
Abstract
Two-thirds of published patients with anti-leucine rich, glioma inactivated 1 (LGI1) encephalitis develop hippocampal sclerosis (HS). It is likely that this contributes to residual cognitive long-term deficits and the risk of epilepsy. Almost all patients harbor anti-LGI1-immunoglobulin G-(IgG-) subclass 4, which is considered a “benign”, non-destructive subclass. In contrast, neuropathological case studies have suggested that the classical complement cascade may contribute to mediotemporal cell death in patients with LGI1 antibodies. IgG subclasses 1, 2, or 3 are required to initiate this cascade. We hypothesized that patients with these anti-LGI1-IgG1/2/3 in addition to IgG4 have a higher risk of developing HS than patients with anti-LGI1-IgG4 alone. We retrospectively assessed all anti-LGI1 encephalitis patients from this center with anti-LGI1-IgG-subclass information and follow-up MRI available. Nine out of 20 patients had developed HS (45%). Volumetric FreeSurfer analysis confirmed the visual HS diagnoses. HS and a lower hippocampal volume were associated with anti-LGI1-IgG1/2/3. All six patients with this IgG subclass status developed HS. There was no association with older or younger age at onset, female sex, longer latency from disease onset to start of immunotherapy, less intense immunotherapy, higher serum titers of LGI1 antibodies, LGI1 antibodies in CSF or higher LGI1-specific antibody indices. There was no association between anti-LGI1-IgG1/2/3 status and neuropsychological performance, epilepsy, or general neurological performance. This confirms our hypothesis that anti-LGI1-IgG1/2/3 in serum puts patients at risk of developing HS. If these findings can be confirmed and clinically corroborated, patients with anti-LGI1-IgG1/2/3 might become candidates for anti-complement-directed immunological treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anti-leucine-rich, glioma-inactivated protein 1 (LGI1) encephalitis is one of the more common forms of autoimmune encephalitis. It mostly manifests as faciobrachial dystonic seizures (FBDS) [45] or limbic encephalitis [1, 32, 47]. Enduring structural hippocampal damage (in the literature termed “hippocampal atrophy”, “hippocampal sclerosis” [HS] or “hippocampal lesion”) is a complication of the condition. According to the existing series, 47–96% of patients develop hippocampal structural damage after median follow-ups of 12–40 months (pooled data: 127/199, 64%) [7, 12, 17, 27, 32, 35, 42, 44]. Neuropathological case studies have documented hippocampal nerve cell loss and gliosis in anti-LGI1 limbic encephalitis [4, 11, 25, 40], i.e., HS [6]. It is plausible that this structural hippocampal damage underlies the residual memory deficits after the active phase of anti-LGI1 encephalitis [12] and possibly the rare development of epilepsy [17].
The following pathogenic mechanism of LGI1 antibodies has been established: in synapses containing presynaptic Kv1.1 potassium channels and postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR), LGI1 antibodies disrupt the interaction between the secreted protein LGI1 and postsynaptic ADAM22 as well as LGI1 and presynaptic ADAM23. This disruption leads to a reduction of Kv1.1 potassium channels and AMPAR densities and thereby causes (i) neural hyperexcitability manifesting as epileptic seizures and (ii) decreased plasticity resulting in memory deficits [34, 35, 38]. Others have observed the internalization of the LGI1-ADAM22/23 complex [38]. Seizures and memory deficits usually improve with immunotherapy and sometimes in parallel to falling antibody titers [37]. It is, therefore, plausible that these pathomechanisms underlie the remediable aspects of anti-LGI1 encephalitis.
For irreversible structural mediotemporal damage and residual cognitive deficits, another mechanism has been described: the classical complement cascade leading to activation of the terminal membrane attack complex, which permits water influx into the target cell and its subsequent death. C9neo, indicative of functional activation of the complement cascade, as well as signs of cell death have been observed in the mediotemporal neurons of two patients with anti-LGI1 limbic encephalitis [4, 25]. Confirmatory evidence for hippocampal and amygdalar C9neo deposition comes from spontaneous limbic encephalitides with LGI1 antibodies in five cats [46]. Complement activation by LGI1 antibodies has also been demonstrated in vitro [20]. This mechanism, however, seems at odds with the observation that almost all patients with anti-LGI1 encephalitis harbor anti-LGI1-immunoglobulin G-(IgG-)4 antibodies [1, 3, 14, 31], so that anti-LGI1 encephalitis has been subsumed under the group of IgG4-mediated neurological autoimmune disorders [19]. IgG4 antibodies are functionally monovalent and bispecific. They are well suited to inhibiting protein-protein-interactions [19], but they do not or only minimally bind C1 as the starter of the classical complement cascade [5, 10, 41]. Complete IgG antibodies of the subclasses 1, 2, or 3 (IgG1/2/3), however, can bind C1 in the order of strength IgG3>IgG1>>IgG2 [2, 21]. In fact, serum anti-LGI1-IgG1/2/3 were found in a relevant proportion of patients by four studies [1, 3, 14, 31], see Table 1.
