
Abstract
Background
Genetic variants are considered to have a crucial impact on the occurrence of ischemic stroke. In clinical routine, the diagnostic value of next-generation sequencing (NGS) in the medical clarification of acute juvenile stroke has not been investigated so far.
Material and methods
We analyzed an exome-based gene panel of 349 genes in 172 clinically well-characterized patients with magnetic resonance imaging (MRI)-proven, juvenile (age ≤ 55 years), ischemic stroke admitted to a single comprehensive stroke center.
Results
Monogenetic diseases causing ischemic stroke were observed in five patients (2.9%): In three patients with lacunar stroke (1.7%), we identified pathogenic variants in NOTCH3 causing cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Hence, CADASIL was identified at a frequency of 12.5% in the lacunar stroke subgroup. Further, in two male patients (1.2%) suffering from lacunar and cardioembolic stroke, pathogenic variants in GLA causing Fabry’s disease were present. Additionally, genetic variants in monogenetic diseases lacking impact on stroke occurrence, variants of unclear significance (VUS) in monogenetic diseases, and (cardiovascular-) risk genes in ischemic stroke were observed in a total of 15 patients (15.7%).
Conclusion
Genetic screening for Fabry’s disease in cardioembolic and lacunar stroke as well as CADASIL in lacunar stroke might be beneficial in routine medical work-up of acute juvenile ischemic stroke.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Genetic variants are considered to have a crucial impact on the occurrence of ischemic stroke. There are several well-defined monogenetic diseases causing ischemic stroke. Among these, cerebral small-vessel vasculopathies in lacunar stroke have been most frequently described. These include inter alia Fabry’s disease, cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), and retinal vasculopathy with cerebral leukoencephalopathy (RCVL) [1]. Furtherly, pathogenic variants in genes associated with cardiomyopathy and arrhythmia in embolic stroke, metabolic disorders in large artery occlusion, and connective tissue diseases in dissections are considered relevant to stroke occurrence [1,2,3,4,5,6,7]. In addition to these monogenetic diseases, genome-wide association studies (GWAS) have identified several genes resulting in a significantly elevated risk for ischemic stroke [2, 8,9,10,11]. The incidence of genetic diseases associated with stroke is controversially discussed and is likely more frequent than previously estimated [7, 12]. So far, whole-exome sequencing (WES) in the medical clarification of juvenile ischemic stroke has been applied within two research studies only: In a cohort of 22 northern European patients with familial clustering stroke [3] and a large study of 10.000 ischemic stroke patients, which is currently performed in China but results are pending [13]. Exome-based gene panel analysis has mainly been performed in cerebral small vessel disease (CSVD) applying small gene panels only [7, 14]. A larger gene panel analysis consisting of 181 genes has so far only been performed in 53 pre-selected Chinese ischemic stroke patients [5]. In clinical practice, genetic analysis is to date not a part of the routine diagnostic workup in juvenile stroke patients admitted to stroke units. German guidelines on diagnostics for acute stroke clarification recommend genetic testing in selected patients based on clinical phenotyping only [15]. However, establishing a molecular diagnosis may have an impact on the diagnostic work-up, secondary prophylactic therapeutic strategies, coping strategies, and genetic counseling of families [16]. Given the broad availability of next-generation sequencing in clinical routine, either by WES or gene panels, we aimed to assess the added diagnostic value of a gene panel analysis as defined by the observed frequency of genetic causes of ischemic stroke in a large cohort of acute juvenile magnetic resonance imaging (MRI)- proven stroke patients admitted to our stroke unit for acute stroke work up.
Material and methods
Written informed consent was obtained from all patients prior to WES and inclusion in our local biobank. The study was approved by the local ethics committee at the Technical University of Munich (project number 204/21S).
