
Abstract
The greatest unmet need in multiple sclerosis (MS) are treatments that delay, prevent or reverse progression. One of the most tractable strategies to achieve this is to therapeutically enhance endogenous remyelination; doing so restores nerve conduction and prevents neurodegeneration. The biology of remyelination—centred on the activation, migration, proliferation and differentiation of oligodendrocyte progenitors—has been increasingly clearly defined and druggable targets have now been identified in preclinical work leading to early phase clinical trials. With some phase 2 studies reporting efficacy, the prospect of licensed remyelinating treatments in MS looks increasingly likely. However, there remain many unanswered questions and recent research has revealed a further dimension of complexity to this process that has refined our view of the barriers to remyelination in humans. In this review, we describe the process of remyelination, why this fails in MS, and the latest research that has given new insights into this process. We also discuss the translation of this research into clinical trials, highlighting the treatments that have been tested to date, and the different methods of detecting remyelination in people.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a chronic, primarily inflammatory, disorder of the central nervous system (CNS) characterised by focal lymphocytic infiltration causing damage to myelin and axons [1,2,3]. Typical clinical features include weakness, sensory loss, diplopia, reduced visual acuity, dysarthria, dysphagia, ataxia, and bladder dysfunction; largely a reflection of the distribution of demyelinating foci throughout the CNS. In 85% of patients, there is an initial period of episodic neurological dysfunction followed by partial or complete recovery (relapsing–remitting MS, RRMS) [4]. Over time, the clinical picture often develops to one of progressive disability (secondary progressive MS, SPMS) [5], while in 15% the illness is progressive from the outset (primary progressive MS, PPMS) [6, 7]. In both SPMS and PPMS, the strongest predictor of the onset of progression is age, typically being seen from around 40 years [8].
There now exists an extensive therapeutic armamentarium against the inflammation of MS [9]. These disease-modifying treatments (DMTs) reduce the incidence and severity of new lesions by limiting the activity and availability of immune cells, manifesting clinically through reductions in relapse rates and disability accrual [10,11,12,13,14,15,16,17,18,19]. However, the “therapeutic window” for treatment with these immunotherapies is limited [20]; best long-term results on disability are seen if an anti-inflammatory treatment is started within 5 years of the first clinical episode of demyelination in relapsing–remitting disease [21]. Furthermore, while the 16 current DMTs are licensed for RRMS, only one—ocrelizumab—is approved for primary progressive disease [22], and even then its effects are so modest that several reimbursement agencies, notably NICE in the UK, declared it only cost-effective in a subset of people with new or contrast-enhancing lesions on MRI and a disease duration of less than 15 years.
Instead, the promotion of regeneration of the myelin sheath, through enhancing the process of endogenous remyelination, has emerged as one of the most amenable prospects to delay, prevent or reverse progression [23]. This is grounded in experimental evidence that demonstrates that the myelin sheath (and its associated oligodendrocytes) does not just facilitate nerve conduction, but is also directly protective against degeneration [24,25,26,27,28,29]. In this review, we describe the biology of remyelination, why this fails in MS, and recent research that has raised questions about current approaches to promoting remyelination. We then consider the therapies that have been, and are being, evaluated and discuss the best approach to measure remyelination in people, which is proving increasingly essential as we translate promising preclinical research into clinical trials.
The role of myelin
Myelination offers a far better way of increasing the conduction velocity of nerve fibres than simply increasing axon size. Myelin increases the transverse, insulating, resistance of the axon membrane, while the voltage-gated sodium and potassium channels are virtually confined to short unmyelinated nodes of Ranvier. The action potential is therefore propagated by the comparatively rapid and energy-efficient process of saltatory conduction [30, 31]. It follows that loss of myelin leads to slower transmission of the action potential, and hence prolonged latency, but can also lead to conduction block [32]. Remyelination is therefore a way to restore saltatory conduction [33], and clinical function [34], after demyelination.
Additionally, oligodendrocytes directly support the neuron, for example through providing lactate for metabolism and generation of ATP [25,26,27]. Pathological studies and animal models also suggest that axonal degeneration is reduced in remyelinated areas [24, 28, 29]. Taken together, the rationale for a remyelinating therapy is to restore function and prevent neurodegeneration.
Meanwhile, it is increasingly apparent that myelin regulation is a dynamic process in which both newly formed oligodendrocytes and pre-existing oligodendrocytes remodel myelin, often in response to activity, to facilitate learning and plasticity [35]. Whether activity-dependent remyelination could be restored by the proposed treatments remains an unanswered question.
Biology of remyelination
Demyelination (induced experimentally or by disease) can be followed by this regenerative response leading to the formation of new myelin sheaths around denuded axons by newly formed oligodendrocytes [23, 36,37,38,39]. Histopathological assessments have highlighted that this can occur extensively in some people with MS [40], but is inadequate in a significant proportion [41, 42]. For example, one study analysed forebrain tissue from 51 MS patients and found widespread remyelination in 20% of individuals, yet 34 cases remyelinated fewer than 25% of their lesions [42]. High inter-subject variability in remyelination capacity is supported by dynamic myelin imaging using positron emission tomography (PET) [43] and, when combined with evidence that those demonstrating more remyelination display lower levels of disability [44], it underscores the therapeutic promise of a remyelinating treatment. Consequently, efforts have been made to understand the mechanisms of remyelination and why this fails in MS, in the hope of defining druggable targets to enhance this process.
Mechanisms of remyelination
While pre-existing mature oligodendrocytes are able to increase the number of internodes they generate, and therefore contribute to recovery after demyelination [45], they do not add to the pool of new myelinogenic oligodendrocytes that are required for remyelination in animals [46]. Thus, remyelination is crucially dependent upon adult oligodendrocyte progenitor cells (aOPCs), derived from neonatal OPCs (nOPCs) [47], which have been shown by genetic fate-mapping to be the cells responsible for generation of the majority of new oligodendrocytes in the adult nervous system [48, 49]. These cells are maintained in sufficient quantities predominantly by their own self-renewal, rather than by replacement from neural stem cell niches in the CNS [50, 51].
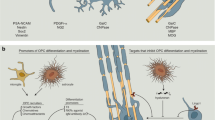
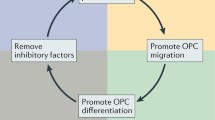
Following damage to myelinated areas, aOPCs must follow a choreographed process of activation, migration, proliferation and differentiation before culminating in the formation of new myelin sheaths [48]. The final product is a compacted layer of myelin that is thinner and shorter than those formed during developmental myelination [52], a fact often used to identify remyelination histologically when the process is studied in animal models (Box 1). Mechanistically, remyelination might fail due to a defect anywhere in this sequence; a paucity of pro-regenerative factors, or excess of inhibitory factors, as can be seen in MS lesions, combined with the intrinsic composition of the aOPC, can limit the capacity to remyelinate [23].
Given that large numbers of aOPCs are seen in chronically demyelinated MS lesions [53], it is often stated that remyelination fails as aOPCs become quiescent and unable to differentiate. As a consequence, increasing research has been deployed to elucidate the key regulators of differentiation [54,55,56,57,58] and identify agents capable of enhancing this process for clinical use, which is discussed further below.
However, it should be acknowledged that differentiation may not universally be the rate-limiting step in humans. It has previously been established that aOPCs do migrate to sites of injury and evenly distribute themselves to facilitate remyelination [59], though they probably do so over short distances [60]. But recently, two papers have also raised questions about the contributions of aOPCs to lesion repair in people. Yeung and colleagues utilised a 14C dating technique to show that oligodendrocytes from shadow plaques in MS brains, areas thought to have undergone at least partial remyelination, could not have been generated from new OPCs [61]. Further, Jäkle et al. reported that at autopsy, OPCs are reduced in number within shadow plaques [62]. Indeed, this latter study also highlighted changes in oligodendrocyte gene expression profiles between areas of normal-appearing white matter of MS brains and healthy controls, implying that the pathology seen in lesions may not reflect the global cellular changes occurring in MS.