We hypothesized that patients harboring anti-LGI1-IgG1/2/3 might be at an increased risk of developing HS and having lower hippocampal volumes. Alternative hypotheses were that predictors of a (differently defined) “poor outcome” might also be associated with the development of HS and lower hippocampal volumes. These factors are: older [24, 31] or younger age at onset [44], female sex [31, 44], longer duration between disease onset and the start of immunotherapy [12, 17, 45], less intense immunotherapy [43], higher titers of serum LGI1 antibodies [37], LGI1 antibodies in the CSF [24, 31], and higher specific antibody indices indicating the intrathecal synthesis of LGI1 antibodies [14]. In addition, we tested the hypothesis that a mediotemporal lesion on the earliest available MRI predicts the development of HS.
Patients and methods
We retrospectively identified all patients with anti-LGI1 encephalitis who were examined and treated between 2011 and 2023 in The Mara and included those with this minimum set of information: anti-LGI1-IgG subclasses in serum at first visit and a follow-up MRI.
Clinical data
The dates of birth, disease onset, making the diagnosis of anti-LGI1 encephalitis and start and types of immunotherapies, as well as antibody titers, were obtained from hospital records. For bivariate associations, the titers were expressed as log2 values to reflect the dilution series, which were multiples of 1:2.
Patients were invited for a prospective outpatient follow-up visit with co-author AR, including brain MRI and neuropsychological investigation. For patients who did not attend, the most recent follow-up data from clinical visits were used. As neuropsychological outcome measures, we noted the results of each patient’s most recent Verbal Learning and Memory Test (VLMT, a derivate of the Rey Auditory Verbal Learning and Memory Test [RAVLT]) [18] with the domains learning (d5, d1–5) and free recall (d7 and d5–d7) in the form of raw values. This type of verbal memory test has proven useful in the cognitive assessment of patients with limbic encephalitis and LGI1 antibodies [12, 50]. We also checked whether epilepsy according to our recent definition [36] was present at most recent follow-up. Co-author AR determined the modified Rankin Score (mRS) [15] and the Clinical Assessment Scale in Autoimmune Encephalitis (CASE) [26] at follow-up before knowing the anti-LGI1-IgG1/2/3 subclass status.
Antibody diagnostics, IgG subclass determination
LGI1 antibody titers in serum and CSF were those of the original clinically motivated investigation of the earliest available samples. Antibodies were detected using commercially available biochips (Euroimmun, Lübeck, Germany). These are assemblies of multiple cell-based assays (CBA) consisting of human embryonic kidney (HEK-293) cells, including cells transfected with a plasmid encoding for LGI1. These were fixed with paraformaldehyde. The indirect immunofluorescence protocol followed the manufacturer’s recommendations (Euroimmun, FA 112d-1005-6, IgG) with modifications: buffer: phosphate-buffered saline (PBS); patient serum diluted to 1:20, CSF undiluted; secondary antibody I (for sensitivity): biotinylated goat-anti-human IgG heavy and light chain (H+L, Jackson ImmunoResearch 109-065-088), 1:100, incubation time 30 min at room temperature (RT), subsequently visualized by incubation with streptavidin coupled with Alexa 594 (Jackson ImmunoResearch 016-580-084), 1:400, 30 min, RT; secondary antibody II (for specificity, simultaneous application with anti-IgG-HL): goat-anti-human antibody against the Fcγ fragment of IgG, conjugated with Alexa Fluor 488 (Jackson Immunoresearch 109-545-098) 1:200, 30 min, RT; nuclear counterstaining with Hoechst 33342, 1:10000; embedding with 1,4-Diazabicyclo[2.2.2]octan. Experienced technicians endpoint-titrated LGI1 antibodies with the anti-Fc secondary antibody in multiples of 1:2. The results were checked by an experienced neurologist (CIB or CGB) and, if necessary, corrected. As different dilution steps were used over the study period, the results were not multiples of each other throughout. We repeated the titration of five LGI1 antibody-positive sera, determined originally ten years ago and stored at – 20 ℃, and obtained results with ≤1 dilution stage difference. This confirmed the stability of the antibodies and consistency of the investigators’ sensitivity.
As part of the diagnostic process, samples were also tested at 1:40 (serum) and 1:2 (CSF) by default on a tissue-based assay in form of unfixed sagittal mouse brain slices containing hippocampus, brain stem, and cerebellum (Euroimmun, Lübeck, Germany) to detect a confirmatory LGI1 neuropil staining [33]. The protocol was identical to that for the CBA.
Anti-LGI1-IgG subclasses in serum were determined in 2023 by CBA with the following secondary antibodies used in a previous study [3]: biotinylated mouse anti-human-IgG1 (Biozol NMB-MAHU-IGG1-BIO), 1:20, 1 h, RT; biotinylated mouse anti-human-IgG2 (Biozol NMB-MAHU-IGG2-BIO), 1:200, 1 h, RT; biotinylated mouse anti-human-IgG3 (Sigma-Aldrich B3523), 1:200, 1 h, RT; biotinylated mouse anti-human-IgG4 (Sigma-Aldrich B3648), 1:200, 1 h, RT. All were subsequently visualized with streptavidin-Alexa 594, 1:200, 30 min, RT. In four cases, not enough serum was left for renewed subclass determination. In those cases, we used the subclass results of a previous study [3], determined in 2017. Even if enough CSF was available, there were hardly any positive IgG subclass stainings in our hands, so we did not include anti-LGI1-IgG subclasses in CSF in this study.
