
Abstract
About 15% of colorectal cancer (CRC) patients have first-degree relatives affected by the same malignancy. However, for most families the cause of familial aggregation of CRC is unknown. To identify novel high-to-moderate-penetrance germline variants underlying CRC susceptibility, we performed whole exome sequencing (WES) on four CRC cases and two unaffected members of a Polish family without any mutation in known CRC predisposition genes. After WES, we used our in-house developed Familial Cancer Variant Prioritization Pipeline and identified two novel variants in the solute carrier family 15 member 4 (SLC15A4) gene. The heterozygous missense variant, p. Y444C, was predicted to affect the phylogenetically conserved PTR2/POT domain and to have a deleterious effect on the function of the encoded peptide/histidine transporter. The other variant was located in the upstream region of the same gene (GRCh37.p13, 12_129308531_C_T; 43 bp upstream of transcription start site, ENST00000266771.5) and it was annotated to affect the promoter region of SLC15A4 as well as binding sites of 17 different transcription factors. Our findings of two distinct variants in the same gene may indicate a synergistic up-regulation of SLC15A4 as the underlying genetic cause and implicate this gene for the first time in genetic inheritance of familial CRC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Several studies have estimated that around 15% of colorectal cancer (CRC) patients show a first-degree family history of colorectal malignancies (Ponz de Leon et al. 1989; Hemminki et al. 2008; Frank et al. 2015; Chau et al. 2016). Analyzing the underlying heritable and environmental factors in twins from Sweden, Denmark, and Finland, Lichtenstein et al. have estimated that genetic factors account for up to 35% of the CRC risk (Lichtenstein et al. 2000). Nevertheless, only a small proportion of familial CRC cases can be traced back to germline mutations in established CRC-predisposing genes. In the present study, we used a family-based whole-exome sequencing approach to fill in this gap and to identify novel CRC predisposition genes with high-to-moderate penetrance germline variants.
The early-identified traditional CRC susceptibility genes include APC and mismatch repair genes (MLH1, MSH2, MSH6, PMS2), MUTYH and SMAD4/BMPR1A. Later on, sequencing studies have identified novel predisposition genes for CRC, such as NTHL1, RNF43, POLE, POLD1, FAN1 and RPS20 (Jasperson et al. 2010; Briggs and Tomlinson 2013; Palles et al. 2013; Gala et al. 2014; Nieminen et al. 2014; Kuiper and Hoogerbrugge 2015; Segui et al. 2015; Weren et al. 2015; Yan et al. 2017; Lorans et al. 2018; Valle et al. 2019). Further candidate genes recently suggested by modern next generation sequencing methods include the solute carrier (SLC) family of membrane transport genes: SLC5A9 (p.G492Afs*13), SLC26A8 (p.R954C) and SLC11A1 (p.P64A) (Hansen et al. 2017; Yu et al. 2018). Additionally, germline deletions affecting the open reading frame of SLC18A1 gene have been reported to increase the risk of CRC and lower SLC18A1 protein expression has been further associated with poor clinical outcome (Zhang et al. 2017).
Despite novel findings of predisposition gene candidates in CRC, there still exist 75% of unexplored familial CRC cases. This proportion of familial CRC with unknown genetic background may be accounted for by two major components: either following a monogenic inheritance model based on a single high-penetrance mutation or a polygenic inheritance model based on the combination of multiple low/moderate-penetrance risk alleles (Zetner and Bisgaard 2017). Assuming the monogenic disease model for CRC cases with strong familial clustering, the identification of rare highly penetrant germline variants within pedigree-based studies constitutes a promising approach for elucidating the remaining genetic burden of familial CRC.
For this purpose, we performed whole exome sequencing (WES) on a Polish family with CRC aggregation over three generations. Sequencing data of four CRC cases and two unaffected family members were subsequently analyzed using our in-house developed Familial Cancer Variant Prioritization Pipeline (FCVPPv2) which was used earlier in identification of variants and pathways involved in several familial cancers (Bandapalli et al. 2018; Kumar et al. 2018; Srivastava et al. 2019; Srivastava et al. 2020a; Srivastava et al. 2020b; Skopelitou et al. 2021; Srivastava et al. 2021). Further in silico analyses resulted in the prioritization of a novel missense variant in the solute carrier family 15 member 4 gene (SLC15A4), encoding a proton-dependent peptide/histidine transporter. By being involved in multiple signaling pathways regulating cytokine production and thus innate immune responses, SLC15A4 has been shown to promote colitis in an in vivo mouse model (Sasawatari et al. 2011; Kobayashi et al. 2014). Since high expression of the encoded membrane transporter has been further reported in the feces of CRC patients as well as in early-stage CRC cell lines, an important role of SLC15A4 in initial inflammation-induced colorectal carcinogenesis has been suggested (Lee et al. 2016). In this study, we conducted in silico analyses as well as further literature search to link the function of the SLC15A4 protein to a genetic basis, potentially contributing to CRC development in the studied family. By identifying and analyzing an additional variant in the upstream region of the SLC15A4 gene showing the same familial segregation, we aimed to expand the theory of high-penetrance monogenic inheritance to a synergistic model of coding and non-coding variants underlying cancer predisposition.
Materials and methods
Patient samples and ethical permissions
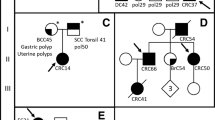
The family with CRC history over three generations was recruited from Poland (Fig. 1a). Six family members were included in our experiments: four siblings diagnosed with CRC, a child of one CRC case with colorectal polyps and a healthy cousin of the CRC cases. The family was screened for alterations in APC, the mismatch repair genes MLH1, MSH2, MSH3, large deletions in EPCAM and POLE p. Leu424Val, POLD1 p.Ser478Asn and NTHL1 p.Gln90* mutations and found to be negative. Collection of blood samples and clinical information from subjects was undertaken with informed written consent in agreement with the tenets of the declaration of Helsinki. The study was approved by the Bioethics Committee of the Pomeranian Medical Academy in Szczecin (protocol code No: BN-001/174/05).