Such results may find explanations in the differences between humans and the rat and mouse models used to study the disease. Taken together with evidence that some MS patients remyelinate better than others [42, 43], it seems clear that a treatment strategy that enhances differentiation alone may not necessarily be sufficient to address remyelination across a population of heterogenous MS patients.
Further, interactions between other glial cells and OPCs are also being increasingly clearly defined. Reactive astrocytes found at the site of demyelination, for example, secrete inhibitors of remyelination such as Endothelin-1 [63] and the recent description of A1 reactive astrocytes, which contribute to the death of oligodendrocytes [64], needs to be incorporated into the current model of remyelination and potential therapeutic targets explored.
In parallel, the demonstration that protein synthesis in OPCs is modulated by axonal action potentials [65] speaks to an underlying symbiosis between the neuron and the cells responsible for its myelination. In the peripheral nervous system, there is a necessary relationship between axon and Schwann cell, exemplified by dependency on the neuron-derived growth factor Neuregulin 1 to drive peripheral nerve myelination [66]. In the CNS, OPCs are able to differentiate even in the absence of axons [67, 68] and, as will be discussed below, in culture are able to myelinate inert axon-like substrates [69, 70]. Yet, while oligodendrocytes have a default ability to differentiate and myelinate axons, this is modulated by axon diameter and activity, implying a requirement for intact axons in vivo [71].
Therefore, it looks increasingly probable that combinations of drugs, acting on different processes, will be required to facilitate remyelination, and that these will be most effective when there is a sufficiently preserved demyelinated axon. This latter point forms the rationale for many phase 2 studies first focussing on people with RRMS, in whom it is anticipated that fewer axons will have degenerated.
Reasons for remyelination failure
To understand why remyelination fails in MS, one must look at two crucial contributory processes—namely those of age and the immune system.
While the immune system is often seen as having a detrimental role in MS, the innate immune system has been shown to be essential in the biology of remyelination [72]. Myelin debris contains inhibitors of aOPC differentiation and so its clearance, by phagocytosis, is an important step in the regeneration of the myelin sheath [73,74,75,76]. Similarly, infiltrating macrophages and activated microglia secrete a myriad of neurotrophic factors, which have direct effects on aOPCs [77]. Indeed, to facilitate robust remyelination in vitro, the polarisation of the macrophage response to an immunoregulatory, “M2”, phenotype is required [78]. It is not clear how these findings relate to the behaviour of monocyte-derived macrophages and microglia, in vivo, yet they emphasise how improving our understanding of subpopulations of macrophages/microglia and lymphocytes in the brain is essential to developing treatments that prevent demyelination while promoting remyelination.
The potential for endogenous remyelination is both age and disease duration dependent: remyelination is greatest in people aged less than 55 years and within the first 10 years of disease onset [39, 79, 80]. Disentangling the relative contribution of age versus duration of lesion demyelination to remyelination failure remains to be done, but clinical evidence would suggest age is especially pertinent as patients reach disability milestones at similar ages whether they have relapsing or progressive symptoms at onset [8, 80]. Similarly, lesional magnetisation transfer ratio (MTR)—a putative marker of remyelination—also shows age-dependent decline [81]. Remyelination is therefore akin to other regenerative processes [82] in becoming less efficient with time [83,84,85,86,87]; understanding age-associated remyelination failure is essential in treatment development.
Mechanistically, in this circumstance, the rate-limiting step is more clearly differentiation of the aOPC, as increasing aOPC recruitment does not lead to enhanced remyelination in aged mice [88]. Studies of how extrinsic factors vary with age have implicated a declining efficiency of the inflammatory response [89]; as noted above, macrophages produce pro-differentiation factors and clear debris by phagocytosis [76, 90, 91], which is essential for remyelination. That this process might be modifiable was demonstrated by the reversal of a deficit in remyelination of an aged mouse by twinning its circulation with a young animal by heterochronic parabiosis [92]. In a similar way, small molecule treatments can be used to promote endogenous remyelination, even in aged animals.
A detailed understanding of the intrinsic age-related changes in aOPCs is a recent development in the field following work by Neumann and colleagues [93]. They demonstrated that aged aOPCs become less responsive to factors that induce differentiation, contributing to the reduced remyelination capacity seen in many non-remyelinating chronic MS lesions [94]. Moreover, RNA sequencing from young and aged aOPCs highlighted a significant contribution from the mTOR pathway. This led to the novel observation that manipulating this pathway in aged rats with caloric restriction (three non-consecutive days of fasting per week over 6 months), or with the AMPK-agonist metformin (over 3 months), reverses the diminished differentiation capacity of aOPCs and restores their ability to remyelinate. As a result, manipulation of intrinsic changes in these stem cells is emerging as a promising treatment strategy.
Finally, there are also anatomical variations to the extent of remyelination within different lesions in the same individual. For example, periventricular lesions are less amenable to remyelination than subcortical lesions [42, 80]. This might reflect an underlying heterogeneity in OPCs or in locational differences in permissibility for their differentiation [95]: there are fewer inhibitors of remyelination in the cortex [96]. It could also be due to the importance of neuronal activity for remyelination, which is more likely to occur closer to the soma. Regional variations in remyelination within an individual is an opportunity to investigate barriers to enhancing remyelination, but also raise questions about which lesions should be tested in clinical trials.
Identification of agents capable of remyelination
An enhanced understanding of the intrinsic and extrinsic regulatory pathways implicated in remyelination has identified a multitude of sensible targets for therapeutic manipulation. An example of this has been the development of opicinumab to inhibit Lingo-1 (leucine-rich repeat and immunoglobulin-like domain-containing nogo receptor-interacting protein 1), a negative regulator of differentiation [97].
Another fruitful technique has been high-throughput screening of libraries of compounds, looking for an effect on aOPC differentiation [98]. One such study focussed on the ability of candidate compounds to promote differentiation of rat optic nerve-derived progenitor cells as evidenced by their production of oligodendrocyte differentiation markers [99]. This revealed that antagonism of muscarinic receptors, with the antihistamine/anticholinergic benzatropine, promotes OPC differentiation in vitro, which translated into a remyelinating effect in both EAE and cuprizone mice models. Similarly, Najm et al. used a flat plate culture system to screen a library of bioactive small molecules, this time on mouse pluripotent epiblast stem cell-derived OPCs. They discovered that the topical corticosteroid, clobetasol, and the anti-fungal, miconazole, as well as benzatropine, lead to a mature oligodendrocyte morphology, and improved remyelination in a lysolecithin-induced mouse model of focal demyelination [100].
A slightly different approach has used concentric wrapping of myelin around micropillars as an end point rather than differentiation per se. Mei et al. assessed the ability of 1000 FDA-approved small molecules to promote OPCs and oligodendrocytes to ensheath these cone-like structures with myelin [69]. In this way, they identified a cluster of compounds with an anti-muscarinic effect: atropine, ipratropium, oxybutynin, trospium, quetiapine, benztropine and clemastine. This work was quickly translated into the positive phase 2 trial of clemastine as a remyelinating therapy [101], discussed below.
Such small molecules may not have their remyelinating effect through an exclusive action at their canonical targets. The closest to a unifying mechanism has been through demonstration that a wide range of these, including clemastine, benztropine, miconazole and ketoconazole, might promote remyelination through altering the sterol landscape in the OPC to favour accumulation of 8,9-unsaturated sterols [102].