Specific LGI1-antibody indices were calculated according to the following formula; in theory, values >1 would suggest intrathecal synthesis, but for reasons of specificity, a cutoff >4 with the use of titers has been suggested [39]:
MRI assessment
The diagnoses “HS” or “no HS” were taken from FGW´s original clinical readings of the individual patient’s most recent MRIs performed at our center (3 Tesla Siemens Magnetom Verio or Vida). The visual diagnosis of HS relied on a reduced hippocampal volume and abnormally high signal on FLAIR/T2 images [48]. FGW re-checked his diagnoses for the purposes of the study and confirmed all of them. Using 3D T1 weighted magnetization-prepared rapid gradient echo (MPRAGE, TR 1900 msec, TE 3 msec, TI 900 msec; voxel dimensions 0.8 x 0.8 x 0.8 mm3) scans of the most recent available MRI studies, hippocampal volumes were automatically segmented (FreeSurfer, version 7.3.2, standard analysis [13]). We added the volumes of left and right hippocampi of the individual patients and divided the result by two to obtain one mean hippocampal volume per patient.
Statistics
The Fisher’s exact test and Mann–Whitney U test were used for group comparisons. Bivariate associations were calculated as Pearson correlation (r), point–biserial correlation (rpbis) or Phi coefficient depending on the constellation of variables. Statistical analyses were performed using IBM SPSS Statistics (version 29) and R software (version 4.3.0, package “epiR”). The significance level was set to α = 0.05.
Ethics
The Ethics committee of the University of Münster approved the study (2020-244-f-S) and patients gave informed consent for the use of their data. In the case of those who could not be reached for consent, the Ethics committee waived it in accordance with the Gesundheitsdatenschutzgesetz Nordrhein-Westfalen (North-Rhine-Westphalian law of healthcare data protection) because it was a retrospective data assessment of personally studied patients.
Results
We identified 26 patients with anti-LGI1 encephalitis. Two patients were excluded because of missing serum for subclass testing, one because the patient did not return for follow-up (so the MRI outcome was not known) and another three patients with serum LGI1 antibody titers <1:100 because their sera did not give any signal upon anti-LGI1-IgG subclass 1–4 testing. This left 20 patients (nine females) for the study with LGI1 antibody serum titers between 1:160 and 1:8000. Among them, 16 patients had confirmatory serum or CSF neuropil staining on the tissue-based assay or were CBA-positive with CSF, as required by authorities in the field [9]. Four patients [nos. 8 (serum titer 1:160), 14 (1:320), 19 (1:160) and 20 (1:160)] did not fulfill these criteria, but the LGI1 antibody positivity was corroborated by the IgG4 positivity, which is unlikely to occur by chance since the IgG4 subclass is estimated as making up for only 5% of the total amount of IgG [23]; more importantly, no better diagnosis than anti-LGI1 encephalitis could be made after a full diagnostic work-up (nos. 14 and 20: FBDS; nos. 8 and 19: limbic encephalitis). Sixteen patients had a prospective follow-up with AR. The individual patient data are summarized in Table 2 and Supplementary Table 1. Immunotherapy regimens are described in Supplementary Information 1.
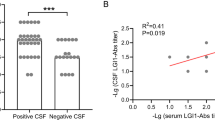
Nine patients (45%) had developed HS (seven unilateral, two bilateral) at the most recent examination, which took place after a median of 31 months after disease onset (range 4–173). All nine patients had IgG4 antibodies, which were the only subclass in three; five patients had in addition anti-LGI1-IgG1 antibodies (two of them with concomitant IgG2 and 3 antibodies) and one patient IgG2 antibodies (Table 2). All six patients with anti-LGI1-IgG1/2/3 antibodies developed HS. Representative case-wise anti-LGI1-IgG subclass stainings and follow-up MRIs are shown in Fig. 1. Anti-LGI1-IgG1/2/3 antibodies were significantly more frequent in patients who later developed HS (p = 0.002, Fisher’s exact text, two-tailed); the sensitivity of the anti-LGI1-IgG1/2/3-subclass status predicting HS was 0.67 (95% confidence interval [CI] 0.30–0.93), the specificity 1.00 (95% CI 0.72–1.00), the positive predictive value 1.00 (95% CI 0.54–1.00), and the negative predictive value 0.79 (95% CI 0.49–0.95). Hippocampal volumes significantly differed between patients with and without anti-LGI1-IgG1/2/3 (p = 0.012, Fig. 2A), as well as those with and without diagnosis HS (p < 0.001, Mann–Whitney U tests, Fig. 2B). The HS had already been detected as early as 7.5 months (median; range 1.4–20.2 months) after disease onset.

