
Abstract
Currarino syndrome (CS) is a peculiar form of caudal regression syndrome [also known as autosomal dominant sacral agenesis (OMIM no. 176450)] characterised by (1) partial absence of the sacrum with intact first sacral vertebra, (2) a pre-sacral mass and (3) anorectal anomalies (Currarino triad). We studied a 3-year-old girl with Currarino triad who had additional systemic features and performed array comparative genomic hybridisation to look for chromosomal abnormalities. This girl had the typical spectrum of anomalies of the CS including (a) partial sacral agenesis (hemisacrum with remnants of only sacral S1–S2 vertebrae and a residual S3 vertebral body) associated with complete coccygeal agenesis, (b) pre-intrasacral dermoid, (c) intra-dural lipoma, (d) ectopic anus and (e) tethered cord. She had, in addition, pre- and post-natal growth impairment (<3rd percentile), severe microcephaly (<−3 SD) with normal gyration pattern and lack of cortical thickening associated with a hypoplastic inferior vermis, facial dysmorphism, sensorineural deafness and decreased serum levels of IGF-1. A de novo 10.3-Mb duplication of 7q34–q35 and an 8.8-Mb deletion on 7q36 were identified in this patient. The Homeobox HLXB9 (CS) gene is contained within the deletion accounting for the CS phenotype including microcephaly. The spectrums of associated abnormalities in the IGF-1 deficiency growth retardation with sensorineural deafness and mental retardation syndrome (OMIM no. 608747) are discussed. To the best of our knowledge, this is the first reported case of a patient with distal 7q chromosomal imbalance and features of CS triad (including microcephaly) and the first documented case of a patient with normal gyration pattern microcephaly. The spectrum of associated anomalies in this newly recognised phenotype complex consists of growth failure, typical facial anomalies with additional (previously unreported) nervous system abnormalities (e.g. sensorineural deafness) and somatomedin C deficiency.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The caudal regression (or caudal agenesis) syndrome is classified into two types depending on the shape and position of the spinal cord terminus: (1) an abrupt and high type and (2) a tethered and low type (types I and II, respectively). Caudal regression is one of the major components of the Currarino syndrome or triad (CS; also known as autosomal dominant sacral agenesis; OMIM no. 176450) [6, 13], in which its classic form consists of (1) partial sacral defects consisting of (partial) sacral agenesis with an intact first sacral vertebra (hemisacrum or “sickle-shaped” sacrum), (2) a pre-sacral mass, including an anterior meningocele, an enteric cyst and/or a pre-sacral teratoma, and (3) congenital anal stenosis or other anorectal malformations (such as imperforate anus with/without anal fistula to the spinal cord or to the urogenital system) [13, 15].
Currently, CS is regarded as a developmental field disorder based on its recognised aetiological heterogeneity and is thought to result from abnormal separation of the neuroectoderm from the endoderm [9]. In contrast to other types of partial or total sacrococcygeal agenesis, the sacral agenesis in CS is typically incomplete and only involves vertebrae S2–S5, a sacral anomaly distinct to this syndrome. CS exhibits variable expressivity, reduced penetrance and remarkable variability of objective findings and subjective complaints, with a clinical spectrum ranging from anomalies localised only to the caudal region to those involving complex malformations of other organ systems including the kidneys, brain and spinal cord [5, 15, 16]. An X-linked (dominant) pattern of inheritance was proposed, though more recent reports demonstrate an autosomal dominant mode of inheritance [1, 8, 9]. CS has recently been associated with mutations of HLXB9, a homeobox gene that maps to chromosome 7q36 [12, 13, 21].
We report a case of a patient with features of CS combined with severe microcephaly, facial dysmorphism, sensorineural deafness and somatomedin deficiency in which we found a complex rearrangement of distal 7q that included deletion of the Homeobox HLBX9 gene. The co-occurrence of a CS-like phenotype and microcephaly has been reported in previous patients [11, 17, 20] (Table 1): Notably, these had a 7q36 deletion with incomplete features of CS combined with tethered cord (in 2/3) and congenital ptosis (2/3). One of these patients had features suggesting the microform of holoprosencephaly [17].