However, these techniques predominantly rely on the assumption that OPC differentiation is the rate-limiting step in remyelination, which might not be as rational as once thought [61, 62, 103]. The micropillar array is also limited in its ability to test the development of functional architecture in the form of nodes and internodes. It follows that combination therapies may be necessary to optimise an effect across the population of MS lesions. Moreover, such efforts will inevitably be hampered by the lack of an animal model that encapsulates the complexity of the MS lesion; there is a risk that agents showing promise in preclinical work do not translate into a beneficial effect in humans or indeed that a potentially useful treatment effect is missed in such models, halting progression towards clinical studies [23].
Remyelination clinical trials
The identification of agents that therapeutically enhance endogenous remyelination in preclinical models has led several to be translated into clinical trials and the possibility of a neuroprotective treatment in MS looks increasingly likely. In Table 1, we summarise the clinical trials that have been performed while considering a few in more detail below.
Clemastine
This is a first generation anti-histamine that was identified in the micropillar array as being capable of stimulating OPCs to differentiate and carry out the first stages of myelination [69]. This was confirmed in a further screen [99] and shown to occur via an off-target anti-muscarinic action, likely a specific effect on the M1 muscarinic receptor [29]. Ensuing work would confirm its remyelinating effect in multiple animal models [29, 69, 104, 105].
As clemastine has been licensed for allergic rhinitis since 1992, it was readily translated into a clinical trial [101]. The ReBUILD study was a single-centre, double-blind, randomised, placebo-controlled, phase 2, crossover trial, which specifically investigated the remyelinating potential of clemastine in patients with RRMS and evidence of chronic demyelinating optic neuropathy. Their inclusion criteria ensured that there was detectable demyelination in the optic pathway (evidenced by a visual evoked potential (VEP) P100 latency > 118 ms in at least one eye), but also sufficient axons to regenerate [with a retinal nerve fibre layer thickness (RNFL) > 70 μm in the qualifying eye when measured with optical coherence tomography (OCT)]. Meanwhile, the remyelination that might be expected in the natural history of optic neuritis was excluded by selecting only those without a history of acute optic neuritis in the qualifying eye within the last 5 years, or in either eye in the last 6 months.
The study design saw participants divided into two groups, but ensured that all had access to the study drug (which is readily available in the USA without prescription). 25 were given 5.36 mg of clemastine twice daily for 90 days followed by placebo for 60 days (group 1), while a further 25 patients were given placebo for 90 days followed by clemastine for 60 days (group 2).
The results of this were rather promising. VEP P100 full-field latency was reduced by 1.7 ms/eye (p = 0.0048) in the crossover model. Furthermore, the clinical effect of clemastine was sustained in group 1 after switching to placebo. Thus, the crossover model underestimates the actual effect, later demonstrated to be a 3.2 ms reduction in P100 latency. Further, there was a significant improvement in a functional outcome, low contrast letter acuity, when the delayed treatment analysis was employed. All the while, the drug was well tolerated, though was associated with fatigue. Secondary end points were negative, however, including MRI assessments of myelin water fraction (MWF), whole brain MTR, white matter MTR, and white matter fractional anisotropy (FA). There was no effect on the Expanded Disability Status Scale (EDSS), a timed 25-foot walk and the 6-min walk test.
This positive trial has provided some optimism about clemastine, though its selective inclusion criteria raise the possibility that the results are not generalisable; 75 patients were excluded based on VEPs. The ReCOVER trial (NCT02521311) will, no doubt, advance things further as it investigates the effect of clemastine (4 mg three times daily for 1 week, followed by 4 mg twice daily until 3 months treatment) in patients with acute optic neuritis. However, its clinical role needs further definition before widespread use: the ReBUILD result requires progression to phase 3 studies, clemastine should be trialled in progressive cohorts, and the possibility of combining treatments, such as with the potential synergistic effect of metformin, requires investigation.
Opicinimab
As mentioned above, Lingo-1 is a negative regulator of oligodendrocyte differentiation and its antagonism has been shown in vitro and in animal models of CNS demyelination to enhance remyelination [106]. The human monoclonal antibody opicinumab (anti-Lingo-1) showed remyelinating activity in preclinical studies [107] and therefore its utility was explored in early clinical trials. After passing safety analyses in a phase 1 trial [108], there was a phase 2, randomised, double-blind, placebo-controlled, clinical trial (RENEW) in patients with a first episode of acute optic neuritis (but not necessarily multiple sclerosis) [109]. The primary outcome measure was the recovery in VEP P100 latency in the affected eye, referenced to the unaffected eye, over 24 weeks of treatment (at 100 mg/kg) after an episode of optic neuritis. It failed in this regard but, when a per-protocol analysis was employed, an improvement of 7.6 ms in the opicinimab group was seen over that in the placebo group. No change was observed in the secondary end points, though this did not include MRI sequences such as MTR.
The SYNERGY trial followed (NCT01864148): a dose-ranging study including 418 people with RRMS and SPMS. The primary outcome measure was a composite of ambulation (25-foot walk), upper extremity function (9-Hole Peg Test), cognition (3-Second Paced Auditory Serial Addition Test; PASAT) and the EDSS. It failed to meet this primary end point, but was presented as showing an increased percentage of responders in those treated with the mid-range doses of 10 and 30 mg/kg [110]. This has led to Biogen proceeding with a refined phase II trial (AFFINITY) in addition to an extension study (RENEWED, NCT02657915) of the RENEW trial.
GSK239512
This H3-receptor antagonist was originally developed to treat Alzheimer’s disease [111, 112] and was put forward as a potential remyelinating agent because H3 negatively regulates oligodendrocyte differentiation [113]. A phase 2, randomised, placebo-controlled study in people with RRMS on interferon or glatiramer acetate revealed a small, but significant, improvement in mean change in post-lesion MTR in gadolinium-enhanced lesions compared to placebo [114].
Bexarotene
Robin Franklin’s group demonstrated that, in aged animals, 9-cis-retinoic acid enhances remyelination through agonism of the nuclear retinoid acid receptor RXR-γ [115]. As a similar effect could be achieved by a pan-RXR agonist licensed to treat skin lymphoma, bexarotene [116], this facilitated translation into a phase 2 clinical trial (CCMR One). Indeed, while an RXR-γ specific agonist might be more desirable, an effect of RXR-α activity on remyelination was also later reported [117].
The CCMR One study utilised MTR to quantify remyelination as a primary measure. However, in contrast to other studies, its focus is on changes in mean lesional MTR between month zero and month six for the lesions selected, for each patient, whose MTR lies below the within-patient median. In this way, it is hoped that the outcome will focus on an effect on lesions that are demyelinated at the baseline visit [118].
Biotin
Biotin is postulated to promote remyelination when given in high doses through its role as a cofactor for carboxylases required for fatty acid synthesis in oligodendrocytes [119]. To date, clinical trials have focussed on cohorts of progressive patients. The MS-SPI study showed a small statistically significant effect in 12.6% of treated participants when using an improvement of either the EDSS or timed 25-foot walk as its outcome [120]. However, the MS-ON study returned a negative result when change in visual acuity was employed as the primary end point [121]. It therefore remains uncertain whether high-dose biotin could be a clinically useful treatment for people with MS—and is under further investigation in MS-SPII—yet these trials also highlight the importance of selection of patient group and outcome measures, which will be discussed further below.