Five exemplary anti-leucine-rich, glioma inactivated 1-(LGI1-)immunoglobulin G subclass studies on HEK cells transfected with LGI1 and the patients’ most recent coronal MRI. The patient numbers in the first column are those in Table 2. MRI sections go through the hippocampi. Affected hippocampi are marked by arrows. Fluid-attenuated inversion recovery (FLAIR) images emphasize the increased signal in the affected hippocampi, while T2 images highlight the hippocampal atrophy. The time specifications give the latencies from disease onset to the respective depicted diagnostics. In the MRI columns, time specifications in brackets indicate when hippocampal sclerosis was detectable for the first time. Bar: 15 µm, valid for all cell-based assays

Hippocampal volumes in relation to anti-leucine rich, glioma inactivated 1-(LGI1-)immunoglobulin G (IgG) subclass status and the visual diagnosis “hippocampal sclerosis”. A Anti-LGI1-IgG1/2/3 status vs. hippocampal volume, p = 0.012. Open squares and circles: cases with the visual diagnosis of “hippocampal sclerosis”. B “Hippocampal sclerosis” vs. hippocampal volume, p < 0.001. Open squares and circles: cases with anti-LGI1-IgG1/2/3 antibodies. Statistical test: Mann–Whitney U Test, two-tailed. Bars: Medians and interquartile ranges. HS hippocampal sclerosis, L left side, R right side
The anti-LGI1-IgG1/2/3 status was significantly associated with HS (Phi=0.72, p<0.001) and with hippocampal volume (rpbis = − 0.46, p=0.041; Table 3). Apart from that, the number of immunotherapies was significantly associated with “HS” and not with “hippocampal volume”; importantly, the association was in the inverse direction to that which had been hypothesized: more (not less) immunotherapies were associated with “HS”. Similarly, the specific LGI1 antibody index correlated with “hippocampal volume” (not with “HS”) but was again inversely correlated to the hypothesis as higher indices were correlated to larger (undamaged) hippocampi. None of the other hypothesized factors was associated with the dependent variables “HS” or “hippocampal volume”: sex, age at onset, latency from disease onset to start of immunotherapy, LGI1 antibody titer in serum or CSF (log2), latency from start of immunotherapy to follow-up MRI, or mediotemporal lesion on the earliest available MRI scan (Table 3).
There was no association of the anti-LGI1-IgG1/2/3 status with verbal learning or memory (data available for 4/6 patients from the LGI1-IgG1/2/3-positive and 11/14 from the negative group after a median follow-up of 52 months, range 4 months–14 years; all p > 0.500, data not shown). In no patient could epilepsy be diagnosed. There was no association with the mRS (rpbis = − 0.18, p = 0.436) or CASE scores (rpbis = − 0.05, p = 0.830).
Discussion
In this sample of 20 patients with anti-LGI1 encephalitis and a 100% rate of anti-LGI1-IgG4 serum antibodies, the additional presence of potentially complement activating anti-LGI1-IgG1/2/3 was the only factor that was associated with HS and reduced hippocampal volumes according to the initial hypotheses.
This fits well with the previous neuropathological observation of mediotemporal Ig deposition and C9neo expression, which indicates the functional activation of the classical complement cascade, together with neural cell death in affected humans [4, 25]. Cats with this disease exhibit the same features [22]. In the cats, a breakdown of the mediotemporal vascular tight junctions with leakage of proteins from the blood stream into the CNS explains the spatial selectivity of the disease process for limbic structures [46]. Alternatively or additionally, anti-LGI1-IgG1/2/3 could exert other downstream IgG effects such as internalization of the LGI1-ADAM22/23 complex [38]. It would need to be demonstrated how this mechanism may lead to HS.
The specificity and positive predictive value of anti-LGI1-IgG1/2/3 positivity for HS development was 100%. Sensitivity and negative predictive values were lower since three patients developed HS in the absence of anti-LGI1-IgG1/2/3. This suggests that either the test is not sensitive enough or that other factors can lead to HS.
Other analyzed factors were sex, age at onset, latency from disease onset to start of immunotherapy, LGI1 antibody titer in serum or CSF, specific LGI1 antibody index, number of immunotherapies, latency from start of immunotherapy to follow-up MRI and mediotemporal lesion on the earliest available MRI scan; none were associated with the dependent variables “HS” or “hippocampal volume”. HS was associated with more immunotherapies, which is probably not causative but rather the consequence of a more severe disease course, likely triggered by the anti-LGI1-IgG1/2/3; also, stronger intrathecal synthesis of the antibody was associated with larger hippocampal volumes, which is counterintuitive.
The MRI diagnosis “HS” is the typical expression of acquired hippocampal damage with nerve cell loss and gliosis [6]. The visual assessment acknowledges volume and signal differences in parallel to detect hippocampal damage with high sensitivity [48]. The FreeSurfer approach is objective but limited to volume measurement and was, therefore, primarily used as a control step of the visual results. Indeed, lower volumes were associated with the diagnosis “HS”, which supports the validity of the visual diagnoses (Figure 2B).
The study that is most comparable to our investigation focused on the intrathecal production of LGI1 antibodies (specifically of anti-LGI1-IgG4). Apart from some differences in methodology, the authors chose mRS and not HS on MRI as their primary outcome variable. The authors found an association for intrathecal LGI1 antibody synthesis but not anti-LGI1-IgG subclass status with mRS or with “MRI”; they did not give details on the latter [14].
Features of anti-LGI1 encephalitis suggesting a pathophysiological action of bivalent, i.e. IgG1/2/3, activity have been observed before: LGI1 antibodies were experimentally able to internalize LGI1-ADAM22/23 complexes, which is impossible for IgG4 [34, 35, 38, 45]; another study found that patients with FBDS and cognitive impairment had a higher proportion of LGI1-IgG1 antibodies compared to those with FBDS only (MRI data were not given) [45].
If our observation is confirmed in other, ideally larger cohorts, if a clinical relevance becomes evident and if more neuropathological evidence supports the complement-hypothesis, the use of complement-inhibiting compounds (like eculizumab, ravulizumab, and zilucoplan, licensed for myasthenia gravis [8]) could become a specific therapeutic option for patients with anti-LGI1-IgG subclasses 1/2/3. Simply giving more standard immunotherapies, however, does not seem to be beneficial.