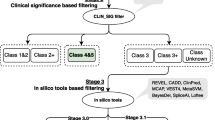
a Pedigree of the studied family with CRC aggregation over three generations and the presence of the missense and upstream variants in the SLC15A4 gene b Graphical overview of the filtering process according to the Familial Cancer Variant Prioritization Pipeline version 2 (FCVPPv2)
Whole exome sequencing, variant calling and annotation
Genomic DNA was isolated with a modified Lahiri and Schnabel method (Lahiri and Schnabel 1993) and WES was performed using Illumina-based small read sequencing. After mapping to the human reference genome (assembly GRCh37 version Hs37d5) by means of BWA (Li and Durbin 2009), duplicates were removed with Picard (http://broadinstitute.github.io/picard/). SAM tools (Li 2011) and Platypus (Rimmer et al. 2014) were used for calling single nucleotide variants (SNVs) as well as short insertions and deletions (indels), respectively. Variants were then annotated by ANNOVAR (Wang et al. 2010), 1000 Genomes Project (Genomes Project et al. 2015), dbSNP (Smigielski et al. 2000) and Exome Aggregation Consortium (ExAC) (Lek et al. 2016). To be further processed, variants should have a quality score of ≥ 20 and a coverage score of ≥ 5 ×, SNVs should pass the strand bias filter (a minimum one read support from both forward and reverse strand) and indels should pass all the Platypus internal filters. Based on minor allele frequencies (MAFs) deduced from the 1000 Genomes Project Phase 3, non-TCGA ExAC data, NHLBI-ESP6500 and local data sets, rare variants with a MAF ≤ 0.1% in the European population were retained for further analysis. We checked for potential sample swaps and family relatedness by pairwise comparison of the shared rare variants.
Coding variant analysis according to the FCVPPv2
The resulting variants were analyzed based on our in-house developed FCVPPv2 (Kumar et al. 2018). First, variants were filtered according to the pedigree segregation of the malignancy. Variants should be present in family members affected by CRC and absent in the healthy family member. Since colorectal polyps at a relatively young age may represent a preliminary stage of familial CRC, the respective family member could be a possible carrier and show either presence or absence of the variant of interest.
Of the coding variants fulfilling the pedigree segregation criteria, the most deleterious 10% were retained for further analysis, represented by a PHRED-like CADD score ≥ 10 (Kircher et al. 2014; Rentzsch et al. 2019). To evaluate the evolutionary conservation as an indicator for functional importance of a genomic position, the following scoring tools were applied with respective cutoff values given in brackets: Genomic Evolutionary Rate Profiling (GERP; ≥ 2.0), PhastCons (> 0.3) and PhyloP score (≥ 3.0) (Cooper et al. 2005; Siepel et al. 2005; Pollard et al. 2010). Next, the intolerance of genes against functional genetic variation was assessed by using three intolerance scores (< 0) based on allele frequency data from our in-house datasets, from NHLBI-ESP6500 and ExAC (Petrovski et al. 2013). In the course of intolerance screening, missense and loss-of-function variants were further annotated by the Z Score (> 0) and pLI score (≥ 0.9), respectively, which were specifically developed by the ExAC consortium for the particular type of variants (Lek et al. 2016). Last, we evaluated the deleteriousness of non-synonymous and splice site SNVs by applying ten different scoring tools accessed from dbNSFP v3.0 (database for nonsynonymous SNPs’ functional predictions): Sorting Tolerant From Intolerant (SIFT), Polymorphism Phenotyping v2 (PolyPhen-2) HumDiv, PolyPhen-2 HumVar, Log ratio test (LRT), MutationTaster, MutationAssessor, Functional Analysis Through Hidden Markov Models (FATHMM), Reliability Index, Variant Effect Scoring Tool version 3 (VEST3) and Protein Variation Effect Analyzer (PROVEAN) (Liu et al. 2016).
Summarizing, variants with a PHRED-like CADD-score of ≥ 10 as well as ≥ 2 out of the three conservational tools, ≥ 60% of the four intolerance scores and ≥ 60% of the 10 deleteriousness scores fulfilling the selection criteria were retained as the top exonic candidates. Allele frequencies were re-evaluated by means of the gnomAD browser (https://gnomad.broadinstitute.org/) (Karczewski et al. 2019). Since the studied CRC family originates from Poland, the non-Finnish European (NFE) population was taken as the representative population on a large scale.
We further assessed the potential of the variants for being cancer drivers in CRC by checking overall somatic alteration frequencies according to cBioPortal and TCGA PanCancer Atlas, comprising data of 594 CRC patients (Cancer Genome Atlas Research et al. 2013; Gao et al. 2013). Moreover, protein expression levels in CRC tissue were accessed from The Human protein atlas (http://www.proteinatlas.org) (Uhlen et al. 2017).
Additional in silico analyses based on protein function and phylogenetic conservation
The potential impact of the top missense variants on protein function was assessed by means of Snap2 (Hecht et al. 2015). Based on a neural network, Snap2 calculates the likelihood of single amino acid substitutions to alter protein function, giving scores between − 100 (low) and + 100 (high). The predicted functional impact is represented in form of heat maps covering each possible amino acid substitution at each position.
Since predictions of the functional impact of variants are based on evolutionary information, we further checked phylogenetic conservation of the top variants among different vertebrate species. Multiple protein sequences of the candidate genes and their orthologs were derived from the National Center for Biotechnology Information (NCBI) (Coordinators 2018) and aligned using COBALT, a constraint-based multiple alignment tool (Papadopoulos and Agarwala 2007). Visualized alignments were manually checked for conservation at the mutation sites and the surrounding regions and percent identity of protein sequences was further calculated by NCBI BLAST (Basic Local Alignment Search Tool). Details of multiple sequence alignment including selected representative species and NCBI accessions of respective genes and their orthologs are summarized in Online Resource 1.
We checked recent literature for established gene-cancer associations, postulated oncogene or tumor-suppressor roles as well as potential cancer-promoting protein functions of the top candidates. Considering the entirety of derived information and in silico analysis results, the candidates showing the most promising impact on protein function or gene regulation were prioritized as the potentially cancer-causing variants in the studied family. Familial segregation of the top-listed variants with the disease was confirmed by visually checking WES data with the help of the Integrative Genomics Viewer (IGV) (Robinson et al. 2017).