Cell-based therapies
Enhancing the activity of endogenous oligodendrocyte progenitors has proved the most accessible strategy to promote remyelination to date, therefore forming the focus of this review. However, other non-ablative cell-based approaches have been generating substantial interest (reviewed in [122]): transplantation of mesenchymal stem cells (MSCs), derived from bone marrow or other tissues, and transplantation of OPCs, derived from foetal tissue, embryonic stem cells or induced pluripotent stem cells (iPSCs) [123, 124] are viable options, but remain experimental. Challenges exist for each with regard to cell production, mode of delivery, tumour-forming potential, and requirements for immune suppression, although using autologous sources may abrogate the need for the latter. One noteworthy, albeit uncontrolled, trial administered bone marrow-derived MSCs to ten patients with progressive MS, noting an improvement in VEP latency of 1.3 ms, interpreting the mechanism for this as a neuroprotective effect through the promotion of myelin repair [125]. Larger phase 2 studies are underway [126], though there are many unresolved barriers to widespread application of a transplant-based approach to MS.
The target population for remyelination therapies
The efficacy of a remyelinating therapy would be greatest early in the course of MS, to stop long-term axonal degeneration and so prevent, or at least slow the onset of, secondary progression. More problematic is how effective such a therapy would be later in the disease course, for instance in progressive multiple sclerosis, when presumably many axons have already degenerated and so there is no scaffold for remyelination. Ultimately, this question can only be resolved by clinical trials. For the moment, we would advocate testing potential remyelinating agents in trials of patients identified as having demyelinated lesions with axons still present, such as those in the ReBUILD trial.
Determining remyelination in clinical trials
One of the foremost challenges to translating promising preclinical findings into clinical studies is uncertainty in the optimum way to demonstrate a remyelinating effect in living individuals. Given that the anticipated benefit to the patient, the prevention or delay to progression, only manifests over years, reliance on standard objective clinical markers of disability, for example the EDSS [127], or indeed on functional measures such as visual acuity, as outcomes may miss a useful therapeutic effect over a comparatively short clinical trial. Moreover, using functional scores to study remyelination specifically is further complicated by other adaptations that occur in nerves in response to injury, such as ion channel redistribution and cortical plasticity/adaptation after demyelination [128, 129]. There follows a reliance on paraclinical measures to determine a treatment effect; with no biomarker of myelin regeneration in biological fluids and the lack of accessible tissue for histological examination, a combination of neurophysiological and imaging-based assessments are required [130].
Neuroimaging
From the imaging perspective, standard MRI measures (such as T2-weighted and gadolinium-enhanced T1-weighted sequences) correlate only modestly with disability and lack the ability to differentiate between pathological correlates of MS: namely inflammation, oedema, axonal loss, demyelination, remyelination and gliosis [131]. As a result, advanced MRI techniques including myelin water fraction (MWF) [132, 133], diffusion tensor imaging (DTI) [134, 135] and magnetisation transfer ratio (MTR)[136, 137], as well as positron emission tomography (PET) [43, 138], have been variably used to measure myelin dynamics.
Magnetisation transfer techniques investigate the exchange of magnetisation between protons in at least two pools: those that are mobile and those associated with macromolecules such as myelin or axonal membranes. Expressed as a ratio (MTR), it provides a quantitative measure of the proportion of protons bound to macromolecular structures relative to those that are free in water and has been demonstrated to correlate with pathological quantification of myelin: demyelinated lesions have a significantly lower MTR than remyelinated lesions [137]. MTR can be used to quantify myelin in several ways. One can use serial measures of mean MTR in white and grey matter [139], in chronic lesions [118] or in acute (gadolinium-enhancing) lesions [81]. Indeed, as not all lesions remyelinate to the same extent, further refinements have been proposed. For example, in the CCMR One study of bexarotene, the primary outcome measure is based on the change in mean MTR of established lesions with a low MTR at the baseline scan, thereby maximising sensitivity to an effect on lesions that are demyelinated at the outset [118]. These aforementioned techniques have been used to show that mean MTR in white and grey matter remains static over time in people with MS treated with alemtuzumab, but deteriorates if not on disease-modifying treatment [139], and that the anti-histamine GSK239512 has a small positive effect on mean MTR in gadolinium-enhancing lesions [114].
Alternatively, DTI provides information about tissue microstructure by measuring water diffusion in vivo. Parameters derived from this such as radial diffusivity (a marker of water motion perpendicular to the axon) as well as the overall fractional anisotropy can be used as a surrogate of myelin content [135, 140]. Meanwhile, MWF indicates the proportion water trapped between myelin bilayers relative to water inside and outside of axons (which have different T1 and T2 relaxation times), and can be used as a proxy for myelin content [132, 141].
Finally PET imaging, alongside a myelin-specific ligand such as Pittsburgh compound B (PiB), might be especially sensitive to changes in myelin [142] and could potentially be used to stratify the patients by their remyelination potential for clinical studies [43]. However, the issues of availability and radiation exposure are likely to limit the role this has to play.
Neurophysiology
Evoked potentials allow for an assessment of nervous conduction along visual, somatosensory, auditory, and motor tracts in a way that correlates with function [143,144,145] and disability [146], but their clinical utility, particularly in diagnostics, has largely been replaced by MRI [147]. However, such indices are proving invaluable as biomarkers in assessing remyelination; in the recent positive phase 2 trial of clemastine, it was a reduction in VEP P100 latency, rather than clinical or imaging markers, that confirmed the biological effect [101, 148].
The pattern reversal VEP represents the average recordable electric potential in the visual cortex in response to the presentation of an alternating checkerboard-patterned stimulus. The main focus is on the positive deflection in the VEP waveform approximately 100 ms after the visual stimulus (the P100), which provides measures in the form of latency (a surrogate of demyelination in the optic pathways) and amplitude (predominantly a function of axonal loss). Following an attack of optic neuritis, VEP latencies are prolonged but a period of recovery follows, most significantly within the first 6 months, but for perhaps as long as 3 years [149, 150]. Meanwhile, in those with chronic stable optic neuropathy, a prolonged P100 latency is seen, which has been shown in longitudinal data to remain stable, or gradually lengthen, with time [151]. As a result, in studies of patients without a recent bout of optic neuritis, improvements in VEP P100 latency can be used as a marker of remyelination; this was the rationale behind the ReBUILD trial [101]. When studies have enrolled patients with acute optic neuritis meanwhile, such as in the RENEW study of opicinumab [109], values for the unaffected contralateral eye have been used as a control and the outcome measure given as the change in latency difference between the two eyes.
The clinical heterogeneity of MS has already been alluded to, which extends beyond a consideration of optic neuritis. Given the emerging importance of neurophysiological measures in such studies, a more robust biomarker in future trials might be a combination of VEPs, motor EPs (MEPs), somatosensory EPs (SEPs), and brainstem auditory EPs (BAEPs) [152]. Such “multimodal” evoked potentials can be combined to give a “global” outcome, which has previously been shown to correlate with disability and inform disease progression [153], and have already been employed in the field of bone marrow-derived cell therapy [126, 154]. Indeed other exploratory, neurophysiological techniques have also been emerging, such as the use of saccadometry [155], most recently showing improved conduction along the medial longitudinal fasciculus in the setting of internuclear ophthalmoparesis treated with fampridine [156].
Other techniques
Optical coherence tomography (OCT) is another tool being employed to indirectly assess remyelinating therapies. By generating high-resolution images of the retina to gauge axonal loss in the retinal nerve fibre layer (RNFL) [157], which is decreased following optic neuritis [158], it provides a measure of neurodegeneration rather than specifically remyelination, but can be used to complement the analysis of the VEP [159]. Similarly, serum neurofilament, a marker of axonal damage that correlates with MS disease activity [160], has been postulated to be a valuable outcome measure for remyelination trials. A subset of participants from the SYNERGY trial (discussed above) showed a trend toward neurofilament light decline among treatment responders [161]; however such measures remain an indirect marker of remyelination and, in our view, should remain exploratory.
