ketcham RA (2015) Technical Note: calculation of stoichiometry from EMP data for apatite and other phases with mixing on monovalent anion sites. Am Mineral 100:1620–1623. https://doi.org/10.2138/am-2015-5171
Knauth LP, Lowe DR (2003) High Archean climatic temperature inferred from oxygen isotope geochemistry of cherts in the 3.5 Ga Swaziland Supergroup, South Africa. Geol Soc Am Bull 115:566–580. https://doi.org/10.1130/0016-7606(2003)115%3C0566:HACTIF%3E2.0.CO;2
Krissansen-Totton J, Arney GN, Catling DC (2018) Constraining the climate and ocean pH of the early earth with a geological carbon cycle model. Proc Natl Acad Sci USA 115:4105–4110. https://doi.org/10.1073/pnas.1721296115
Kröner A, Anhaeusser CR, Hoffmann JE et al (2016) Chronology of the oldest supracrustal sequences in the Palaeoarchaean Barberton Greenstone Belt, South Africa and Swaziland. Precambrian Res 279:123–143. https://doi.org/10.1016/J.PRECAMRES.2016.04.007
Lepland A, Arrhenius G, Cornell D (2002) Apatite in early Archean Isua supracrustal rocks, southern West Greenland: its origin, association with graphite and potential as a biomarker. Precambrian Res 118:221–241. https://doi.org/10.1016/S0301-9268(02)00106-7
Lepland A, Joosu L, Kirsimäe K et al (2013) Potential influence of sulphur bacteria on Palaeoproterozoic phosphogenesis. Nat Geosci 7:20–24. https://doi.org/10.1038/ngeo2005
Libowitzky E, Rossman GR (1996) Principles of quantitative absorbance measurements in anisotropic crystals. Phys Chem Miner 23:319–327
Liu X, Byrne RH (1997) Rare earth and yttrium phosphate solubilities in aqueous solution. Geochim Cosmochim Act 61:1625–1633
Lowe DR, Byerly GR (2007) Chap. 5.3 an overview of the geology of the Barberton Greenstone Belt and Vicinity: implications for early crustal development. Dev Precambrian Geol 15:481–526. https://doi.org/10.1016/S0166-2635(07)15053-2
Macleod G, McKeown C, Hall AJ, Russell MJ (1994) Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Orig Life Evol Biosph 24:19–41. https://doi.org/10.1007/BF01582037
Mojzsis SJ, Arrhenius G, McKeegan KD et al (1996) Evidence for life on Earth before 3,800 million years ago. Nature 384:55–59. https://doi.org/10.1038/384055a0
Moorbath S, O’Nions RK, Pankhurst RJ (1973) Early Archaean Age for the Isua Iron formation, West Greenland. Nature 245:138–139. https://doi.org/10.1038/245138a0
Morad S, Felitsyn S (2001) Identification of primary Ce-anomaly signatures in fossil biogenic apatite: implication for the Cambrian oceanic anoxia and phosphogenesis. Sediment Geol 143:259–264. https://doi.org/10.1016/S0037-0738(01)00093-8
Neuser VRD, Bruhn F, Götze J et al (1995) Kathodolumineszenz: Methodik und Anwendung. Zentralblatt für Geol Paläontologie Tl I 1/2:287–306
Nishizawa M, Terada K, Ueda Y, Sano Y (2004) Ion microprobe U-Pb dating and REE analysis of apatite from kerogen-rich silica dike from North Pole area, Pilbara Craton, Western Australia. Geochem J 38:243–254. https://doi.org/10.1109/LPT.2005.860032
Nishizawa M, Takahata N, Terada K et al (2005) Rare-earth element, lead, carbon, and nitrogen geochemistry of apatite-bearing metasediments from the similar to 3.8 Ga Isua supracrustal belt, West Greenland. Int Geol Rev 47:952–970. https://doi.org/10.2747/0020-6814.47.9.952
Nutman AP, Bennett VC, Friend CRL, Rosing MT (1997) ∼ 3710 and ≥ 3790 Ma volcanic sequences in the Isua (Greenland) supracrustal belt; structural and Nd isotope implications. Chem Geol 141:271–287. https://doi.org/10.1016/S0009-2541(97)00084-3