Patient cohort
For patient identification, our local neurological biobank was applied. The neurological biobank of the Technical University of Munich is a registered biobank sampling patient biomaterial including DNA probes from patients with different neurological diseases [17]. In a retrospective approach, we identified all samples of included ischemic stroke patients treated from 2013 to 2018 at our comprehensive stroke care center. An ischemic stroke was diagnosed following clinical examination and confirmed by diffusion-weighted MRI sequences (DWI) in each case as part of standard care. We included patients in genetic analysis suffering from an ischemic stroke with a first stroke event up to and including 55 years of age, which will be characterized as juvenile stroke in the following [18]. Transient ischemic attacks (TIA) and patients with hemorrhagic stroke were not considered for analysis. No other in- or exclusion criteria were applied. Clinical baseline data were retrospectively retrieved from medical records.
Standard treatment protocol
All included patients received standard stroke care, treatment and etiologic work-up according to national guidelines and following local standard operating procedures. This included Holter-ECG monitoring, sonographic examination of the blood supplying cervical and cranial vessels, cardiac echocardiography and laboratory examinations including lipid profile [19]. Stroke etiology was classified according to the international Trial of Org 10,172 in Acute Stroke Treatment criteria (TOAST) depending on diagnostic findings [20]. Family history was evaluated according to the letter of discharge and regarded as positive in cases of first-grade relatives suffering from ischemic stroke aged up to 55 years.
Whole exome sequencing
WES was performed at the Regeneron Genetics Center (New York, USA) as previously described [21]. Generated sequences corresponding to a 20-fold coverage in > 80% of target bases. Sufficient quality was ensured by imposing quality control exclusions based on contamination score (contamination > 5% via verifyBamID software & heterozygous/homozygous ratio), gender concordance, sample duplication, and exome-genotype concordance.
Definition of gene panel
Based on Ilinca et al.’s and Fang et al.’s suggestions for a gene panel on mendelian stroke, we defined a new panel consisting of 349 genes (Supplementary Table) [4, 5]. In 2020, Fang et al. proposed a panel consisting of 181 genes resulting in monogenetic diseases associated with stroke and 265 genes influencing the risk of stroke [5]. In 2018, Ilinca et al. described a panel of 120 genes with documented impact on stroke etiology in at least one patient, 62 genes with possible impact on stroke occurrence but lacking case report, and 32 risk genes detected by GWAS [4]. We combined both panels and added recent findings according to a search in PubMed. Consequently, the applied panel combined known monogenetic disorders causing ischemic stroke, genes resulting in a higher occurrence of cardiovascular risk factors, susceptibility genes for stroke and cardiovascular risk factors, and identified gene loci according to recent GWAS [2, 4, 5, 9, 11, 20].
Genetic analysis
WES data analysis was performed using the megSAP pipeline (https://github.com/imgag/megSAP). For variant analysis, the GSvar graphical user interface was used (https://github.com/imgag/ngs-bits/tree/master/doc/GSvar) [22]. Applying the gene panel described above, variants were evaluated according to their impact, allelic frequency in control databases (gnomAD and 1000 Genomes projects), and presence in HGMD and ClinVar sources [23, 24]. Copy Number Variations and structure variants were assessed. The pathogenicity of the identified variants was determined according to the American College of Medical Genetics and Genomics (ACMG) guidelines [25]. Accordingly, we subsumed our findings into two subgroups: Pathogenic or likely pathogenic variants in monogenetic diseases causing ischemic stroke and additional findings in genetic analysis. Ethnicity of the patients was estimated by comparing the frequencies of uncorrelated single nucleotide polymorphisms (SNPs) of our patients with individuals of major continental ancestries (European, African, and East Asian) from the 1000 Genomes panel version 3.
Statistical analysis
All statistical analyses were performed using SPSS (IBM, SPSS Statistics 28.0.1). Alpha error was set at 5%.
Results
Patient cohort
In total, 172 patients were identified with MRI-proven ischemic stroke aged up to 55 years. Baseline patient and stroke characteristics are depicted in Table 1. Most frequently observed were strokes of undetermined etiology (n = 77; 41.0%). A positive family history was documented in 9.9% (n = 17).