Conclusions
People living with multiple sclerosis in resource-rich regions now have access to a range of anti-inflammatory treatments which promise long-term disease modification if given early in the course of the relapsing–remitting phase of the disease. However, even the most effective of these treatments leaves significant numbers of axons demyelinated and vulnerable to degeneration, which is the substrate of progressive disability. Finding remyelinating therapies, with the potential to both restore function and prevent axon degeneration is therefore an urgent clinical need.
A host of strategies have been identified from preclinical research, and some have now been translated to early phase clinical trials. Two of these have yielded positive results on surrogate measures, such as VEP latency or lesional MTR, whose clinical validity is yet to be demonstrated. There have also been setbacks, such as the failure of anti-Lingo-1 antibodies in phase 2 trials to replicate their in vitro efficacy.
In this review, we have highlighted a number of unresolved questions the scientific community faces. First, to ensure we take the right drugs forward, we must continue to develop an understanding of the barriers to remyelination, specifically in humans, and better appreciate how biology observed in animal models of remyelination translates into human disease. Second, we need to identify which is the best, most reliable, measure of remyelination to use in clinical trials. Third, we must clarify when is the best time to initiate a remyelinating treatment in the disease course. Finally, we need to consider which is the most appropriate test population for our trials. Ultimately, more work is required to reach a reality of neurologists prescribing neuroprotective treatments to people with MS; however, advances in the last decade have made this look increasingly probable.
References
Compston A, Coles A (2008) Multiple sclerosis. The Lancet. 372(9648):1502–1517
Reich DS, Lucchinetti CF, Calabresi PA (2018) Multiple sclerosis. N Engl J Med. 378(2):169–180
Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O (2018) Multiple sclerosis. The Lancet. 391(10130):1622–1636
O’Connor P (2002) Key issues in the diagnosis and treatment of multiple sclerosis. Neurology. 59(6 suppl 3):S1
University of California SFM-ET, Cree BAC, Gourraud P-A, Oksenberg JR, Bevan C, Crabtree-Hartman E et al. (2016) Long-term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 80(4):499–510
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ et al (2014) Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 83(3):278–286
Miller DH, Leary SM (2007) Primary-progressive multiple sclerosis. The Lancet Neurol. 6(10):903–912
Confavreux C, Vukusic S (2006) Age at disability milestones in multiple sclerosis. Brain 129(3):595–605
Scolding N, Barnes D, Cader S, Chataway J, Chaudhuri A, Coles A et al (2015) Association of British Neurologists: revised (2015) guidelines for prescribing disease-modifying treatments in multiple sclerosis. Pract Neurol. 15(4):273
Ebers GC (1998) Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. The Lancet. 352(9139):1498–1504
Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP et al (1995) Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis. Neurology. 45(7):1268
Confavreux C, O’Connor P, Comi G, Freedman MS, Miller AE, Olsson TP et al (2014) Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Neurol. 13(3):247–256
Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K et al (2012) Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 367(12):1098–1107
Calabresi PA, Radue E-W, Goodin D, Jeffery D, Rammohan KW, Reder AT et al (2014) Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet Neurol. 13(6):545–556
Giovannoni G, Comi G, Cook S, Rammohan K, Rieckmann P, Sørensen PS et al (2010) A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 362(5):416–426
Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH et al (2006) A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 354(9):899–910
Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ et al (2012) Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. The Lancet. 380(9856):1829–1839
Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung H-P et al (2012) Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. The Lancet. 380(9856):1819–1828
Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung H-P, Hemmer B et al (2016) Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 376(3):221–234
Coles AJ, Cox A, Le Page E, Jones J, Trip SA, Deans J et al (2006) The window of therapeutic opportunity in multiple sclerosis. J Neurol 253(1):98–108
Brown JWL, Coles A, Horakova D, Havrdova E, Izquierdo G, Prat A et al (2019) Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA 321(2):175–187
Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G et al (2016) Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 376(3):209–220
Franklin RJM, French-Constant C (2017) Regenerating CNS myelin—from mechanisms to experimental medicines. Nat Rev Neurosci 18:753
Irvine KA, Blakemore WF (2008) Remyelination protects axons from demyelination-associated axon degeneration. Brain 131(6):1464–1477
Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J et al (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485(7399):517–521
Morrison BM, Lee Y, Rothstein JD (2013) Oligodendroglia: metabolic supporters of axons. Trends Cell Biol 23(12):644–651
Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN et al (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487(7408):443–448
Kornek B, Storch MK, Weissert R, Wallstroem E, Stefferl A, Olsson T et al (2000) Multiple sclerosis and chronic autoimmune encephalomyelitis: a comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am J Pathol. 157(1):267–276
Mei F, Lehmann-Horn K, Shen Y-AA, Rankin KA, Stebbins KJ, Lorrain DS et al. (2016) Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 5:e18246
Smith K, McDonald W, Blakemore W (1979) Restoration of secure conduction by central demyelination. Trans Am Neurol Assoc. 104:25–29
Smith K, Blakemore W, McDonald W (1981) The restoration of conduction by central remyelination. Brain 104(2):383–404
Smith Kenneth J, McDonald WI (1999) The pathophysiology of multiple sclerosis⋮ the mechanisms underlying the production of symptoms and the natural history of the disease. Philos Trans R Soc Lond B Biol Sci 354(1390):1649–1673
Smith KJ, Blakemore WF, McDonald WI (1979) Central remyelination restores secure conduction. Nature 280(5721):395–396
Duncan ID, Brower A, Kondo Y, Curlee JF Jr, Schultz RD (2009) Extensive remyelination of the CNS leads to functional recovery. Proc Natl Acad Sci USA 106(16):6832–6836
Sampaio-Baptista C, Johansen-Berg H (2017) White matter plasticity in the adult brain. Neuron 96(6):1239–1251
Bunge MB, Bunge RP, Ris H (1961) Ultrastructural study of remyelination in an experimental lesion in adult cat spinal cord. J Biophys Biochem Cytol 10(1):67–94
Périer O, Grégoire A (1965) Electron microscopic features of multiple sclerosis lesions. Brain 88(5):937–952
Franklin RJM (2002) Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci 1(3):705
Franklin RJM, French-Constant C, Edgar JM, Smith KJ (2012) Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol 8:624
Patani R, Balaratnam M, Vora A, Reynolds R (2007) Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol 33(3):277–287
Prineas JW, Barnard RO, Kwon EE, Sharer LR, Cho E-S (1993) Multiple sclerosis: remyelination of nascent lesions. Ann Neurol 33(2):137–151
Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H et al (2006) Remyelination is extensive in a subset of multiple sclerosis patients. Brain 129(12):3165–3172
Bodini B, Veronese M, García-Lorenzo D, Battaglini M, Poirion E, Chardain A et al (2016) Dynamic imaging of individual remyelination profiles in multiple sclerosis. Ann Neurol 79(5):726–738
Bramow S, Frischer JM, Lassmann H, Koch-Henriksen N, Lucchinetti CF, Sørensen PS et al (2010) Demyelination versus remyelination in progressive multiple sclerosis. Brain 133(10):2983–2998
Jeffries MA, Urbanek K, Torres L, Wendell SG, Rubio ME, Fyffe-Maricich SL (2016) ERK1/2 Activation in preexisting oligodendrocytes of adult mice drives new myelin synthesis and enhanced CNS function. J Neurosci. 36(35):9186–9200
Crawford AH, Tripathi RB, Foerster S, McKenzie I, Kougioumtzidou E, Grist M et al (2016) Pre-Existing mature oligodendrocytes do not contribute to remyelination following toxin-induced spinal cord demyelination. Am J Pathol. 186(3):511–516
French-Constant C, Raff MC (1986) Proliferating bipotential glial progenitor cells in adult rat optic nerve. Nature. 319:499
Zawadzka M, Rivers LE, Fancy SPJ, Zhao C, Tripathi R, Jamen F et al (2010) CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 6(6):578–590
Tripathi RB, Rivers LE, Young KM, Jamen F, Richardson WD (2010) NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J Neurosci. 30(48):16383–16390
Psachoulia K, Jamen F, Young KM, Richardson WD (2009) Cell cycle dynamics of NG2 cells in the postnatal and ageing brain. Neuron Glia Biol. 5(3–4):57–67
Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A (2006) Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 26(30):7907
Blakemore WF (1974) Pattern of remyelination in the CNS. Nature 249(5457):577–578
Wolswijk G (1998) Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 18(2):601
Moyon S, Huynh JL, Dutta D, Zhang F, Ma D, Yoo S et al (2016) Functional characterization of DNA methylation in the oligodendrocyte lineage. Cell Rep. 15(4):748–760
He D, Wang J, Lu Y, Deng Y, Zhao C, Xu L et al (2017) lncRNA functional networks in oligodendrocytes reveal stage-specific myelination control by an lncOL1/Suz12 complex in the CNS. Neuron 93(2):362–378
Moyon S, Ma D, Huynh JL, Coutts DJC, Zhao C, Casaccia P, et al. (2017) Efficient remyelination requires DNA methylation. eNeuro 4(2): Eneuro.0336-16.2017.
Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB et al (2009) Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138(1):172–185
Duncan GJ, Plemel JR, Assinck P, Manesh SB, Muir FGW, Hirata R et al (2017) Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol 134(3):403–422
Hughes EG, Kang SH, Fukaya M, Bergles DE (2013) Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci 16(6):668–676
Franklin RJM, Gilson JM, Blakemore WF (1997) Local recruitment of remyelinating cells in the repair of demyelination in the central nervous system. J Neurosci Res 50(2):337–344
Yeung MSY, Djelloul M, Steiner E, Bernard S, Salehpour M, Possnert G et al (2019) Dynamics of oligodendrocyte generation in multiple sclerosis. Nature 566(7745):538–542
Jäkel S, Agirre E, Mendanha Falcão A, van Bruggen D, Lee KW, Knuesel I et al (2019) Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566(7745):543–547
Hammond TR, Gadea A, Dupree J, Kerninon C, Nait-Oumesmar B, Aguirre A et al (2014) Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 81(3):588–602
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 18(541):481
Wake H, Lee PR, Fields RD (2011) Control of local protein synthesis and initial events in myelination by action potentials. Science 333(6049):1647
Birchmeier C, Nave K (2008) Neuregulin-1, a key axonal signal that drives Schwann cell growth and differentiation. Glia. 56(14):1491–1497
Ueda H, Levine JM, Miller RH, Trapp BD (1999) Rat optic nerve oligodendrocytes develop in the absence of viable retinal ganglion cell axons. J Cell Biol. 146(6):1365
Almeida R, Lyons D (2016) Oligodendrocyte development in the absence of their target axons in vivo. PLoS ONE 11(10):e0164432
Mei F, Fancy SPJ, Shen Y-AA, Niu J, Zhao C, Presley B et al. (2014) Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat Med 20(8):954–960
Lee S, Leach MK, Redmond SA, Chong SYC, Mellon SH, Tuck SJ et al (2012) A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat Methods 15(9):917
Klingseisen A, Lyons DA (2017) Axonal regulation of central nervous system myelination: structure and function. Neuroscientist. 24(1):7–21
Rawji KS, Yong VW (2013) The benefits and detriments of macrophages/microglia in models of multiple sclerosis. Clin Dev Immunol 2013:948976–948976
Döring A, Sloka S, Lau L, Mishra M, van Minnen J, Zhang X et al (2015) Stimulation of monocytes, macrophages, and microglia by amphotericin B and macrophage colony-stimulating factor promotes remyelination. J Neurosci. 35(3):1136
Robinson S, Miller RH (1999) Contact with central nervous system myelin inhibits oligodendrocyte progenitor maturation. Dev Biol. 216(1):359–368
Plemel JR, Manesh SB, Sparling JS, Tetzlaff W (2013) Myelin inhibits oligodendroglial maturation and regulates oligodendrocytic transcription factor expression. Glia. 61(9):1471–1487
Kotter MR, Li W-W, Zhao C, Franklin RJM (2006) Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 26(1):328
Yong VW, Rivest S (2009) Taking advantage of the systemic immune system to cure brain diseases. Neuron 64(1):55–60
Miron VE, Boyd A, Zhao J-W, Yuen TJ, Ruckh JM, Shadrach JL et al (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16(9):1211–1218
Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I et al (2015) Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 78(5):710–721
Goldschmidt T, Antel J, König FB, Brück W, Kuhlmann T (2009) Remyelination capacity of the MS brain decreases with disease chronicity. Neurology. 72(22):1914
Brown RA, Narayanan S, Arnold DL (2013) Segmentation of magnetization transfer ratio lesions for longitudinal analysis of demyelination and remyelination in multiple sclerosis. NeuroImage. 1(66):103–109
Oh J, Lee YD, Wagers AJ (2014) Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med 20(8):870–880
Shields SA, Gilson JM, Blakemore WF, Franklin RJM (1999) Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia. 28(1):77–83
Hampton DW, Innes N, Merkler D, Zhao C, Franklin RJM, Chandran S (2012) Focal immune-mediated white matter demyelination reveals an age-associated increase in axonal vulnerability and decreased remyelination efficiency. Am J Pathol. 180(5):1897–1905
Sim FJ, Zhao C, Penderis J, Franklin RJM (2002) The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 22(7):2451
van Wijngaarden P, Franklin RJM (2013) Ageing stem and progenitor cells: implications for rejuvenation of the central nervous system. Development. 140(12):2562
Rist JM, Franklin RJM (2008) Taking ageing into account in remyelination-based therapies for multiple sclerosis. J Neurol Sci 274(1):64–67
Woodruff RH, Fruttiger M, Richardson WD, Franklin RJM (2004) Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci 25(2):252–262
Zhao C, Li W-W, Franklin RJM (2006) Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol Aging 27(9):1298–1307
Hinks GL, Franklin RJM (1999) Distinctive patterns of PDGF-A, FGF-2, IGF-I, and TGF-β1 gene expression during remyelination of experimentally-induced spinal cord demyelination. Mol Cell Neurosci 14(2):153–168
Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJM (2001) Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia. 35(3):204–212
Ruckh JM, Zhao J-W, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ et al (2012) Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 10(1):96–103
Neumann B, Baror R, van Wijngaarden P, Franklin RJ (2017) Remyelination of regenerating axons. Acta Ophthalmola 95(S259). https://doi.org/10.1111/j.1755-3768.2017.03525
Kuhlmann T, Miron V, Cuo Q, Wegner C, Antel J, Brück W (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131(7):1749–1758
Kitada M, Rowitch DH (2006) Transcription factor co-expression patterns indicate heterogeneity of oligodendroglial subpopulations in adult spinal cord. Glia. 54(1):35–46
Chang A, Staugaitis SM, Dutta R, Batt CE, Easley KE, Chomyk AM et al (2012) Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol 72(6):918–926
Mi S, Blake Pepinsky R, Cadavid D (2013) Blocking LINGO-1 as a therapy to promote CNS repair: from concept to the clinic. CNS Drugs 27(7):493–503
Bove RM, Green AJ (2017) Remyelinating pharmacotherapies in multiple sclerosis. Neurotherapeutics. 14(4):894–904
Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ et al (2013) A regenerative approach to the treatment of multiple sclerosis. Nature 502(7471):327–332
Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC et al (2015) Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 522(7555):216–220
Green AJ, Gelfand JM, Cree BA, Bevan C, Boscardin WJ, Mei F et al (2017) Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. The Lancet. 390(10111):2481–2489