Analysis of regulatory elements and prediction of transcription factor binding sites in the non-coding regions
To assess the biological function and to identify potentially active regulatory regions, the chromatin state of specific genomic positions was predicted by the updated version of CADD (v1.6). For this purpose, CADD v1.6 provides chromHmm and Segway data, which annotate the chromatin state based on large-scale functional genomics datasets such as ChIP-seq data (Ernst and Kellis 2012; Hoffman et al. 2012; Roadmap Epigenomics et al. 2015). Using the intersect function of the Bedtools as well as FANTOM5 and SEA databases, we further scanned for potentially affected regulatory elements such as promoters, enhancers and super-enhancers (Lizio et al. 2015; Wei et al. 2016). Moreover, transcription factor binding sites (TFBSs) were predicted by means of Jaspar2020 with the default relative profile score threshold of 80% and compared between wild-type and mutant sequence (Fornes et al. 2020). Details on the regulatory annotations are provided in a systematic review of in silico prioritization of non-coding variants (Lee et al. 2018).
Results
Application of the FCVPPv2 results in the prioritization of two coding variants in PTGES and SLC15A4 genes
The studied family was diagnosed with CRC over three generations, as represented in the pedigree (Fig. 1a). Four siblings affected by CRC in the second generation at the age of < 60 years were considered as cases and should therefore carry the variant of interest. Similarly, the daughter (IV2) of one of the cases (III2) developed colorectal polyps at the relatively young age of 29 years, potentially representing a preliminary stage of familial CRC. Considering the option of having inherited the variant of interest, IV2 was defined as a possible carrier and may present the variant as well. In contrast, an unaffected first cousin of the four CRC cases of a similar age and with healthy parents served as a control and should thus not carry the variant.
Analysis of WES data was performed using our in-house developed FCVPPv2, as visually summarized in Fig. 1b. Of the totally identified 11,076 variants with a MAF ≤ 0.1%, only 135 variants fulfilled the pedigree segregation criteria. Exclusion of intergenic and intronic variants resulted in 28 variants in the coding region and 43 variants located in the non-coding region near transcription start and end sites (5′ and 3′ untranslated regions, upstream and downstream regions). Due to their less pathogenic character, synonymous variants were excluded, leaving 17 missense or nonsense variants for further analysis. 12 of the remaining coding variants reached a PHRED-like CADD score ≥ 10, representing the most deleterious 10% of the variants in the human genome. Application of conservational, intolerance and deleteriousness scores further narrowed down the number of variants to 9, 4 and 2, respectively. The two final missense variants were located in solute carrier family 15 member 4 gene (SLC15A4, p.Y444C) and prostaglandin E synthase gene (PTGES, p.A133T) and are summarized with respective analysis results in Table 1.
PTGES encodes a glutathione-dependent synthase catalyzing the oxidoreduction to prostaglandin E2. By playing a role in inflammatory responses, fever and pain, PTGES protein has been reported to be involved in inflammatory diseases such as collagen-induced arthritis and gastritis (Gudis et al. 2005; Korotkova et al. 2011). Similarly, the gene product encoded by SLC15A4 regulates innate immune responses. Being a proton-dependent peptide/histidine transporter, SLC15A4 protein controls the transport of various molecules from the inside of endosomes to the cytosol and has been associated inter alia with systemic lupus erythematosus (Wang et al. 2013; Lee et al. 2014; Zuo et al. 2014; Zhang et al. 2016).
According to the gnomAD browser, both top-listed variants showed very low allele frequencies in the general NFE population: the PTGES variant was annotated with a frequency of around 8.4 × 10–5 and the SLC15A4 variant even less with 0 counts in 113,688 alleles. Moreover, only one allele of the SLC15A4 variant has been reported in the worldwide population accessed by gnomAD browser, counting in total 251,362 alleles (Karczewski et al. 2019). For further validation of allele frequencies, population data of Trans-Omics for Precision Medicine (TOPMed), integrating large-scale whole genome sequencing data, was checked and did not report the identified SLC15A4 variant, confirming again its low allele frequency (Li et al. 2020; Taliun et al. 2021).
Higher alteration frequency and protein expression of SLC15A4 in CRC compared to PTGES
We next checked available CRC patient data for overall somatic gene alteration frequencies to assess the potential of the top candidates for being cancer drivers in CRC. cBioPortal recorded six somatic missense mutations in the SLC15A4 gene (frequency = 1.01%, Fig. 2a) and only two somatic mutations in the PTGES gene (frequency = 0.34%, Fig. 3a) identified within 594 colorectal adenocarcinoma samples from the TCGA PanCancer Atlas. Regarding the overall somatic alteration frequency in all listed cancers, SLC15A4 showed a generally higher frequency with up to 5.48% in uterine cancer (Online Resource 2a), whereas the maximum alteration frequency of the PTGES gene was only 1.7%, also in uterine cancer (Online Resource 2b) (Cancer Genome Atlas Research et al. 2013; Gao et al. 2013). Besides genetic alterations documented in CRC, we checked protein expression levels in CRC samples. According to the Human Protein Atlas, 4 out of 12 investigated CRC samples showed a medium expression of the SLC15A4 protein, whereas 0 out of 11 CRC samples showed a high or medium expression of the PTGES protein (Uhlen et al. 2017).