Patients with pathogenic variants in monogenetic diseases causing ischemic stroke
In five European patients, we identified an underlying monogenetic disease causing an ischemic stroke. Patient characteristics are depicted in Table 2.
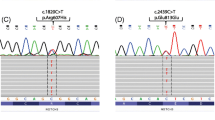
In three patients with lacunar stroke (patient number 1–3), we detected pathogenic, heterozygous missense variants in NOTCH3 causing CADASIL (c.1672C > T, p.Arg558Cys; c.544C > T, p.Arg182Cys; c.872G > A, p.Cys291Tyr) [3, 26,27,28]. CADASIL had already been genetically proven in one case (patient 1) and was suspected in another case (patient 2) based on clinical and cerebral imaging characteristics. In patient 3, CADASIL was not suspected by the treating clinicians lacking both a positive family history and migraine. MRI characteristic showed periventricular white matter hyperintensities, which were solely attributed to the presence of cardiovascular risk factors prior to WES. MRI findings in FLAIR and T2* weighted sequence (“heme” sequence) of each patient are shown in Fig. 1.

MRI images of the three CADASIL patients identified in our cohort. Patient 1 refers to the known CADASIL disease. 1.1: FLAIR sequence showing lacunar defects and extensive confluent white matter lesions, 1.2: FLAIR sequence, white matter lesions without emphasize on temporopolar region, 1.3: SWI sequence showing extensive microbleeds both thalamic and cortical. Patient 2 refers to the clinically suspected patient. 2.1: FLAIR sequence with extensive white matter lesions. 2.2: FLAIR Sequence, no emphasize on temporopolar, 2.3: T2* (“heme”) sequence showing supratentorial cortical microbleeds as well as thalamic microbleeds. Patient 3 refers to the clinically not suspected patient. 3.1 FLAIR sequence showing temporal white matter lesions and a lacunar defect in the thalamus, 3.2: FLAIR sequence, temporal white matter lesions, 3.3: T2* sequence, no microbleeds were shown
In two further male patients, a hemizygous pathogenic variant in GLA was detected (c.782dup, p.Trp262LeufsTer3; c.547 + 1G > A). Fabry’s disease was known in both patients. One patient suffered from vertebrobasilar lacunar stroke (patient 4). The other one (patient 5) suffered from cardioembolic stroke, in association with cardiomyopathy and atrial fibrillation, being treated with insufficient oral anticoagulants.
Patients with additional findings in genetic analysis
In total, we detected additional findings in 27 European patients. In one patient, two variants in cardiovascular risk factors became apparent. In contrast to the aforementioned patients, the detected variants were either pathogenic variants lacking association with stroke etiology, risk genes in ischemic stroke, or sufficient evidence for pathogenicity in stroke occurrence was missing. We consequently divided all observed variants into four subgroups:
-
(I)
Likely pathogenic and pathogenic variants in genes resulting in monogenetic diseases that did not explain the occurrence of stroke (n = 2; 1.2%) (TNNI3, KCQN1),
-
(II)
Variants of uncertain significance (VUS) that cannot conclude or exclude a monogenetic disease associated with stroke (n = 6; 3.5%) (JAK2, COL4A1, COL5A1, NOTCH3),
-
(III)
Variants in genes associated with an increased risk of stroke (n = 5; 2.9%) (RNF213, ABCC6, PROS1), and
-
(IV)
Variants in genes associated with cardiovascular risk factors (n = 15, 8.7%) (ABCA1, APOB, LPL).
The observed additional findings are listed in Table 3 depicting genetic and clinical information.
Discussion
We present an exome-based large gene panel analysis of patients with MRI-proven acute juvenile ischemic stroke representing a real-world study population admitted to a single comprehensive stroke care center. As a major finding, our data shows the clinically relevant frequency of CADASIL and Fabry’s disease in juvenile ischemic stroke patients.