Case report
This 3-year-old girl was the second born to unrelated Italian parents.
In her family history, there were two previous miscarriages: one voluntary (due to severe, systemic foetal malformations) and the other spontaneous (occurred at the third gestational month). The proband’s father, aged 32 years, has an asymptomatic total posterior sacral canal schisis; the proband’s sister, aged 5 years, has an asymptomatic partial schisis of the sacrum canal. In both, the evaluation for further signs fitting with the CS phenotype was negative.
The proband was born at term by caesarean section after a pregnancy characterised by hyperemesis, repeated threatened abortion, intra-uterine growth retardation and threatened pre-term delivery. The birth weight was 1,740 g, the birth length was 43 cm, and the head circumference was 28 cm. At birth, she was admitted to a neonatal intensive care unit elsewhere in Italy because of respiratory distress: at that time, the heart ultrasound examination revealed a patent inter-atrial septum and a patent ductus arteriosus. She was hospitalised for 2 months. After discharge, she was followed up as an outpatient for psychomotor delay. During follow-up, she developed frequent episodes of cyanosis and recurrent respiratory tract infections. An auditory-evoked potential study revealed moderately severe sensorineural deafness (66 dB).
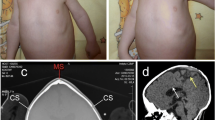
At age 18 months, she was admitted to a tertiary care hospital for a diagnostic work-up. Upon general and neurological examination, there was severe microcephaly (head circumference was 38 cm), convergent strabismus and axial hypotonia; she was unable to sit and did not vocalise. Chest X-rays, as well as heart and abdominal ultrasound examinations, was negative. Her bone age was 8.5 months. On central nervous system magnetic resonance imaging (MRI), the following features were recorded: microencephaly with normal gyration pattern, lack of cortical thickening, normal corpus callosum and myelination and associated with a hypoplastic inferior vermis (Fig. 1a, b). An X-ray study of the spine revealed partial sacrococcygeal agenesis, and the spinal MRI confirmed the presence of the first two sacral elements as well as a rudimentary S3 vertebra, whereas the S4 and S5 vertebras and the coccyx were absent. The spinal cord was low in position and tethered to an intra-dural lipoma, which in turn was connected to a dermoid that extended from the spinal canal to the pre-sacral space (Fig. 1c–f). To exclude dysganglionosis, a rectal biopsy was performed, which yielded normal findings. In the meantime, the patient underwent gastrointestinal surgery (Nissen fundoplication) for gastroesophageal reflux. At this time, DNA analysis for the HLBX9 gene mutations was negative.

MRI study of the central nervous system in the proband (a–f). Sagittal T2-weighted (a) and axial T2-weighted (b) images show microcephaly with normal gyration pattern. Note the disproportion between the size of the face and that of the brain (a), which is a typical feature of microcephaly. The inferior cerebellar vermis is hypoplastic (black arrows; a) with corresponding enlargement of the cisterna magna; the lateral ventricles are somewhat dysmorphic (b). Sagittal T2-weighted (c) and contrast enhanced sagittal T1-weighted (d) images of the lumbosacral spine show partial sacrococcygeal agenesis with a rudiment of S3 as final visible vertebra; the spinal cord is low in position (arrowhead) and tethered to an intra-dural lipoma (d; thin white arrow), which in turn is contiguous with a intra-sacral mass (a dermoid; c, d; thick black arrows), which extends inferiorly around the extremity of the dysgenetic sacrum; the mass is very slightly hypointense to CSF on T2-weighted images (c) and hyperintense on T1-weighted images without evidence of contrast enhancement (d). Sagittal diffusion-weighted images (e, f) with the corresponding ADC map show restricted diffusion (arrow), consistent with dermoid
The patient was readmitted at age 21 and 24 months for further diagnostic work-up, and she underwent surgery for the cord tethering with partial removal of the spinal masses.