In silico analysis results of the SLC15A4 variant p.Y444C a Graphical overview of the SLC15A4 protein with the PTR2 domain. Somatic mutations identified in CRC were extracted from cBioPortal (www.cbioportal.org) on 13th of December 2020 using the TCGA PanCancer data and are represented by dark pins. The germline missense variant identified in the studied CRC family is highlighted in the form of a yellow pin. b Snap2 heatmap depicting the functional impact of amino acid substitutions. The missense mutation p.Y444C is highlighted by grey boxes. c Extract of multiple sequence alignment of amino acids 430–460 of SLC15A4 and orthologs. The mutation site is highlighted by a yellow box

In silico analysis results of the PTGES variant p.A133T a Graphical overview of the PTGES protein with MAPEG domain. Somatic mutations identified in CRC are extracted from cBioPortal (www.cbioportal.org) on 13th of December 2020 using the TCGA PanCancer data and are represented by dark pins. The germline missense variant identified in the studied CRC family is highlighted in the form a yellow pin. b Snap2 heatmap depicting the functional impact of amino acid substitutions. The missense mutation p.A133T is highlighted by grey boxes. c Extract of multiple sequence alignment of amino acids 120–150 of PTGES and orthologs. The mutation site is highlighted by a yellow box
In silico analyses predict functional consequences of the SLC15A4 variant on protein level
The identified SLC15A4 variant p.Y444C was predicted to affect the PTR2 (peptide transport) domain (p.104–495) of the POT (proton-dependent oligopeptide transporter) family (Fig. 2a) and in particular a non-cytoplasmic loop of the SLC15A4 transporter protein, which comprises in total 12 transmembrane domains according to Interpro (Blum et al. 2020). Analysis of the potential impact of the SLC15A4 missense variant on protein function by means of Snap2 resulted in a predicted effect score of 44 with an accuracy of 71% (Fig. 2b). In contrast, the missense variant p.A133T in the PTGES gene, affecting the cytoplasmic part of the MAPEG (membrane-associated proteins in eicosanoid and glutathione metabolism) domain (p.17–146) of the PTGES protein (Fig. 3a), was annotated by Snap2 with a score of 16 and an accuracy of 59% (Fig. 3b) (Hecht et al. 2015). Due to a higher effect score and accuracy of the prediction in cross-validation, the functional impact of the SLC15A4 variant was expected to be of higher relevance.
Sequence alignment of the orthologs showed for both variants a universally conserved position with an overall high conservation of the surrounding region among the selected vertebrate species (Figs. 2c, 3c, respectively). Focusing on the five directly adjacent upstream and downstream amino acid positions, multiple sequence alignment resulted in 95.86% identity of the SLC15A4 and 82.64% identity of the PTGES gene with their orthologs. Based on this observation, a higher phylogenetic conservation in the region surrounding the mutation site can be assumed for SLC15A4.
The entirety of the in silico analyses led to the prioritization of the missense variant in SLC15A4 gene (p.Y444C). Familial segregation of this variant was manually checked and confirmed by applying IGV on the WES data.
Identification of an additional variant at an active transcription start site of SLC15A4 gene
We checked the WES data of the studied family for further variants affecting the same gene of interest. Interestingly, one additional variant in the upstream region of the SLC15A4 gene showing the same familial segregation as the missense variant (present in the cases and the possible carrier) was identified (12_129308531_C_T; 43 bp upstream of transcription start site, ENST00000266771.5). Functional annotation of the non-coding variant was derived from CADD v1.6 providing a PHRED-like CADD-score of 11.38 (Kircher et al. 2014; Rentzsch et al. 2019). Moreover, the variant was annotated to be located at an active transcription start site according to ChromHmm (TssA, Score = 0.969) and Segway (TSS) (Ernst and Kellis 2012; Hoffman et al. 2012). CADD v1.6 further calculated 52 different overlapping ChIP TFBSs covered by the upstream variant and 115 TFBS peaks when summed over different cell types and tissue.
Using the intersect function of the Bedtools, the non-coding variant was predicted to affect the promoter (129,308,487.129308588) of the SLC15A4 gene. All described analysis results of the SLC15A4 upstream variant are summarized in Table 2.
In order to identify those transcription factors for which the binding may be affected the most by the variant, we used Jaspar2020 for prediction and comparison of the TFBSs for the wild-type and the mutant sequence of the SLC15A4 upstream region (Fornes et al. 2020). Whereas most of the identified TFBS were shared by both sequences, nine transcription factors were predicted to bind only to the wild-type sequence, indicating a TFBS disruption by the variant, and eight were predicted to bind only to the mutant sequence, indicating a TFBS creation by the variant (Table 3). One of the identified transcription factors, whose binding site was disrupted was STAT1 which has been established as a favorable prognostic marker in several types of cancers, including CRC (Klampfer 2008; Simpson et al. 2010; Gordziel et al. 2013). Moreover, STAT1 has been proposed as a tumor suppressor particularly in colitis‐associated CRC (Crncec et al. 2018), in turn suggesting a carcinogenic potential of its disruption by the identified upstream variant.
Discussion
Performing WES on a family with CRC aggregation and applying our in-house developed FCVPPv2, we identified two novel heterozygous variants in the SLC15A4 gene that segregated with the disease in the family. The missense variant, p. Y444C, was predicted to affect the phylogenetically conserved PTR2/POT domain and to have a deleterious effect on the function of the encoded peptide/histidine transporter. The other variant was located in the upstream region of the same gene and it was annotated to affect the promoter region of SLC15A4 as well as binding sites of several transcription factors. Our findings of two distinct variants in the same gene may indicate a synergistic up-regulation of SLC15A4 as the underlying genetic cause and implicate this gene for the first time in genetic inheritance of familial CRC.
SLC15A4 belongs to the family of the proton-coupled oligopeptide transporters (POTs) that enable the transfer of histidine and oligopeptides derived from degradation products from inside of the endosome to the cytosol. Since proton dependency implies higher transport activity at low pH levels, endosomal acidification during the maturation to lysosomes is required for substrate uptake by the SLC15A4 transporter (Yamashita et al. 1997; Bhardwaj et al. 2006).
Well-established examples of SLC15A4 substrates are the NOD1 ligands L-Ala-D-Glu-meso-diaminopimelic acid (Tri-DAP) and γ-D-Glu-meso-diaminopimelic acid (iE-DAP), components of the cell wall peptidoglycan of primarily Gram-negative bacteria (Lee et al. 2009; Sasawatari et al. 2011). NOD1 stimulation by DAP induces the activation of nuclear factor-κB and mitogen-activated protein (MAP) kinases and thus the transcription of various genes responsible for innate and adaptive immune responses (Hayden and Ghosh 2004; Franchi et al. 2009). Knockdown of SLC15A4 in HEK293T cells has been shown to lead to decreased nuclear factor-κB activation by the NOD1 ligands (Lee et al. 2009), which was supported by in vivo experiments resulting in loss of Tri-DAP–induced cytokine production in SLC15A4-deficient mice. The same study has further reported an association of SLC15A4 with toll like receptor 9 (TLR9) functions: SLC15A4-deficient dendritic cells showed decreased TLR9-mediated cytokine production which was traced back by the authors to high lysosomal histidine concentrations in the absence of SLC15A4. By being required for TLR9- as well as NOD1-mediated cytokine production, SLC15A4 has been shown to promote Th1-dependent colitis in vivo (Sasawatari et al. 2011).