Hubler Z, Allimuthu D, Bederman I, Elitt MS, Madhavan M, Allan KC et al (2018) Accumulation of 8,9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 560(7718):372–376
Boyd A, Zhang H, Williams A (2013) Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol 125(6):841–859
Liu J, Dupree JL, Gacias M, Frawley R, Sikder T, Naik P et al (2016) Clemastine enhances myelination in the prefrontal cortex and rescues behavioral changes in socially isolated mice. J Neurosci. 36(3):957–962
Li Z, He Y, Fan S, Sun B (2015) Clemastine rescues behavioral changes and enhances remyelination in the cuprizone mouse model of demyelination. Neurosci Bull. 31(5):617–625
Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z et al (2005) LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci 15(8):745
Zhang Y, Zhang YP, Pepinsky B, Huang G, Shields LBE, Shields CB et al (2015) Inhibition of LINGO-1 promotes functional recovery after experimental spinal cord demyelination. Exp Neurol 1(266):68–73
Tran JQ, Rana J, Barkhof F, Melamed I, Gevorkyan H, Wattjes MP et al. (2014) Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol Neuroimmunol Neuroinflamm 1(2):e18–e18
Cadavid D, Balcer L, Galetta S, Aktas O, Ziemssen T, Vanopdenbosch L et al (2017) Safety and efficacy of opicinumab in acute optic neuritis (RENEW): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 16(3):189–199
Mellion M, Edwards KR, Hupperts R, Drulović J, Montalban X, Hartung H-P et al. (2017) Efficacy Results from the Phase 2b SYNERGY Study: treatment of disabling multiple sclerosis with the Anti-LINGO-1 monoclonal antibody opicinumab (S33.004). Neurology 88(16 Supplement):S33.004
Grove Richard A, Harrington Conn M, Mahler Andreas, Beresford Isabel, Maruff Paul, Lowy Martin T et al (2014) A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res 11(1):47–58
Nathan Pradeep J, Boardley Rebecca, Scott Nicola, Berges Alienor, Maruff Paul, Sivananthan Tharani et al (2013) The safety, tolerability, pharmacokinetics and cognitive effects of GSK239512, a selective histamine H3 receptor antagonist in patients with mild to moderate Alzheimer’s disease: a preliminary investigation. Curr Alzheimer Res 10(3):240–251
Chen Y, Zhen W, Guo T, Zhao Y, Liu A, Rubio JP et al (2017) Histamine Receptor 3 negatively regulates oligodendrocyte differentiation and remyelination. PLoS ONE 12(12):e0189380–e0189380
Schwartzbach CJ, Grove RA, Brown R, Tompson D, Then Bergh F, Arnold DL (2017) Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing-remitting multiple sclerosis: a randomised, single-blind, phase II study. J Neurol 264(2):304–315
Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W et al (2011) Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci 14(1):45–53
Heck MC, Wagner CE, Shahani PH, MacNeill M, Grozic A, Darwaiz T et al (2016) Modeling, synthesis, and biological evaluation of potential retinoid X receptor (RXR)-selective agonists: analogues of 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic acid (bexarotene) and 6-(ethyl(5,5,8,8-tetrahydronaphthalen-2-yl)amino)nicotinic acid (NEt-TMN). J Med Chem. 59(19):8924–8940
Natrajan MS, de la Fuente AG, Crawford AH, Linehan E, Nuñez V, Johnson KR et al (2015) Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain 138(Pt 12):3581–3597
Altmann DR, Button T, Schmierer K, Hunter K, Tozer DJ, Wheeler-Kingshott CA et al (2014) Sample sizes for lesion magnetisation transfer ratio outcomes in remyelination trials for multiple sclerosis. Mult Scler Relat Disord. 3(2):237–243
Sedel F, Bernard D, Mock DM, Tourbah A (2016) Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology 1(110):644–653
Tourbah A, Lebrun-Frenay C, Edan G, Clanet M, Papeix C, Vukusic S et al (2016) MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: A randomised, double-blind, placebo-controlled study. Mult Scler. 22(13):1719–1731
Tourbah A, Gout O, Vighetto A, Deburghgraeve V, Pelletier J, Papeix C et al. (2018) MD1003 (high-dose pharmaceutical-grade biotin) for the treatment of chronic visual loss related to optic neuritis in multiple sclerosis: a randomized, double-blind: Placebo-Controlled Study. CNS Drugs 32(7), 661–6ss72.
Scolding NJ, Pasquini M, Reingold SC, Cohen JA (2017) International conference on cell-based therapies for multiple sclerosis: cell-based therapeutic strategies for multiple sclerosis. Brain 140(11):2776–2796
Goldman SA, Nedergaard M, Windrem MS (2012) Glial progenitor cell-based treatment and modeling of neurological disease. Science 338(6106):491
Franklin RJM, Goldman SA (2015) Glia disease and repair—remyelination. cold spring harbor perspectives in biology 7(7). https://cshperspectives.cshlp.org/content/7/7/a020594.abstract. Accessed date 01 Jul 2015
Connick P, Kolappan M, Crawley C, Webber DJ, Patani R, Michell AW et al (2012) Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 11(2):150–156
Rice CM, Marks DI, Ben-Shlomo Y, Evangelou N, Morgan PS, Metcalfe C et al (2015) Assessment of bone marrow-derived cellular therapy in progressive multiple sclerosis (ACTiMuS): study protocol for a randomised controlled trial. Trials. 16(1):463
Kurtzke JF (1983) Rating neurologic impairment in multiple sclerosis. Neurology. 33(11):1444
Waxman SG (2006) Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 1(7):932
Tomassini V, Matthews PM, Thompson AJ, Fuglø D, Geurts JJ, Johansen-Berg H et al (2012) Neuroplasticity and functional recovery in multiple sclerosis. Nat Rev Neurol. 18(8):635
Plemel JR, Liu W-Q, Yong VW (2017) Remyelination therapies: a new direction and challenge in multiple sclerosis. Nat Rev Drug Discov 7(16):617
Mallik S, Samson RS, Wheeler-Kingshott CAM, Miller DH (2014) Imaging outcomes for trials of remyelination in multiple sclerosis. J Neurol Neurosurg Psychiatry. 85(12):1396
Laule C, Kozlowski P, Leung E, Li DKB, MacKay AL, Moore GRW (2008) Myelin water imaging of multiple sclerosis at 7 T: correlations with histopathology. NeuroImage. 40(4):1575–1580
O’Muircheartaigh J, Vavasour I, Ljungberg E, Li DKB, Rauscher A, Levesque V et al. (2019) Quantitative neuroimaging measures of myelin in the healthy brain and in multiple sclerosis. Human Brain Mapp https://doi.org/10.1002/hbm.24510. Accessed date 15 Jan 2019
Klawiter EC, Schmidt RE, Trinkaus K, Liang H-F, Budde MD, Naismith RT et al (2011) Radial diffusivity predicts demyelination in ex vivo multiple sclerosis spinal cords. NeuroImage. 55(4):1454–1460
Song S-K, Yoshino J, Le TQ, Lin S-J, Sun S-W, Cross AH et al (2005) Demyelination increases radial diffusivity in corpus callosum of mouse brain. NeuroImage. 26(1):132–140
Henkelman RM, Stanisz GJ, Graham SJ (2001) Magnetization transfer in MRI: a review. NMR Biomed 14(2):57–64