Pan Y, Fleet ME (2002) Compositions of the Apatite-group minerals: substitution mechanisms and controlling factors. Rev Mineral Geochem 48:13–49. https://doi.org/10.2138/rmg.2002.48.2
Pan Y, Fleet ME, Macrae ND (1993) Oriented monazite inclusions in apatite porphyroblasts from the Hemlo gold deposit, Ontario, Canada. Mineral Mag 57:697–707
Puchelt H, Emmermann R (1976) Bearing of rare earth patterns of apatites from igneous and metamorphic rocks. Earth Planet Sci Lett 31:279–286
Pyle JM, Spear FS, Wark DA (2002) Electron microprobe analysis of REE in apatite, monazite and xenotime: protocols and pitfalls. Rev Mineral Geochem 48:337–362. https://doi.org/10.2138/rmg.2002.48.8
Qiu Y, Jiang H, Kovács I et al (2018) Quantitative analysis of H-species in anisotropic minerals by unpolarized infrared spectroscopy: an experimental evaluation. Am Mineral 103:1761–1769. https://doi.org/10.2138/am-2018-6620
Reynard B, Lécuyer C, Grandjean P (1999) Crystal-chemical controls on rare-earth element concentrations in fossil biogenic apatites and implications for paleoenvironmental reconstructions. Chem Geol 155:233–241. https://doi.org/10.1016/S0009-2541(98)00169-7
Rollinson H (2003) Metamorphic history suggested by garnet-growth chronologies in the Isua Greenstone Belt, West Greenland. Precambrian Res 126:181–196. https://doi.org/10.1016/S0301-9268(03)00094-9
Ronde CE, De Channer DM, K Faure, Bray CJ et al (1997) Fluid chemistry of Archean seafloor hydrothermal vents: Implications for the composition of circa 3.2 Ga seawater. Geochim Cosmochim Acta 61:4025–4042
Rosing MT, Rose NM, Bridgwater D, Thomsen HS (1996) Earliest part of Earth’s stratigraphic record: a reappraisal of the> 3.7 Ga Isua (Greenland) supracrustal sequence. Geology 24:43–46. https://doi.org/10.1130/0091-7613(1996)024%3C0043:EPOESS%3E2.3.CO;2
Rouchon V, Orberger B (2008) Origin and mechanisms of K-Si-metasomatism of ca. 3.4–3.3 Ga volcaniclastic deposits and implications for Archean seawater evolution: examples from cherts of Kittys Gap (Pilbara craton, Australia) and Msauli (Barberton Greenstone Belt, South Africa). Precambrian Res 165:169–189. https://doi.org/10.1016/j.precamres.2008.06.003
Shields G, Stille P (2001) Diagenetic constraints on the use of cerium anomalies as palaeoseawater redox proxies: an isotopic and REE study of Cambrian phosphorites. Chem Geol 175:29–48. https://doi.org/10.1016/S0009-2541(00)00362-4
Słaby E, Koch-Müller M, Förster HJ et al (2016) Determination of volatile concentrations in fluorapatite of Martian shergottite NWA 2975 by combining synchrotron FTIR, Raman spectroscopy, EMPA, and TEM, and inferences on the volatile budget of the apatite host-magma. Meteorit Planet Sci 51:390–406. https://doi.org/10.1111/maps.12598
Sleep NH, Zahnle K (2001) Carbon dioxide cycling and implications for climate on ancient Earth. J Geophys Res Planets 106:1373–1399. https://doi.org/10.1029/2000JE001247
Stormer JC, Pierson ML, Tacker RC (1993) Variation of F and Cl X-ray intensity due to anisotropic diffusion in apatite during electron microprobe analysis. Am Mineral 78:641–648
Tacker RC (2004) Hydroxyl ordering in igneous apatite. Am Mineral 89:1411–1421. https://doi.org/10.2138/am-2004-1008
Tostevin R, Shields GA, Tarbuck GM et al (2016) Effective use of cerium anomalies as a redox proxy in carbonate-dominated marine settings. Chem Geol 438:146–162. https://doi.org/10.1016/J.CHEMGEO.2016.06.027