Since chronic intestinal inflammation has been associated with increased CRC risk, potentially mediated by oxidative DNA damage and innate and adaptive immune responses (Feagins et al. 2009; Ullman and Itzkowitz 2011), SLC15A4 may further play an important role in the initial inflammation-induced colorectal carcinogenesis (https://www.ebi.ac.uk/gwas/efotraits/EFO_0003767; accessed on March 5th, 2021). Based on these findings, we are suggesting a role in CRC susceptibility as well for genetic variation of SLC15A4.
Performing WES on a family with CRC aggregation and applying our in-house developed FCVPPv2, we were able to identify a novel heterozygous variant in the coding region of the SLC15A4 gene. By being present in all four CRC-affected siblings as well as one direct descendant with colorectal polyps, the identified missense variant in SLC15A4 shows segregation with the disease and a potential for medium-to-high-penetrance susceptibility to CRC in the studied family. Considering the very low allele frequency of the variant in the NFE population of 0 counts in 113,688 alleles, the proposed association of the identified genetic variation with familial CRC is further supported. In silico analyses based on evolutionary conservation, intolerance against functional genetic alterations and deleteriousness led to the prediction of pathogenicity for the missense variant. Snap2 further predicted an effect on protein function by the missense variant leading to the amino acid substitution Y444C in SLC15A4. Considering all analyses, we propose an up-regulating mode of action for the identified missense variant on SLC15A4 protein level.
Interestingly, we identified another variant with the same familial segregation in the upstream region of the SLC15A4 gene (12_129308531_C_T; 43 bp upstream of transcription start site, ENST00000266771.5). GnomAD browser reported an allele frequency of 3.754 × 10–3 in the NFE population. Taking this relatively high frequency into account, high penetrance and thus strong functional consequence of the upstream variant by itself may not be expected. Nevertheless, synergistic effects of both variants occurring in the same gene have to be considered: The upstream variant may have an enhancing impact on SLC15A4 protein expression, potentially of minor relevance when solely occurring but which may reinforce the postulated up-regulating mode of action of the SLC15A4 coding variant in the course of colorectal carcinogenesis. In order to confirm the proposed mode of function, we assessed the upstream variant for potentially influencing gene transcription. According to our analysis, the upstream variant was annotated to be located at an active transcription start site affecting the promoter region of the SLC15A4 gene. In particular, binding sites of 17 different transcription factors were predicted to be exclusive for either the wild type or the mutant sequence due to the identified upstream variant, representing a potential mechanism of enhancing gene transcription. Whether the variant potentially destroys TFBSs for transcriptional repressors or creates new TFBSs for transcriptional activators, remains unclear and requires further functional experiments. By providing a list of TFBSs and potential transcriptional repressors or activators, including the tumor suppressor STAT1, we aim to lay the foundation for functional validation of the regulatory impact of the upstream variant and instigate further research in this field. Thus, we hope to facilitate a better understanding of the identified upstream variant in the context of SLC15A4 gene regulation in particular and of the postulated synergistic model of coding and non-coding variants in cancer predisposition in general.
Certainly, the confined number of analyzed family members and particularly healthy controls has to be taken into account as a statistical limitation of this study when finally interpreting the described results. Due to lack of availability of additional blood samples, the inclusion of further family members in our analyses was not feasible to increase the statistical power. We met this limitation to some extent by considering the allele frequencies of the identified variants in large populations according to gnomAD (Karczewski et al. 2019) and TOPMed data (Li et al. 2020; Taliun et al. 2021). Further validation of the identified variants has been provided by the large-scale WES data of UK Biobank, reporting statistically significant gene-phenotype associations of the SLC15A4 gene and the clinical phenotypes of malignant neoplasms in the colon and rectum (Wang et al. 2021).
By identifying germline variants in the SLC15A4 gene in familial CRC, we implicated this gene for the first time in genetic inheritance of a malignancy, expanding its role from a potential CRC marker in quantitative fecal tests to a potential marker of CRC susceptibility in genetic testing. However, the results of this study need to be further replicated in validation cohorts and validated using experimental approaches in cell lines.
Data availability
Unfortunately, for reasons of ethics and patient confidentiality, we are not able to provide the sequencing data into a public database. The data underlying the results presented in the study are available from the corresponding author or from Dr. Asta Försti (Email: a.foersti@kitz-heidelberg.de).
Code availability
Not applicable.