Limitations
The number of patients in this series is limited. This results from the fact that we intended to have as precise and comparably collected MRI and clinical data as possible, which are difficult to obtain in laboratory-based studies. There is the broad range of follow-up latencies but no statistical difference between anti-LGI1-IgG1/2/3-positive and -negative patients. The retrospective nature of the study may have introduced bias. To minimize it, co-authors assessing the MRIs (FGW, MM) and the clinical outcomes (AR) were blind for the subclass status of the patients. In 4/20 cases, LGI1 serum antibodies were neither confirmed by a serum or CSF neuropil pattern on the tissue-based assay nor by CSF positivity on the CBA. Limited sensitivity for LGI1 antibodies in CSF has been documented with the Euroimmun assay [28]; this, however, was not the case with serum in one dedicated study [33]. For the validity of the LGI1 serum positivity, we relied on the typical clinical presentation and anti-LGI1-IgG4 reactivity.
It is possible that our IgG subclass determination is not sufficiently sensitive; the positivity rates for the anti-LGI1-IgG subclasses lies, however, well in the range of previous studies (with the French group standing out by a particularly high rate of IgG1), see Table 1. In the present study, the negative anti-LGI1-IgG1-4 staining in sera with low titer antibodies and in CSF would speak in favor of insufficient sensitivity; other groups had the same difficulties [14, 16]. At present, there is no other method with which the CBA subclass results could be validated or improved in sensitivity.
The lack of an association of anti-LGI1-IgG1/2/3 with a poorer verbal memory outcome is probably related to the fact that the left hippocampus in the HS group was not always affected, which subsides verbal memory [49]. On the other hand, memory performance can deteriorate in this ageing population even in the absence of HS. Neuropsychological studies in autoimmune encephalitis in general, and even in the more circumscribed domain of limbic encephalitis, have been considered essential for the follow-up of patients to inform treatment decisions [50]. Systematic, group-wise studies of affected individuals, however, have been less discriminative and informative than, e.g., in the context of epilepsy surgery. For example, specific associations of autoantibody types with memory performance or other neuropsychological domains could not be found acutely or in the chronic phase [29, 30]. Reasons for this may be the small numbers of patients, the dynamic nature of autoimmune encephalitis, the right-sided, left-sided or bilateral affection within one antibody group (as in the present study) and the higher age of individuals with anti-LGI1 encephalitis with potentially concomitant and overlapping deficits [31]. Such deficits may have existed pre-morbidly, may have evolved for non-encephalitis-related reasons with age, or both. Of note, Finke et al. found correlations between some MRI measures of hippocampal subfield atrophy and domains of the RAVLT [12]. No association between the IgG subclass status and clinical outcome was reported by Muñiz-Castrillo [31]
While neuropsychological measures may, therefore, be too sensitive to detect differences in a small sample, the mRS and CASE scores may not be sensitive enough, especially in the psychological domain. Also, none of our patients developed epilepsy [36]; therefore, with this parameter, too, no difference between anti-LGI1-IgG1/2/3-positive and -negative patients could be detected.
Conclusion
Anti-LGI1-IgG1/2/3 antibodies carry a high risk of the development of HS. The neuropathological evidence of activation of the classical complement with spatially related mediotemporal nerve cell loss in humans and in cats is a plausible link to neural death and hippocampal atrophy. HS may contribute to a poorer outcome of anti-LGI1 encephalitis, even though this could not be demonstrated in our limited number of patients. If our findings are corroborated, if the clinical relevance can be demonstrated and if further neuropathological work confirms the complement hypothesis, anti-complement-directed compounds may become useful in patients with anti-LGI1-IgG1/2/3 antibodies.
Data availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
References
Ariño H, Armangue T, Petit-Pedrol M, Sabater L, Martinez-Hernandez E, Hara M, Lancaster E, Saiz A, Dalmau J, Graus F (2016) Anti-LGI1-associated cognitive impairment: presentation and long-term outcome. Neurology 87:759–765. https://doi.org/10.1212/WNL.0000000000003009