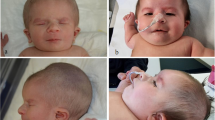
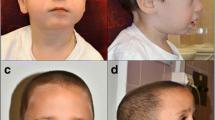
Upon admission at the University Children’s Hospital of Catania, at age 28 months, the girl’s general conditions were fair: her weight was 7 kg, height was 72 cm, and head circumference was 39.5 cm (all below the third percentile). Her head was extremely small (<−3 SD) with a sloping forehead; she had low-set hair; her ears were large; there was right palpebral ptosis, hypothelorism, epicanthal folds and bilateral hypoplasia of the lateral sides of the eyebrows; her nasal root was depressed; the maxilla was prominent with mild micro-retrognathia, and she had a small, heart-shaped mouth with a V-shaped depression in the chin; the dentition was normal except for two large maxillary central incisors (Fig. 2); the hands were small with short thumbs; there were remnants of a sacrococcygeal tail and partial syndactyly of the second and third toes bilaterally. Neurological examination revealed a severely retarded child with mild generalised muscular hypotonia. She was unable to vocalise and could not sit, and she kept her feet in the plantar position. Patellar reflexes were brisk.

The child at age 3 years: note the two large maxillary central incisors
Laboratory analysis, including routine serum and urinalysis, full autoimmune profiles and coagulation studies, were all within the normal limits. The endocrine status, including FT3, FT4, TSH, FSH, LH, PRL, cortisol and GH levels tested from a single blood sample (without stimulation), were within the normal limits. IGF-1 levels, tested by ELISA, was 1.2 ng/ml (normal values for children aged 0 to 5 years of 3.7–150 ng/ml). Metabolic studies, including plasma ammonia, lactate, amino acids, serum transferrine (by means of isoelectric focusing) and urine organic acids, were normal. Electrocardiogram, electroencephalogram and heart ultrasound examination were all normal. Full ophthalmologic examination was normal. Visual and auditory evoked potentials showed decreased latencies; the electroretinogram was within normal limits.
Repeated blood and urinalyses, including endocrine status and IGF-1 levels (in a single blood sample and without stimulation), at age 36 months, confirmed the previous findings.
Sequencing of the HLXB9 gene was normal. However, array comparative genomic hybridisation using a 19 K BAC array [4, 7] demonstrated a de novo 10.3-Mb duplication of 7q34–q35 (chr7: 138,293,371–148,443,994) as well as an 8.8-Mb deletion on 7q36 (chr7: 148,472,027–157,265,994). Fluorescence in situ hybridisation (FISH) confirmed the boundaries of the abnormalities, and microsatellite analysis determined that the abnormalities occurred de novo on the paternal chromosome. The deleted region contains 70 RefSeq genes including HLXB9, SHH and EN2, while the duplication contains 82 genes (Table 1)
.
Discussion
In our patient, the deletion of HLXB9 must account for the CS, while loss or gain of other genes probably explains other aspects of her phenotype [14]. The most striking components of her phenotype beyond the CS were severe congenital microcephaly, hypotelorism, cerebellar vermis hypoplasia and hearing loss. The microcephaly is in the range seen with autosomal recessive primary microcephaly or with the severe intra-uterine growth deficiency plus microcephaly syndrome such as Seckel syndrome. However, the complex malformation phenotype presented by our patient did not fit with the above conditions. Heterozygous loss of SHH is a well-known cause of holoprosencephaly [19], but many patients with SHH mutations have less severe phenotypes, often designated as microforms. Both hypotelorism and congenital microcephaly (usually in the range of −2 to −4 SD) without holoprosencephaly are well-known microforms associated with SHH mutations [10, 19], so that loss of SHH could be likely responsible for all or part of these features. But the microcephaly in our patient was more severe than typically seen with SHH mutations, so we suspect a contribution from one or more other genes or an effect of the haploinsufficiency for the HLXB9 gene. While no humans with EN2 mutations have been reported, the mouse knockout has severe cerebellar hypoplasia [18], so that deletion of EN2 may at least partly account for the cerebellar vermis hypoplasia.