Veizer J, Hoefs J, Lowe DR, Thurston PC (1989) Geochemistry of Precambrian carbonates: II. Archean greenstone belts and Archean sea water. Geochim Cosmochim Acta 53:859–871. https://doi.org/10.1016/0016-7037(89)90031-8
Viljoen MJ, Viljoen RP (1969) An introduction to the geology of the Barberton granite-greenstone terrain. Geol Soc South Africa Spec Publ 2:9–28
Walsh MW (1992) Microfossils and possible microfossils from the early archean onverwacht group, Barberton mountain land, South Africa. Precambrian Res 54:271–293. https://doi.org/10.1016/0301-9268(92)90074-X
Walsh MM, Lowe DR (1999) Modes of accumulation of carbonaceous matter in the early Archean: A petrographic and geochemical study of the carbonaceous cherts of the Swaziland Supergroup. In: Lowe DR, Byerly GR (eds) Geologic evolution of the Barberton greenstone belt. Geological Society of America, South Africa
Wang KL, Zhang Y, Naab FU (2011) Calibration for IR measurements of OH in apatite. Am Mineral 96:1392–1397. https://doi.org/10.2138/am.2011.3756
Webster JD, Piccoli PM (2015) Magmatic Apatite: a powerful, yet deceptive. Mineral Elem 11:177–182. https://doi.org/10.2113/gselements.11.3.177
Westall F, de Wit MJ, Dann J et al (2001) Early Archean fossil bacteria and biofilms in hydrothermally-influenced sediments from the Barberton greenstone belt, South Africa. Precambrian Res 106:93–116. https://doi.org/10.1016/S0301-9268(00)00127-3
Wirth R (2004) Focused ion beam (FIB): a novel technology for advanced application of micro- and nanoanalysis in geosciences and applied mineralogy. Eur J Mineral 16:863–876. https://doi.org/10.1127/0935-1221/2004/0016-0863
Wirth R (2009) Focused ion beam (FIB) combined with SEM and TEM: Advanced analytical tools for studies of chemical composition, microstructure and crystal structure in geomaterials on a nanometre scale. Chem Geol 261:217–229. https://doi.org/10.1016/j.chemgeo.2008.05.019
Wudarska A, Wiedenbeck M, Słaby E et al (2018) Halogen chemistry and hydrogen isotopes of apatite from the> 3.7 Ga Isua supracrustal belt, SW Greenland. Precambrian Res 310:153–164. https://doi.org/10.1016/J.PRECAMRES.2018.02.021
Zhang J, Nozaki Y (1996) Rare earth elements and yttrium in seawater: ICP-MS determinations in the East Caroline, Coral Sea, and South Fiji basins of the western South Pacific Ocean. Geochim Cosmochim Acta 60:4631–4644. https://doi.org/10.1016/S0016-7037(96)00276-1
Acknowledgements
We appreciate thoughtful and constructive reviews by two anonymous reviewers who provided inspiring advice; J. Hoefs is gratefully acknowledged for efficient editorial handling and very helpful, stimulating comments. The research was funded by Polish NCN Grant no. 2013/11/B/ST10/04753 and supported by the EU COST Action TD1308 “ORIGINS”.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Jochen Hoefs.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
410_2019_1560_MOESM1_ESM.xlsx
Supplementary Table S2 Electron probe microanalyses of apatite from Barberton greenstone belt using second protocol (dedicated for trace elements) (XLSX 175 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Birski, Ł., Słaby, E., Wirth, R. et al. Archaean phosphates: a case study of transformation processes in apatite from the Barberton greenstone belt. Contrib Mineral Petrol 174, 25 (2019). https://doi.org/10.1007/s00410-019-1560-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00410-019-1560-z