References
Bandapalli OR, Paramasivam N, Giangiobbe S, Kumar A, Benisch W, Engert A, Witzens-Harig M, Schlesner M, Hemminki K, Forsti A (2018) Whole genome sequencing reveals DICER1 as a candidate predisposing gene in familial Hodgkin lymphoma. Int J Cancer 143:2076–2078
Bhardwaj RK, Herrera-Ruiz D, Eltoukhy N, Saad M, Knipp GT (2006) The functional evaluation of human peptide/histidine transporter 1 (hPHT1) in transiently transfected COS-7 cells. Eur J Pharm Sci 27:533–542
Blum M, Chang HY, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, Nuka G, Paysan-Lafosse T, Qureshi M, Raj S, Richardson L, Salazar GA, Williams L, Bork P, Bridge A, Gough J, Haft DH, Letunic I, Marchler-Bauer A, Mi H, Natale DA, Necci M, Orengo CA, Pandurangan AP, Rivoire C, Sigrist CJA, Sillitoe I, Thanki N, Thomas PD, Tosatto SCE, Wu CH, Bateman A, Finn RD (2020) The interpro protein families and domains database: 20 years on. Nucleic Acids Res 49:D344–D354
Briggs S, Tomlinson I (2013) Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol 230:148–153
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM (2013) The cancer genome atlas pan-cancer analysis project. Nat Genet 45:1113–1120
Chau R, Jenkins MA, Buchanan DD, Ait Ouakrim D, Giles GG, Casey G, Gallinger S, Haile RW, Le Marchand L, Newcomb PA, Lindor NM, Hopper JL, Win AK (2016) Determining the familial risk distribution of colorectal cancer: a data mining approach. Fam Cancer 15:241–251
Cooper GM, Stone EA, Asimenos G, Program NCS, Green ED, Batzoglou S, Sidow A (2005) Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15:901–913
Coordinators NR (2018) Database resources of the national center for biotechnology information. Nucleic Acids Res 46:D8–D13
Crncec I, Modak M, Gordziel C, Svinka J, Scharf I, Moritsch S, Pathria P, Schlederer M, Kenner L, Timelthaler G, Muller M, Strobl B, Casanova E, Bayer E, Mohr T, Stockl J, Friedrich K, Eferl R (2018) STAT1 is a sex-specific tumor suppressor in colitis-associated colorectal cancer. Mol Oncol 12:514–528
Ernst J, Kellis M (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9:215–216
Feagins LA, Souza RF, Spechler SJ (2009) Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol 6:297–305
Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, Santana-Garcia W, Tan G, Cheneby J, Ballester B, Parcy F, Sandelin A, Lenhard B, Wasserman WW, Mathelier A (2020) JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48:D87–D92
Franchi L, Warner N, Viani K, Nunez G (2009) Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev 227:106–128
Frank C, Fallah M, Sundquist J, Hemminki A, Hemminki K (2015) Population landscape of familial cancer. Sci Rep 5:12891
Gala MK, Mizukami Y, Le LP, Moriichi K, Austin T, Yamamoto M, Lauwers GY, Bardeesy N, Chung DC (2014) Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 146:520–529
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:p11
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR (2015) A global reference for human genetic variation. Nature 526:68–74
Gordziel C, Bratsch J, Moriggl R, Knosel T, Friedrich K (2013) Both STAT1 and STAT3 are favourable prognostic determinants in colorectal carcinoma. Br J Cancer 109:138–146
Gudis K, Tatsuguchi A, Wada K, Futagami S, Nagata K, Hiratsuka T, Shinji Y, Miyake K, Tsukui T, Fukuda Y, Sakamoto C (2005) Microsomal prostaglandin E synthase (mPGES)-1, mPGES-2 and cytosolic PGES expression in human gastritis and gastric ulcer tissue. Lab Invest 85:225–236
Hansen MF, Johansen J, Sylvander AE, Bjornevoll I, Talseth-Palmer BA, Lavik LAS, Xavier A, Engebretsen LF, Scott RJ, Drablos F, Sjursen W (2017) Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch Syndrome. Clin Genet 92:405–414
Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18:2195–2224
Hecht M, Bromberg Y, Rost B (2015) Better prediction of functional effects for sequence variants. BMC Genomics 16:S1
Hemminki K, Sundquist J, Bermejo JL (2008) How common is familial cancer? Ann Oncol 19:163–167
Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS (2012) Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 9:473–476
Jasperson KW, Tuohy TM, Neklason DW, Burt RW (2010) Hereditary and familial colon cancer. Gastroenterology 138:2044–2058
Karczewski K, Francioli L, Tiao G, Cummings B, Alföldi J, Wang Q, Collins R, Laricchia K, Ganna A, Birnbaum D, Gauthier L, Brand H, Solomonson M, Watts N, Rhodes D, Singer-Berk M, Seaby E, Kosmicki J, Walters R, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong J, Samocha K, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria A, Minikel EV, Weisburd B, Lek M, Ware J, Vittal C, Armean I, Bergelson L, Cibulskis K, Connolly K, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski M, Neale B, Daly M, MacArthur D, The Genome Aggregation Database C (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. In. bioRxiv
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315
Klampfer L (2008) The role of signal transducers and activators of transcription in colon cancer. Front Biosci 13:2888–2899
Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, Furuyama-Tanaka K, Karyu H, Sugiura Y, Shimizu Y, Hosaka T, Goto M, Kato N, Okamura T, Suematsu M, Yokoyama S, Toyama-Sorimachi N (2014) The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity 41:375–388
Korotkova M, Daha NA, Seddighzadeh M, Ding B, Catrina AI, Lindblad S, Huizinga TW, Toes RE, Alfredsson L, Klareskog L, Jakobsson PJ, Padyukov L (2011) Variants of gene for microsomal prostaglandin E2 synthase show association with disease and severe inflammation in rheumatoid arthritis. Eur J Hum Genet 19:908–914
Kuiper RP, Hoogerbrugge N (2015) NTHL1 defines novel cancer syndrome. Oncotarget 6:34069–34070