Augener W, Grey HM, Cooper NR, Muller-Eberhard HJ (1971) The reaction of monomeric and aggregated immunoglobulins with C1. Immunochemistry 8:1011–1020. https://doi.org/10.1016/0019-2791(71)90489-7
Bien CG, Bien CI, Dogan Onugoren M, De Simoni D, Eigler V, Haensch C-A, Holtkamp M, Ismail FS, Kurthen M, Melzer N, Mayer K, von Podewils F, Rauschka H, Rossetti AO, Schäbitz W-R, Simova O, Witt K, Höftberger R, May TW (2020) Routine diagnostics for neural antibodies, clinical correlates, treatment and functional outcome. J Neurol 267:2101–2114. https://doi.org/10.1007/s00415-020-09814-3
Bien CG, Vincent A, Barnett MH, Becker AJ, Blumcke I, Graus F, Jellinger KA, Reuss DE, Ribalta T, Schlegel J, Sutton I, Lassmann H, Bauer J (2012) Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 135:1622–1638. https://doi.org/10.1093/brain/aws082
Bindon CI, Hale G, Bruggemann M, Waldmann H (1988) Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med 168:127–142. https://doi.org/10.1084/jem.168.1.127
Blümcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A, Bernasconi N, Bien CG, Cendes F, Coras R, Cross JH, Jacques TS, Kahane P, Mathern GW, Miyata H, Moshé SL, Oz B, Özkara Ç, Perucca E, Sisodiya S, Wiebe S, Spreafico R (2013) International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE commission on diagnostic methods. Epilepsia 54:1315–1329. https://doi.org/10.1111/epi.12220
Celicanin M, Blaabjerg M, Maersk-Moller C, Beniczky S, Marner L, Thomsen C, Bach FW, Kondziella D, Andersen H, Somnier F, Illes Z, Pinborg LH (2017) Autoimmune encephalitis associated with voltage-gated potassium channels-complex and leucine-rich glioma-inactivated 1 antibodies—a national cohort study. Eur J Neurol 24:999–1005. https://doi.org/10.1111/ene.13324
Dalakas MC (2022) Role of complement, anti-complement therapeutics, and other targeted immunotherapies in myasthenia gravis. Expert Rev Clin Immunol 18:691–701. https://doi.org/10.1080/1744666x.2022.2082946
Dalmau J, Graus F (2023) Autoimmune encephalitis: Misdiagnosis, misconceptions, and how to avoid them. JAMA Neurol 80:12–14. https://doi.org/10.1001/jamaneurol.2022.4154
Damelang T, de Taeye SW, Rentenaar R, Roya-Kouchaki K, de Boer E, Derksen NIL, van Kessel K, Lissenberg-Thunnissen S, Rooijakkers SHM, Jongerius I, Mebius MM, Schuurman J, Labrijn AF, Vidarsson G, Rispens T (2023) The influence of human IgG subclass and allotype on complement activation. J Immunol 211:1725–1735. https://doi.org/10.4049/jimmunol.2300307
Dunstan EJ, Winer JB (2006) Autoimmune limbic encephalitis causing fits, rapidly progressive confusion and hyponatraemia. Age Ageing. 35:536–537. https://doi.org/10.1093/ageing/afl045
Finke C, Prüss H, Heine J, Reuter S, Kopp UA, Wegner F, Then Bergh F, Koch S, Jansen O, Münte T, Deuschl G, Ruprecht K, Stöcker W, Wandinger KP, Paul F, Bartsch T (2017) Evaluation of cognitive deficits and structural hippocampal damage in encephalitis with leucine-rich, glioma-inactivated 1 antibodies. JAMA Neurol 74:50–59. https://doi.org/10.1001/jamaneurol.2016.4226
Fischl B (2012) FreeSurfer Neuroimage 62:774–781. https://doi.org/10.1016/j.neuroimage.2012.01.021
Gadoth A, Zekeridou A, Klein CJ, Thoreson CJ, Majed M, Dubey D, Flanagan EP, McKeon A, Jenkins SM, Lennon VA, Pittock SJ (2018) Elevated LGI1-IgG CSF index predicts worse neurological outcome. Ann Clin Transl Neurol 5:646–650. https://doi.org/10.1002/acn3.561
Graus F, Vega F, Delattre JY, Bonaventura I, Rene R, Arbaiza D, Tolosa E (1992) Plasmapheresis and antineoplastic treatment in CNS paraneoplastic syndromes with antineuronal autoantibodies. Neurology 42:536–540. https://doi.org/10.1212/WNL.42.3.536
Greguletz P, Plötz M, Baade-Büttner C, Bien CG, Eisenhut K, Geis C, Handreka R, Klausewitz J, Körtvelyessy P, Kovac S, Kraft A, Lewerenz J, Malter M, Nagel M, von Podewils F, Prüß H, Rada A, Rau J, Rauer S, Rößling R, Seifert-Held T, Siebenbrodt K, Sühs KW, Tauber SC, Thaler F, Wagner J, Wickel J, Leypoldt F, Rittner HL, Sommer C, Villmann C, Doppler K (2024) Different pain phenotypes are associated with anti-Caspr2 autoantibodies. J Neurol 271:2736–2744. https://doi.org/10.1007/s00415-024-12224-4
Guery D, Cousyn L, Navarro V, Picard G, Rogemond V, Bani-Sadr A, Shor N, Joubert B, Muñiz-Castrillo S, Honnorat J, Rheims S (2022) Long-term evolution and prognostic factors of epilepsy in limbic encephalitis with LGI1 antibodies. J Neurol 269:5061–5069. https://doi.org/10.1007/s00415-022-11162-3
Helmstaedter C, Lendt M, Lux S (2000) VLMT: Verbaler Lern- und Merkfähigkeitstest. Testhandbuch. Hogrefe, Göttingen.