With a frequency of 12.5% in lacunar stroke, the occurrence of CADASIL in our cohort is more prevalent than described in prior studies: Tan et al. detected CADASIL in eleven patients in their selected patient cohort of 950 lacunar stroke patients ≤ 70 years of age resulting in a total frequency of 1.2% [7]. Compared to our data, the difference in the observed frequency could be attributable to the varying age inclusion criteria. In another study, Ilinca et al. detected one pathogenic NOTCH3 variant in their cohort of 22 juvenile stroke patients ≤ 56 years with familial clustering of ischemic stroke corresponding to a frequency of 4.5% [3]. In contrast to these highly preselected studies, our data show that CADASIL needs to be considered in a comparatively unselected cohort and everyday juvenile stroke care.
Furtherly, three considerations for the indication of genetic testing on NOTCH3 were apparent. Firstly, given the case of CADASIL with negative family history in our patient cohort and as de-novo mutations in CADASIL have been reported before, genetic testing on NOTCH3 solely based on family history may be insufficient [29]. Secondly, imaging findings varied regarding localization of white matter lesions as well as the occurrence of microbleeds and, as shown in one case, combined with the presence of cardiovascular risk factors may bias clinical judgement [6]. Additionally, ethnicity has been shown to have an impact on the clinical as well as the radiological phenotype of CADASIL: The Asian population was found to present less migraine and seizures, but more intracerebral hemorrhage and a difference in the localization of white matter hyperintensities [30, 31]. Our data hence encourages genetic testing on NOTCH3 in all patients suffering from juvenile lacunar stroke.
Regarding Fabry’s disease, the frequency of 1.2% observed in our patient cohort confirms previous findings: A meta-analysis including nine studies by Shi et al. showed a prevalence of 0.4–2.6% of Fabry’s disease in juvenile stroke [32]. Typically, as observed in one patient with the p.Trp262LeufsTer3 variant, patients with Fabry’s disease present microvascular changes on MRI scans [33]. Cardioembolic stroke has additionally been reported in Fabry’s disease [33,34,35]. Our data underline the importance of screening for Fabry’s disease in lacunar juvenile stroke as well as cardioembolic and stroke of undetermined etiology.
We did not detect pathogenic variants in other genes associated with monogenetic diseases causing stroke. This is in accordance with previously published studies: In 950 patients Tan et al. detected only three cases with pathogenic variants in other monogenetic diseases causing stroke (HTRA1, COL4A1) apart from NOTCH3 [7]. Similarly, Coste et al. showed that COL4A1 and COL4A2 as well as APP, TREX1 and HTRA1 were much less frequent than pathogenic variants in NOTCH3 in patients suffering from cerebral small vessel disease (CSVD) advised on genetic testing [14].
In addition to the described pathogenic variants, we detected heterozygous VUS in four genes associated with monogenetic diseases causing an ischemic stroke (Table 3/II). A novel JAK2 variant (p.Arg1063His) was described in a patient with embolic stroke of undetermined etiology (ESUS) and familial clustering of ischemic juvenile stroke [3]. As prothrombotic status with and without erythrocytosis in patients carrying JAK2 variants has been shown, this variant might be considered relevant in ischemic stroke etiology [3]. In our patient cohort, the variant could be detected in a total of three patients (1.7%), which is more frequent than listed in controls (0.4–0.7%, Table 2). However, the phenotype of patients in our cohort was diverse and only one patient suffered from ESUS. Concluding, the impact of this variant on stroke etiology requires future characterization. Further, in one patient with a dissection of the internal carotid artery, we detected a VUS in COL5A1 (p.Pro1436Leu), a gene associated with fibromuscular dysplasia and artery dissection. In a retrospective approach, the impact of the variant on stroke etiology cannot be proven. Furtherly, we detected a VUS in COL4A1 (p.Thr1657Met) previously described as a novel variant in CSVD [7]. However, the stroke phenotype did not match. Lastly, a VUS in NOTCH3 (p.Gly1710Asp) was found in a patient with lacunar stroke, which has been reported in association with cerebral white matter lesions but not ischemic stroke [36, 37]. Considering the lack of a pathognomonic cysteine change and the conflicting interpretation of prediction tools (SIFT, Polyphen2), the variant seemed less likely relevant in stroke etiology.