Kumar A, Bandapalli OR, Paramasivam N, Giangiobbe S, Diquigiovanni C, Bonora E, Eils R, Schlesner M, Hemminki K, Forsti A (2018) Familial cancer variant prioritization pipeline version 2 (FCVPPv2) applied to a papillary thyroid cancer family. Sci Rep 8:11635
Lahiri DK, Schnabel B (1993) DNA isolation by a rapid method from human blood samples: effects of MgCl2, EDTA, storage time, and temperature on DNA yield and quality. Biochem Genet 31:321–328
Lee J, Tattoli I, Wojtal KA, Vavricka SR, Philpott DJ, Girardin SE (2009) pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J Biol Chem 284:23818–23829
Lee HS, Kim T, Bang SY, Na YJ, Kim I, Kim K, Kim JH, Chung YJ, Shin HD, Kang YM, Shim SC, Suh CH, Park YB, Kim JS, Kang C, Bae SC (2014) Ethnic specificity of lupus-associated loci identified in a genome-wide association study in Korean women. Ann Rheum Dis 73:1240–1245
Lee CL, Huang CJ, Yang SH, Chang CC, Huang CC, Chien CC, Yang RN (2016) Discovery of genes from feces correlated with colorectal cancer progression. Oncol Lett 12:3378–3384
Lee PH, Lee C, Li X, Wee B, Dwivedi T, Daly M (2018) Principles and methods of in-silico prioritization of non-coding regulatory variants. Hum Genet 137:15–30
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25:1754–1760
Li X, Li Z, Zhou H, Gaynor SM, Liu Y, Chen H, Sun R, Dey R, Arnett DK, Aslibekyan S, Ballantyne CM, Bielak LF, Blangero J, Boerwinkle E, Bowden DW, Broome JG, Conomos MP, Correa A, Cupples LA, Curran JE, Freedman BI, Guo X, Hindy G, Irvin MR, Kardia SLR, Kathiresan S, Khan AT, Kooperberg CL, Laurie CC, Liu XS, Mahaney MC, Manichaikul AW, Martin LW, Mathias RA, McGarvey ST, Mitchell BD, Montasser ME, Moore JE, Morrison AC, O’Connell JR, Palmer ND, Pampana A, Peralta JM, Peyser PA, Psaty BM, Redline S, Rice KM, Rich SS, Smith JA, Tiwari HK, Tsai MY, Vasan RS, Wang FF, Weeks DE, Weng Z, Wilson JG, Yanek LR, Neale BM, Sunyaev SR, Abecasis GR, Rotter JI, Willer CJ, Peloso GM, Natarajan P, Lin X (2020) Dynamic incorporation of multiple in silico functional annotations empowers rare variant association analysis of large whole-genome sequencing studies at scale. Nat Genet 52:969–983
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K (2000) Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343:78–85
Liu X, Wu C, Li C, Boerwinkle E (2016) dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 37:235–241
Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F, Ishikawa-Kato S, Mungall CJ, Arner E, Baillie JK, Bertin N, Bono H, de Hoon M, Diehl AD, Dimont E, Freeman TC, Fujieda K, Hide W, Kaliyaperumal R, Katayama T, Lassmann T, Meehan TF, Nishikata K, Ono H, Rehli M, Sandelin A, Schultes EA, t Hoen PA, Tatum Z, Thompson M, Toyoda T, Wright DW, Daub CO, Itoh M, Carninci P, Hayashizaki Y, Forrest AR, Kawaji H, consortium F, (2015) Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol 16:22
Lorans M, Dow E, Macrae FA, Winship IM, Buchanan DD (2018) Update on hereditary colorectal cancer: improving the clinical utility of multigene panel testing. Clin Colorectal Cancer 17:e293–e305
Nieminen TT, O’Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, Ellonen P, Abdel-Rahman WM, Valo S, Mecklin JP, Jarvinen HJ, Gleizes PE, Peltomaki P (2014) Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 147(595–598):e595
Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, Ma Y, Kaur K, Dobbins S, Barclay E, Gorman M, Martin L, Kovac MB, Humphray S, Lucassen A, Holmes CC, Bentley D, Donnelly P, Taylor J, Petridis C, Roylance R, Sawyer EJ, Kerr DJ, Clark S, Grimes J, Kearsey SE, Thomas HJ, McVean G, Houlston RS, Tomlinson I (2013) Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45:136–144
Papadopoulos JS, Agarwala R (2007) COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23:1073–1079
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB (2013) Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9:e1003709
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20:110–121
Ponz de Leon M, Sassatelli R, Sacchetti C, Zanghieri G, Scalmati A, Roncucci L (1989) Familial aggregation of tumors in the three-year experience of a population-based colorectal cancer registry. Cancer Res 49:4344–4348
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47:D886–D894
Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SR, Wilkie AO, McVean G, Lunter G (2014) Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet 46:912–918
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M (2015) Integrative analysis of 111 reference human epigenomes. Nature 518:317–330
Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP (2017) Variant review with the integrative genomics viewer. Cancer Res 77:e31–e34
Sasawatari S, Okamura T, Kasumi E, Tanaka-Furuyama K, Yanobu-Takanashi R, Shirasawa S, Kato N, Toyama-Sorimachi N (2011) The solute carrier family 15A4 regulates TLR9 and NOD1 functions in the innate immune system and promotes colitis in mice. Gastroenterology 140:1513–1525
Segui N, Mina LB, Lazaro C, Sanz-Pamplona R, Pons T, Navarro M, Bellido F, Lopez-Doriga A, Valdes-Mas R, Pineda M, Guino E, Vidal A, Soto JL, Caldes T, Duran M, Urioste M, Rueda D, Brunet J, Balbin M, Blay P, Iglesias S, Garre P, Lastra E, Sanchez-Heras AB, Valencia A, Moreno V, Pujana MA, Villanueva A, Blanco I, Capella G, Surralles J, Puente XS, Valle L (2015) Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology 149:563–566
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15:1034–1050
Simpson JA, Al-Attar A, Watson NF, Scholefield JH, Ilyas M, Durrant LG (2010) Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 59:926–933