Notably, the present child had low IGF-1 levels with otherwise normal endocrine status (tested in a single blood sample without stimulation). Decreased IGF-1 levels (isolated IGF-1 deficiency) is known to produce severe pre- as well as post-natal growth impairment with other systemic and neurological abnormalities (OMIM no. 608747; also known as “IGF-1 deficiency, growth retardation with sensorineural deafness and mental retardation”) [2, 22] secondary to abnormalities in the IGF-1 gene at chromosome 12q22-q24.1 (OMIM no. 147440). Woods et al. [22] reported on a case of a 15-year-old child born of consanguineous parents affected by this condition: this boy had severe (pre-natal and post-natal) symmetric growth failure (which continued throughout infancy and childhood), profound bilateral sensorineural deafness and moderately delayed motor development, with behavioural difficulties including hyperactivity and short attention span and mild facial dysmorphism. Bonapace et al. [2] reported a case of a similar patient with IGF-1 deficiency who had poor foetal growth: this child, who was born to second cousins both at the fifth percentile for height, continued to show poor growth and delayed bone age, as well as delayed psychomotor development and sensorineural deafness. The patient of Wood et al. [22] placed on recombinant human IGF-1 treatment improved linear growth and insulin sensitivity [3, 23].
In summary, the key features in our patient included (1) type 2 caudal regression consisting of partial sacral agenesis (presence of only S1–S2 vertebrae and a residual S3 vertebral body) and total coccygeal agenesis; (2) sacral intra- and pre-canalicular dermoid associated with an intra-canalicular lipoma and a tethered cord; (3) severe congenital microcephaly (39.5 cm at age 3 years) with a normal gyral pattern and normal cortical thickness on MRI; (4) inferior cerebellar vermis hypoplasia; (5) bilateral sensorineural hearing loss; (6) facial anomalies including hypotelorism, bilateral hypoplasia of the lateral aspect of the eyebrows, right ptosis, prominent upper mandibula and normal dentition (besides single central incisors); (7) small hands with short thumbs; and (8) isolated somatomedin C deficiency.
To the best of our knowledge, this is the first reported case of a patient with features of the complete CS triad and microcephaly. The spectrum of associated anomalies in this newly recognised phenotype complex consists of typical facial anomalies with additional (and previously unreported) nervous system abnormalities (e.g. sensorineural deafness). This patient documents the highly variable nature of this complex anomaly and highlights its possible link with microcephaly.
Abbreviations
- a-CGH:
-
Array comparative genomic hybridisation
- AMA:
-
Anti-muscle antibodies
- ANA:
-
Anti-nuclear antibodies
- ASMA:
-
Anti-smooth muscle antibodies
- BAC:
-
Bacterial artificial chromosomes
- CNS:
-
Central nervous system
- CS:
-
Currarino syndrome
- ECG:
-
Electrocardiogram
- EEG:
-
Electroencephalogram
- ERG:
-
Electroretinogram
- FISH:
-
Fluorescence in situ hybridisation
- FSH:
-
Follicle-stimulating hormone
- FT3:
-
Free thyroxin 3
- FT4:
-
Free thyroxin 4
- GH:
-
Growth hormone
- HLBX9:
-
Homeobox gene HB9
- IGF-1:
-
Insulin growth factor-1
- LH:
-
Luteinising hormone
- MRI:
-
Magnetic resonance imaging
- OMIM:
-
Online Mendelian Inheritance in Men
- PRL:
-
Prolactin
- S:
-
Sacral (vertebrae)
- SD:
-
Standard deviation
- TSH:
-
Thyroid-stimulating hormone
References
Belloni E, Martucciello G, Verderio D et al (2000) Involvement of the HLXB9 homeobox gene in Currarino syndrome. Am J Hum Genet 66:312–319
Bonapace G, Concolino D, Formicola S, Strisciuglio P (2003) A novel mutation in a patient with insulin-like growth factor (IGF1) deficiency. J Med Genet 40:913–917