Incidental findings in monogenetic analysis became apparent in two patients (1.2%) (Table 3/I): In one possibly pre-symptomatic patient, we detected a pathogenic variant in TNNI3 (p.Ser166Phe) causing autosomal dominant hypertrophic cardiomyopathy [38]. In another patient with Long-QT syndrome, we detected a pathogenic loss-of-function variant in KCNQ1 (p.Gln530Ter) [39]. In follow-up event recorder documentation, atrial fibrillation was not detected, a phenotype associated with gain-of-function variants in this gene. Compared to literature, the observed frequency of incidental findings in genetic testing can be expected [40]. Nevertheless, it addresses the ethical responsibility in genetic counselling and stresses the medical and possibly long-term impact of an incidental genetic finding on the patient and their family.
Risk genes for ischemic stroke became apparent in five patients (Table 3/III). In three patients, we detected pathogenic variants in ABCC6 (p.Arg1141*, p.Arg391Gly, p.Asn411Ser) [10, 41]. Bi-allelic variants are known to cause autosomal recessive pseudoxanthoma elasticum [42], a connective tissue disease resulting inter alia in arterial calcification with a high incidence of cardiovascular events including cerebral ischemia. In patients with heterozygous variants in this gene, an elevated odds ratio (OR) of 4.9 for ischemic stroke has been shown [10]. Secondly, in a patient with patent foramen ovale (PFO) and clinically assumed paradox embolic stroke, we detected a likely pathogenic variant in PROS1, which is in the heterozygous state associated with thrombophilia due to protein S deficiency (p.Thr78Met) [43]. Due to the retrospective analysis, protein S activity was not measured in our patient and causative connection cannot be proven. Lastly, in one patient, a heterozygous variant (p.Arg4019Cys) in RNF213, a susceptibility gene in Moyamoya disease, was present [44, 45]. However, the patient did not present angiographic signs of Moyamoya disease and incomplete penetrance has been shown before [45].
Summarizing, the frequent finding of additional genetic variants in 15.7% of our patient cohort emphasizes the possibility that rarer genetic diseases may account for juvenile ischemic stroke and highlights the relevance of future genetic research in stroke etiology. It has been shown before, that a genomic risk score may outperform classic risk factors concerning the predictive values of stroke recurrence [46]. The broad spectrum of genetic findings furtherly represents the diverse etiologies of ischemic stroke and stresses the necessity of individual stroke care and follow-up.
The strength of our study is a real-world study population. This implies that results may be applicable to other stroke centers. Limitations of this study refer to its retrospective approach. This implies that the genetic data of family members were not available for analysis. Further, the study was limited due to the small size of the study population and etiologic subgroups. The monocentric design and the dependency on our local biobank cannot exclude a possible selection bias. Lastly, the impossibility to detect intronic variants by WES is of importance.
To our knowledge, there are currently no recommendations on standardized genetic testing in stroke or even juvenile stroke patients [15]. Our data showed that CADASIL and Fabry’s disease constitute a frequent etiology of acute juvenile ischemic stroke and should hence already be considered in the initial work-up of patients. Given the divers stroke etiologies observed, screening for Fabry’s disease should be considered in all patients suffering from juvenile stroke of cardioembolic, lacunar, and undetermined etiology. Additionally, we suggest routine genetic testing for CADASIL in all patients suffering from a juvenile lacunar stroke. As described above, the presence of common cardiovascular risk factors may conceal the underlying genetic disease, consequently deteriorate the medical clarification of lacunar strokes, and interfere with the definition of clinically applicable red flags [6]. Only in cases with a more refined phenotype indicating a monogenetic stroke etiology based on characteristic extra- and intracerebral features or conclusive family history, a gene panel analysis based on NGS may have an additional diagnostic benefit. In those patients, exome and genome sequencing has two advantages over panel-based approaches targeting a predefined set of genes: Apart from the methodological advantages in detecting certain types of genetic variations, there is the prospect of identifying novel disease genes.