Huijbers MG, Querol LA, Niks EH, Plomp JJ, van der Maarel SM, Graus F, Dalmau J, Illa I, Verschuuren JJ (2015) The expanding field of IgG4-mediated neurological autoimmune disorders. Eur J Neurol 22:1151–1161. https://doi.org/10.1111/ene.12758
Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C, Zuliani L, Watanabe O, Lang B, Buckley C, Vincent A (2012) Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 72:241–255. https://doi.org/10.1002/ana.23577
Isenman D, Dorrington K, Painter R (1975) The structure and function of immunoglobulin domains: II. The importance of interchain disulfide bonds and the possible role of molecular flexibility in the interaction between immunoglobulin G and complement. J Immunol 114:1726–1729
Klang A, Schmidt P, Kneissl S, Bago Z, Vincent A, Lang B, Moloney T, Bien CG, Halasz P, Bauer J, Pakozdy A (2014) IgG and complement deposition and neuronal loss in cats and humans with epilepsy and voltage-gated potassium channel complex antibodies. J Neuropathol Exp Neurol 73:403–413. https://doi.org/10.1097/NEN.0000000000000063
Koneczny I (2018) A new classification system for IgG4 autoantibodies. Front Immunol 9:97. https://doi.org/10.3389/fimmu.2018.00097
Kong X, Gong X, Li A, Liu Y, Li X, Li J, Zhou D, Hong Z (2023) Efficacy of immunotherapy and prognosis in anti-LGI1 encephalitis patients: a meta-analysis. Ann Clin Transl Neurol 10:1578–1589. https://doi.org/10.1002/acn3.51847
Kuehn JC, Scheuerle A, Bauer J, Becker AJ, Wirtz R, Lewerenz J (2020) A 64-year-old patient with a mesiotemporal mass and symptomatic epilepsy. Brain Pathol 30:413–414. https://doi.org/10.1111/bpa.12818
Lim J-A, Lee S-T, Moon J, Jun J-S, Kim T-J, Shin Y-W, Abdullah S, Byun J-I, Sunwoo J-S, Kim KT, Yang T-W, Lee W-J, Moon H-J, Kim DW, Lim BC, Cho YW, Yang T-H, Kim HJ, Kim Y-S, Koo YS, Park B, Jung K-H, Kim M, Park K-I, Jung K-Y, Chu K, Lee SK (2019) Development of the clinical assessment scale in autoimmune encephalitis. Ann Neurol 85:352–358. https://doi.org/10.1002/ana.25421
Malter MP, Frisch C, Schoene-Bake JC, Helmstaedter C, Wandinger KP, Stoecker W, Urbach H, Surges R, Elger CE, Vincent AV, Bien CG (2014) Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol 261:1695–1705. https://doi.org/10.1007/s00415-014-7408-6
McCracken L, Zhang J, Greene M, Crivaro A, Gonzalez J, Kamoun M, Lancaster E (2017) Improving the antibody-based evaluation of autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm 4:e404. https://doi.org/10.1212/NXI.0000000000000404
Mueller C, Langenbruch L, Rau JMH, Brix T, Strippel C, Dik A, Golombeck KS, Mönig C, Johnen A, Räuber S, Wiendl H, Meuth SG, Bölte J, Kovac S, Melzer N (2022) Neuropsychological performance in autoimmune limbic encephalitis: evidence from an immunotherapy-naïve cohort. Arch Clin Neuropsychol 37:738–752. https://doi.org/10.1093/arclin/acac001
Mueller C, Langenbruch LM, Rau JMH, Brix T, Strippel C, Dik A, Golombeck KS, Moenig C, Raeuber SJ, Kovac S, Wiendl H, Meuth SG, Bölte J, Johnen A, Melzer N (2021) Determinants of cognition in autoimmune limbic encephalitis—a retrospective cohort study. Hippocampus 31:1092–1103. https://doi.org/10.1002/hipo.23375
Muñiz-Castrillo S, Haesebaert J, Thomas L, Vogrig A, Pinto A-L, Picard G, Blanc C, Do L-D, Joubert B, Berzero G, Psimaras D, Alentorn A, Rogemond V, Dubois V, Ambati A, Tamouza R, Mignot E, Honnorat J (2021) Clinical and prognostic value of immunogenetic characteristics in anti-LGI1 encephalitis. Neurol Neuroimmunol Neuroinflamm 8:e974. https://doi.org/10.1212/nxi.0000000000000974
Muñoz-Lopetegi A, Guasp M, Prades L, Martínez-Hernández E, Rosa-Justícia M, Patricio V, Armangué T, Rami L, Borràs R, Castro-Fornieles J, Compte A, Gaig C, Santamaria J, Dalmau J (2024) Neurological, psychiatric, and sleep investigations after treatment of anti-leucine-rich glioma-inactivated protein 1 (LGI1) encephalitis in Spain: a prospective cohort study. Lancet Neurol 23:256–266. https://doi.org/10.1016/s1474-4422(23)00463-5
Muñoz-Sánchez G, Planagumà J, Naranjo L, Couso R, Sabater L, Guasp M, Martínez-Hernández E, Graus F, Dalmau J, Ruiz-García R (2022) The diagnosis of anti-LGI1 encephalitis varies with the type of immunodetection assay and sample examined. Front Immunol 13:1069368. https://doi.org/10.3389/fimmu.2022.1069368
Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, Watanabe M, Watanabe O, Fukata M (2013) Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci 33:18161–18174. https://doi.org/10.1523/JNEUROSCI.3506-13.2013
Petit-Pedrol M, Sell J, Planaguma J, Mannara F, Radosevic M, Haselmann H, Ceanga M, Sabater L, Spatola M, Soto D, Gasull X, Dalmau J, Geis C (2018) LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 141:3144–3159. https://doi.org/10.1093/brain/awy253
Rada A, Bien CG (2023) What is autoimmune encephalitis-associated epilepsy? Proposal of a practical definition. Epilepsia 64:2249–2255. https://doi.org/10.1111/epi.17699