Schmierer K, Scaravilli F, Altmann DR, Barker GJ, Miller DH (2004) Magnetization transfer ratio and myelin in postmortem multiple sclerosis brain. Ann Neurol 56(3):407–415
Stankoff B, Poirion E, Tonietto M, Bodini B (2018) Exploring the heterogeneity of MS lesions using positron emission tomography: a reappraisal of their contribution to disability. Brain Pathol 28(5):723–734
Button T, Altmann D, Tozer D, Dalton C, Hunter K, Compston A et al (2012) Magnetization transfer imaging in multiple sclerosis treated with alemtuzumab. Mult Scler. 19(2):241–244
Schmierer K, Wheeler-Kingshott CAM, Boulby PA, Scaravilli F, Altmann DR, Barker GJ et al (2007) Diffusion tensor imaging of post mortem multiple sclerosis brain. NeuroImage. 35(2):467–477
MacKay AL, Vavasour IM, Rauscher A, Kolind SH, Mädler B, Moore GRW et al (2009) MR relaxation in multiple sclerosis. Neuroimaging Clin N Am 19(1):1–26
Stankoff B, Freeman L, Aigrot M-S, Chardain A, Dollé F, Williams A et al (2011) Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-11C]-2-(4-methylaminophenyl)-6-hydroxybenzothiazole. Ann Neurol 69(4):673–680
Leocani L, Martinelli V, Natali-Sora MG, Rovaris M, Comi G (2003) Somatosensory evoked potentials and sensory involvement in multiple sclerosis: comparison with clinical findings and quantitative sensory tests. Mult Scler. 9(3):275–279
Fukutake T, Kuwabara S, Kaneko M, Kojima S, Hattori T (1998) Sensory impairments in spinal multiple sclerosis: a combined clinical, magnetic resonance imaging and somatosensory evoked potential study. Clin Neurol Neurosurg 100(3):199–204
Weinstock-Guttman B, Baier M, Stockton R, Weinstock A, Justinger T, Munschauer F et al (2003) Pattern reversal visual evoked potentials as a measure of visual pathway pathology in multiple sclerosis. Mult Scler. 9(5):529–534
Borggrefe-Chappuis A, Schindler C, Kappos L, Fuhr P (2001) Visual and motor evoked potentials in the course of multiple sclerosis. Brain 124(11):2162–2168
Hutchinson M (2013) Evoked potentials are of little use in the diagnosis or monitoring of MS: commentary. Mult Scler. 19(14):1824–1825
Silbermann E, Wooliscroft L, Bourdette D (2018) Using the anterior visual system to assess neuroprotection and remyelination in multiple sclerosis trials. Curr Neurol Neurosci Rep. 18(8):49
Brusa A, Jones SJ, Kapoor R, Miller DH, Plant GT (1999) Long-term recovery and fellow eye deterioration after optic neuritis, determined by serial visual evoked potentials. J Neurol 246(9):776–782
Brusa A, Jones SJ, Plant GT (2001) Long-term remyelination after optic neuritisA 2-year visual evoked potential and psychophysical serial study. Brain 124(3):468–479
Niklas A, Sebraoui H, Heß E, Wagner A, Then Bergh F (2009) Outcome measures for trials of remyelinating agents in multiple sclerosis: retrospective longitudinal analysis of visual evoked potential latency. Mult Scler. 15(1):68–74
Hardmeier M, Leocani L, Fuhr P (2017) A new role for evoked potentials in MS? Repurposing evoked potentials as biomarkers for clinical trials in MS. Mult Scler. 23(10):1309–1319
Leocani L, Rovaris M, Boneschi FM, Medaglini S, Rossi P, Martinelli V et al (2006) Multimodal evoked potentials to assess the evolution of multiple sclerosis: a longitudinal study. J Neurol Neurosurg Psychiatry 77(9):1030–1035
Rice CM, Mallam EA, Whone AL, Walsh P, Brooks DJ, Kane N et al (2010) Safety and feasibility of autologous bone marrow cellular therapy in relapsing-progressive multiple sclerosis. Clin Pharmacol Ther 87(6):679–685
Fielding J, Clough M, Beh S, Millist L, Sears D, Frohman AN et al (2015) Ocular motor signatures of cognitive dysfunction in multiple sclerosis. Nat Rev Neurol. 15(11):637
Kanhai KMS, Nij-Bijvank JA, Wagenaar YL, Klaassen ES, Lim K, Bergheanu SC et al. (2019) Treatment of internuclear ophthalmoparesis in multiple sclerosis with fampridine: a randomized double-blind, placebo-controlled cross-over trial. CNS Neurosci Therap https://doi.org/10.1111/cns.13096. Accessed date 12 Feb 2019
Green AJ, Hauser SL, Allen IV, Lyness R, McQuaid S (2010) Ocular pathology in multiple sclerosis: retinal atrophy and inflammation irrespective of disease duration. Brain 133(6):1591–1601
Talman LS, Bisker ER, Sackel DJ, Long DA Jr, Galetta KM, Ratchford JN et al (2010) Longitudinal study of vision and retinal nerve fiber layer thickness in multiple sclerosis. Ann Neurol 67(6):749–760
Martínez-Lapiscina EH, Sanchez-Dalmau B, Fraga-Pumar E, Ortiz-Perez S, Tercero-Uribe AI, Torres-Torres R et al (2014) The visual pathway as a model to understand brain damage in multiple sclerosis. Mult Scler. 20(13):1678–1685
Kuhle J, Kropshofer H, Haering DA, Kundu U, Meinert R, Barro C et al (2019) Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology. 92(10):e1007
Sharma A et al. (2018) Characterization of serum neurofilament, a biomarker for axonal damage, in the SYNERGY study as a complement to opicinumab zreatment effect in MS. ePosters. Mult Scler 24(2 suppl):738–980 (Abstract EP1571)
Blakemore WF, Franklin RJM (2008) Remyelination in experimental models of toxin-induced demyelination. In: Rodriguez M (ed) Advances in multiple sclerosis and experimental demyelinating diseases. Springer, Berlin Heidelberg, pp 193–212. https://doi.org/10.1007/978-3-540-73677-6_8
Matsushima GK, Morell P (2001) The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 11(1):107–116
Kipp M, Nyamoya S, Hochstrasser T, Amor S (2017) Multiple sclerosis animal models: a clinical and histopathological perspective. Brain Pathol 27(2):123–137
Zivadinov R, Dwyer MG, Markovic-Plese S, Kennedy C, Bergsland N, Ramasamy DP et al (2014) Effect of treatment with interferon beta-1a on changes in voxel-wise magnetization transfer ratio in normal appearing brain tissue and lesions of patients with relapsing-remitting multiple sclerosis: a 24-week, controlled pilot study. PLoS ONE 9(3):e91098
Zivadinov R, Dwyer M, Hussein S, Carl E, Kennedy C, Andrews M et al (2011) Voxel-wise magnetization transfer imaging study of effects of natalizumab and IFNβ-1a in multiple sclerosis. Mult Scler. 18(8):1125–1134
Eisen A, Greenberg BM, Bowen JD, Arnold DL, Caggiano AO (2017) A double-blind, placebo-controlled, single ascending-dose study of remyelinating antibody rHIgM22 in people with multiple sclerosis. Mult Scler J Exp Transl Clin. 3(4):2055217317743097–2055217317743097
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cunniffe, N., Coles, A. Promoting remyelination in multiple sclerosis. J Neurol 268, 30–44 (2021). https://doi.org/10.1007/s00415-019-09421-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09421-x