References
Bersano A et al (2020) Heritable and non-heritable uncommon causes of stroke. J Neurol 268(8):2780–2807
Malik R et al (2018) Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 50(4):524–537
Ilinca A et al (2020) Whole-exome sequencing in 22 young ischemic stroke patients with familial clustering of stroke. Stroke 51(4):1056–1063
Ilinca A et al (2019) A stroke gene panel for whole-exome sequencing. Eur J Hum Genet 27(2):317–324
Fang F et al (2020) Gene panel for Mendelian strokes. Stroke Vasc Neurol 5(4):416–421
Mancuso M et al (2020) Monogenic cerebral small-vessel diseases: diagnosis and therapy. Consensus recommendations of the European Academy of Neurology. Eur J Neurol 27(6):909–927
Tan RYY et al (2019) How common are single gene mutations as a cause for lacunar stroke? A targeted gene panel study. Neurology 93(22):e2007–e2020
NINDS Stroke Genetics Network (SiGN); International Stroke Genetics Consortium (ISGC). Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study. Lancet Neurol 15(2):174–184
Auer PL et al (2015) Rare and coding region genetic variants associated with risk of ischemic stroke: the NHLBI exome sequence project. JAMA Neurol 72(7):781–788
De Vilder EYG et al (2018) Pathogenic variants in the ABCC6 gene are associated with an increased risk for ischemic stroke. Brain Pathol (Zurich, Switzerland) 28(6):822–831
Malik R et al (2017) Common coding variant in SERPINA1 increases the risk for large artery stroke. Proc Natl Acad Sci USA 114(14):3613–3618
Rutten JW et al (2019) The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1–6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet Med 21(3):676–682
Cheng S et al (2021) Whole genome sequencing of 10K patients with acute ischaemic stroke or transient ischaemic attack: design, methods and baseline patient characteristics. Stroke Vascular Neurol 6(2):291–297
Coste T et al (2021) Heterozygous HTRA1 nonsense or frameshift mutations are pathogenic. Brain 144(9):2616–2624
Hennerici M. G., Kern R. et al (2017) S1-Leitlinie Diagnostik akuter zerebrovaskulärer Erkrankungen. In: Deutsche Gesellschaft für Neurologie, Hrsg. Leitlinien für Diagnostik und Therapie in der Neurologie. www.dgn.org/leitlinien. Accessed 13 Nov 2022
Ortiz A et al (2018) Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab 123(4):416–427
Gasperi C et al (2020) Genetic determinants of the humoral immune response in MS. Neurol (R) Neuroimmunol Neuroinflamm 7(5):e827
Ferro JM, Massaro AR, Mas J-L (2010) Aetiological diagnosis of ischaemic stroke in young adults. Lancet Neurol 9(11):1085–1096
Ikenberg B et al (2019) Neurosonography after mechanical thrombectomy for acute stroke treatment. J Neuroimaging 29(3):364–370
Adams HP Jr et al (1993) Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 24(1):35–41
Van Hout CV et al (2020) Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586(7831):749–756
Falb RJ et al (2021) Bi-allelic loss-of-function variants in KIF21A cause severe fetal akinesia with arthrogryposis multiplex. J Med Genet
Karczewski KJ et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443
Auton A et al (2015) A global reference for human genetic variation. Nature 526(7571):68–74
Richards S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–423
Mukai M et al (2020) Genotype-phenotype correlations and effect of mutation location in Japanese CADASIL patients. J Hum Genet 65(8):637–646
Liu X et al (2015) The genetic spectrum and the evaluation of CADASIL screening scale in Chinese patients with NOTCH3 mutations. J Neurol Sci 354(1):63–69
Joutel A et al (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383(6602):707–710