Camacho-Hubner C, Woods KA, Miraki-Moud F et al (1999) Effects of recombinant human insulin-like growth factor I (IGF-I) therapy on the growth hormone system of a patient with a partial IGF-I gene deletion. J Clin Endocr Metab 84:1611–1616
Christian SL, Brune CW, Sudi J et al (2008) Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry 63:1111–1117
Crétolle C, Pelet A, Sanlaville D et al (2008) Spectrum of HLXB9 gene mutations in Currarino syndrome and genotype-phenotype correlation. Hum Mutat 29:903–910
Currarino G, Coln D, Votteler T (1981) Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol 137:395–398
de Ravel TJ, Devriendt K, Fryns JP, Vermeesch JR (2007) What’s new in karyotyping. The move towards array comparative genomic hybridization (CGH). Eur J Pediatr 166:637–643
Garcia-Barcelo M, So MT, Lau DK et al (2006) Population differences in the polyalanine domain and 6 new mutations in HLXB9 in patients with Currarino syndrome. Clin Chem 52:46–52
Hagan DM, Ross AJ, Strachan T et al (2000) Mutation analysis and embryonic expression of the HLXB9 Currarino syndrome gene. Am J Hum Genet 66:1504–1515
Heussler HS, Suri M, Young ID et al (2002) Extreme variability of expression of a Sonic Hedgehog mutation: attention difficulties and holoprosencephaly. Arch Dis Child 86:293–296
Horn D, Tonnies H, Neitzel H et al (2004) Minimal clinical expression of the holoprosencephaly spectrum and of Currarino syndrome due to different cytogenetic rearrangements deleting the Sonic Hedgehog gene and the HLXB9 gene at 7q36.3. Am J Med Genet 128A:85–92
Kim IS, Oh SY, Choi SJ et al (2007) Clinical and genetic analysis of HLXB9 gene in Korean patients with Currarino syndrome. J Hum Genet 52:698–701
Lerone M, Bolino A, Martucciello G (1997) The genetics of anorectal malformations: a complex matter. Semin Pediatr Surg 6:170–179
Lynch SA, Bond PM, Copp AJ et al (1995) A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q36. Nat Genet 11:93–95
Lynch SA, Wang Y, Strachan T et al (2000) Autosomal dominant sacral agenesis: Currarino syndrome. J Med Genet 37:561–566
Martucciello G, Torre M, Belloni E et al (2004) Currarino syndrome: proposal of a diagnostic and therapeutic protocol. J Pediatr Surg 39:1305–1311
Masuno M, Fukushima Y, Sugio Y et al (1990) Two unrelated cases of single maxillary central incisor with 7q terminal deletion. Jpn J Hum Genet 35:311–317
Millen KJ, Wurst W, Herrup K et al (1994) Abnormal embryonic cerebellar development and patterning of postnatal foliation in two mouse Engrailed-2 mutants. Development 120:695–706
Nanni L, Ming JE, Bocian M et al (1999) The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet 8:2479–2488
Rodriguez L, Cuadrado Perez I et al (2002) Terminal deletion of the chromosome 7 (q36-qter) in an infant with sacral agenesis and anterior myelomeningocele. Am J Med Genet 110:73–77
Ross AJ, Ruiz-Perez V, Wang Y et al (1998) A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet 20:358–361
Woods KA, Camacho-Hubner C, Savage MO, Clark AJL (1996) Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. New Engl J Med 335:1363–1367
Woods KA, Camacho-Hubner C, Bergman RN, Carter D, Clark AJL, Savane MO (2000) Effects of insulin-like growth factor I (IGF-I) therapy on body composition and insulin resistance in IGF-I gene deletion. J Clin Endocr Metab 85:1407–1411
Acknowledgements
We wish to thank Mr Francesco Marino (CNR, Catania) for skilful technical assistance.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
We have no financial or nonfinancial interest to disclose.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pavone, P., Ruggieri, M., Lombardo, I. et al. Microcephaly, sensorineural deafness and Currarino triad with duplication–deletion of distal 7q. Eur J Pediatr 169, 475–481 (2010). https://doi.org/10.1007/s00431-009-1061-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-009-1061-6