Rada A, Birnbacher R, Gobbi C, Kurthen M, Ludolph A, Naumann M, Neirich U, von Oertzen TJ, Ransmayr G, Riepe M, Schimmel M, Schwartz O, Surges R, Bien CG (2021) Seizures associated with antibodies against cell surface antigens are acute symptomatic and not indicative of epilepsy: insights from long-term data. J Neurol 268:1059–1069. https://doi.org/10.1007/s00415-020-10250-6
Ramberger M, Berretta A, Tan JMM, Sun B, Michael S, Yeo T, Theorell J, Bashford-Rogers R, Paneva S, O’Dowd V, Dedi N, Topia S, Griffin R, Ramirez-Franco J, El Far O, Baulac S, Leite MI, Sen A, Jeans A, McMillan D, Marshall D, Anthony D, Lightwood D, Waters P, Irani SR (2020) Distinctive binding properties of human monoclonal LGI1 autoantibodies determine pathogenic mechanisms. Brain 143:1731–1745. https://doi.org/10.1093/brain/awaa104
Reiber H, Lange P (1991) Quantification of virus-specific antibodies in cerebrospinal fluid and serum: sensitive and specific detection of antibody synthesis in brain. Clin Chem 37:1153–1160
Schultze-Amberger J, Pehl D, Stenzel W (2012) LGI-1-positive limbic encephalitis: a clinicopathological study. J Neurol 259:2478–2480. https://doi.org/10.1007/s00415-012-6559-6
Schumaker VN, Calcott MA, Spiegelberg HL, Mueller-Eberhard HJ (1976) Ultracentrifuge studies of the binding of IgG of different subclasses to the Clq subunit of the first component of complement. Biochemistry 15:5175–5181
Shao X, Fan S, Luo H, Wong TY, Zhang W, Guan H, Qiu A (2021) Brain magnetic resonance imaging characteristics of anti-leucine-rich glioma-inactivated 1 encephalitis and their clinical relevance: a single-center study in China. Front Neurol 11:618109. https://doi.org/10.3389/fneur.2020.618109
Shin Y-W, Lee S-T, Shin J-W, Moon J, Lim J-A, Byun J-I, Kim T-J, Lee K-J, Kim Y-S, Park K-I, Jung K-H, Lee SK, Chu K (2013) VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 265:75–81. https://doi.org/10.1016/j.jneuroim.2013.10.005
Smith KM, Dubey D, Liebo GB, Flanagan EP, Britton JW (2021) Clinical course and features of seizures associated with LGI1-antibody encephalitis. Neurology 97:e1141–e1149. https://doi.org/10.1212/WNL.0000000000012465
Thompson J, Bi M, Murchison AG, Makuch M, Bien CG, Chu K, Farooque P, Gelfand JM, Geschwind MD, Hirsch LJ, Somerville E, Lang B, Vincent A, Leite MI, Waters P, Irani SR, Faciobrachial Dystonic Seizures Study Group (2018) The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain 141:348–356. https://doi.org/10.1093/brain/awx323
Tröscher AR, Klang A, French M, Quemada-Garrido L, Kneissl SM, Bien CG, Pákozdy Á, Bauer J (2017) Selective limbic blood–brain barrier breakdown in a feline model of limbic encephalitis with LGI1 antibodies. Front Immunol 8:1364. https://doi.org/10.3389/fimmu.2017.01364
van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MA, van Coevorden-Hameete MH, Wirtz PW, Schreurs MW, Sillevis Smitt PA, Titulaer MJ (2016) Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology 87:1449–1456. https://doi.org/10.1212/WNL.0000000000003173
Wieser HG, ILAE commission on neurosurgery for epilepsy (2004) ILAE commission report: mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 45:695–714. https://doi.org/10.1111/j.0013-9580.2004.09004.x
Witt JA, Coras R, Schramm J, Becker AJ, Elger CE, Blumcke I, Helmstaedter C (2014) The overall pathological status of the left hippocampus determines preoperative verbal memory performance in left mesial temporal lobe epilepsy. Hippocampus 24:446–454. https://doi.org/10.1002/hipo.22238
Witt JA, Helmstaedter C (2021) Neuropsychological evaluations in limbic encephalitis. Brain Sci 11:576. https://doi.org/10.3390/brainsci11050576
Funding
Open Access funding enabled and organized by Projekt DEAL. JB was supported by the Austrian Science Fund, project number P 34864-B.
Author information
Authors and Affiliations
Contributions
Conceptualization: Christian G. Bien; methodology: Christian G. Bien, Friedrich G. Woermann; formal analysis and investigation: all authors; writing—original draft preparation: Christian G. Bien; writing—review and editing: all authors; funding acquisition: not applicable; resources: Christian G. Bien; supervision: Christian G. Bien.
Corresponding author
Ethics declarations
Conflicts of interest
None of the authors report a conflict of interest.
Ethical approval
The ethics committee of the Medical Faculty of the University of Münster, which is in charge for us, has approved this study and waived patient consent because of the mainly retrospective nature of this work on patients personally investigated and treated by the authors (no. 2020-244-f-S). Nevertheless, 16/20 patients who were seen prospectively for follow-up in addition signed a consent form. All investigations have been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bien, C.G., Rada, A., Mertens, M. et al. LGI1 encephalitis: potentially complement-activating anti-LGI1-IgG subclasses 1/2/3 are associated with the development of hippocampal sclerosis. J Neurol 271, 6325–6335 (2024). https://doi.org/10.1007/s00415-024-12594-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-024-12594-9