Coto E et al (2006) A new de novo Notch3 mutation causing CADASIL. Eur J Neurol 13(6):628–631
Kim Y, Lee S-H (2019) Novel characteristics of race-specific genetic functions in Korean CADASIL. Medicina 55(9):521
Liao YC et al (2015) Characterization of CADASIL among the Han Chinese in Taiwan: distinct genotypic and phenotypic profiles. PLoS One 10(8):e0136501
Shi Q et al (2014) Prevalence of Fabry disease in stroke patients—a systematic review and meta-analysis. J Stroke Cerebrovasc Dis 23(5):985–992
Kolodny E et al (2015) Cerebrovascular involvement in Fabry disease. Stroke 46(1):302–313
Lenders M et al (2015) Thromboembolic events in Fabry disease and the impact of factor V Leiden. Neurology 84(10):1009–1016
Rolfs A et al (2005) Prevalence of Fabry disease in patients with cryptogenic stroke: a prospective study. Lancet 366(9499):1794–1796
Schmidt H et al (2011) Genetic variants of the NOTCH3 gene in the elderly and magnetic resonance imaging correlates of age-related cerebral small vessel disease. Brain 134(Pt 11):3384–3397
Ross OA et al (2013) NOTCH3 variants and risk of ischemic stroke. PLoS One 8(9):e75035–e75035
van Lint FHM et al (2019) Large next-generation sequencing gene panels in genetic heart disease: yield of pathogenic variants and variants of unknown significance. Neth Heart J 27(6):304–309
Chen YH et al (2003) KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 299(5604):251–254
Elfatih A et al (2021) Frequency and management of medically actionable incidental findings from genome and exome sequencing data: a systematic review. Physiol Genomics 53(9):373–384
Legrand A et al (2017) Mutation spectrum in the ABCC6 gene and genotype-phenotype correlations in a French cohort with pseudoxanthoma elasticum. Genet Med 19(8):909–917
Kauw F et al (2017) Cerebral disease in a nationwide Dutch pseudoxanthoma elasticum cohort with a systematic review of the literature. J Neurol Sci 373:167–172
Do MD et al (2021) Recurrent PROC and novel PROS1 mutations in Vietnamese patients diagnosed with idiopathic deep venous thrombosis. Int J Lab Hematol 43(2):266–272
Kobayashi H et al (2016) RNF213 rare variants in Slovakian and Czech Moyamoya disease patients. PLoS One 11(10):e0164759–e0164759
Cecchi AC et al (2014) RNF213 Rare variants in an ethnically diverse population with Moyamoya disease. Stroke 45(11):3200–3207
Abraham G et al (2019) Genomic risk score offers predictive performance comparable to clinical risk factors for ischaemic stroke. Nat Commun 10(1):5819
Acknowledgements
The Authors thank our colleagues from the Department of Neuroradiology, Klinikum rechts der Isar, for the collaboration and acquisition of MRI Data within clinical routine and the provision of data for this study.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
All authors have read and approved the submitted manuscript, the manuscript has not been submitted elsewhere nor published elsewhere in whole or in part.
Corresponding author
Ethics declarations
Conflicts of interest
BH received funding from Regeneron Pharmaceuticals for genetic research in Multiple Sclerosis. The other authors declare that they have no conflict of interest.
Additional information
The members of the “Regeneron Genetics Center” in supplementary material “Banner author list and contribution statements”.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Härtl, J., Hartberger, J., Wunderlich, S. et al. Exome-based gene panel analysis in a cohort of acute juvenile ischemic stroke patients:relevance of NOTCH3 and GLA variants. J Neurol 270, 1501–1511 (2023). https://doi.org/10.1007/s00415-022-11401-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-022-11401-7