Skopelitou DM, Srivastava B, Kumar A, Kuswick A, Dymerska M, Paramasivam D, Schlesner N, Lubinski M, Hemminki J, Försti K, Bandapalli A, Obul R (2021) Whole exome sequencing identifies APCDD1 and HDAC5 genes as potentially cancer predisposing in familial colorectal cancer. Int. J. Mol. Sci. 22:1837
Smigielski EM, Sirotkin K, Ward M, Sherry ST (2000) dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res 28:352–355
Srivastava A, Kumar A, Giangiobbe S, Bonora E, Hemminki K, Forsti A, Bandapalli OR (2019) Whole genome sequencing of familial non-medullary thyroid cancer identifies germline alterations in MAPK/ERK and PI3K/AKT signaling pathways. Biomolecules 9:605
Srivastava A, Giangiobbe S, Kumar A, Paramasivam N, Dymerska D, Behnisch W, Witzens-Harig M, Lubinski J, Hemminki K, Forsti A, Bandapalli OR (2020a) Identification of familial hodgkin lymphoma predisposing genes using whole genome sequencing. Front Bioeng Biotechnol 8:179
Srivastava A, Miao B, Skopelitou D, Kumar V, Kumar A, Paramasivam N, Bonora E, Hemminki K, Forsti A, Bandapalli OR (2020) A germline mutation in the POT1 gene is a candidate for familial non-medullary thyroid cancer. Cancers (Basel) 12:1441
Srivastava A, Giangiobbe S, Skopelitou D, Miao B, Paramasivam N, Diquigiovanni C, Bonora E, Hemminki K, Försti A, Bandapalli OR (2021) Whole genome sequencing prioritizes CHEK2, EWSR1, and TIAM1 as possible predisposition genes for familial non-medullary thyroid cancer. Front Endocrinol. https://doi.org/10.3389/fendo.2021.600682
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, Kaplan S, Dahary D, Warshawsky D, Guan-Golan Y, Kohn A, Rappaport N, Safran M, Lancet D (2016) The genecards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. https://doi.org/10.1002/cpbi.5
Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, Pitsillides AN, LeFaive J, Lee SB, Tian X, Browning BL, Das S, Emde AK, Clarke WE, Loesch DP, Shetty AC, Blackwell TW, Smith AV, Wong Q, Liu X, Conomos MP, Bobo DM, Aguet F, Albert C, Alonso A, Ardlie KG, Arking DE, Aslibekyan S, Auer PL, Barnard J, Barr RG, Barwick L, Becker LC, Beer RL, Benjamin EJ, Bielak LF, Blangero J, Boehnke M, Bowden DW, Brody JA, Burchard EG, Cade BE, Casella JF, Chalazan B, Chasman DI, Chen YI, Cho MH, Choi SH, Chung MK, Clish CB, Correa A, Curran JE, Custer B, Darbar D, Daya M, de Andrade M, DeMeo DL, Dutcher SK, Ellinor PT, Emery LS, Eng C, Fatkin D, Fingerlin T, Forer L, Fornage M, Franceschini N, Fuchsberger C, Fullerton SM, Germer S, Gladwin MT, Gottlieb DJ, Guo X, Hall ME, He J, Heard-Costa NL, Heckbert SR, Irvin MR, Johnsen JM, Johnson AD, Kaplan R, Kardia SLR, Kelly T, Kelly S, Kenny EE, Kiel DP, Klemmer R, Konkle BA, Kooperberg C, Kottgen A, Lange LA, Lasky-Su J, Levy D, Lin X, Lin KH, Liu C, Loos RJF, Garman L, Gerszten R, Lubitz SA, Lunetta KL, Mak ACY, Manichaikul A, Manning AK, Mathias RA, McManus DD, McGarvey ST, Meigs JB, Meyers DA, Mikulla JL, Minear MA, Mitchell BD, Mohanty S, Montasser ME, Montgomery C, Morrison AC, Murabito JM, Natale A, Natarajan P, Nelson SC, North KE, O’Connell JR, Palmer ND, Pankratz N, Peloso GM, Peyser PA, Pleiness J, Post WS, Psaty BM, Rao DC, Redline S, Reiner AP, Roden D, Rotter JI, Ruczinski I, Sarnowski C, Schoenherr S, Schwartz DA, Seo JS, Seshadri S, Sheehan VA, Sheu WH, Shoemaker MB, Smith NL, Smith JA, Sotoodehnia N, Stilp AM, Tang W, Taylor KD, Telen M, Thornton TA, Tracy RP, Van Den Berg DJ, Vasan RS, Viaud-Martinez KA, Vrieze S, Weeks DE, Weir BS, Weiss ST, Weng LC, Willer CJ, Zhang Y, Zhao X, Arnett DK, Ashley-Koch AE, Barnes KC, Boerwinkle E, Gabriel S, Gibbs R, Rice KM, Rich SS, Silverman EK, Qasba P, Gan W, Papanicolaou GJ, Nickerson DA, Browning SR, Zody MC, Zollner S, Wilson JG, Cupples LA, Laurie CC, Jaquish CE, Hernandez RD, O’Connor TD, Abecasis GR (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590:290–299
Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnstrom H, Glimelius B, Sjoblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F (2017) A pathology atlas of the human cancer transcriptome. Science 357:eaan2507
Ullman TA, Itzkowitz SH (2011) Intestinal inflammation and cancer. Gastroenterology 140:1807–1816
Valle L, de Voer RM, Goldberg Y, Sjursen W, Forsti A, Ruiz-Ponte C, Caldes T, Garre P, Olsen MF, Nordling M, Castellvi-Bel S, Hemminki K (2019) Update on genetic predisposition to colorectal cancer and polyposis. Mol Aspects Med 69:10–26
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164
Wang C, Ahlford A, Jarvinen TM, Nordmark G, Eloranta ML, Gunnarsson I, Svenungsson E, Padyukov L, Sturfelt G, Jonsen A, Bengtsson AA, Truedsson L, Eriksson C, Rantapaa-Dahlqvist S, Sjowall C, Julkunen H, Criswell LA, Graham RR, Behrens TW, Kere J, Ronnblom L, Syvanen AC, Sandling JK (2013) Genes identified in Asian SLE GWASs are also associated with SLE in Caucasian populations. Eur J Hum Genet 21:994–999
Wang Q, Dhindsa RS, Carss K, Harper AR, Nag A, Tachmazidou I, Vitsios D, Deevi SVV, Mackay A, Muthas D, Huhn M, Monkley S, Olsson H, AstraZeneca Genomics I, Wasilewski S, Smith KR, March R, Platt A, Haefliger C, Petrovski S (2021) Rare variant contribution to human disease in 281,104 UK biobank exomes. Nature 597:527–532
Wei Y, Zhang S, Shang S, Zhang B, Li S, Wang X, Wang F, Su J, Wu Q, Liu H, Zhang Y (2016) SEA: a super-enhancer archive. Nucleic Acids Res 44:D172-179
Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, van Zelst-Stams WA, Jongmans MC, Gilissen C, Hehir-Kwa JY, Hoischen A, Shendure J, Boyle EA, Kamping EJ, Nagtegaal ID, Tops BB, Nagengast FM, Geurts van Kessel A, van Krieken JH, Kuiper RP, Hoogerbrugge N (2015) A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 47:668–671
Yamashita T, Shimada S, Guo W, Sato K, Kohmura E, Hayakawa T, Takagi T, Tohyama M (1997) Cloning and functional expression of a brain peptide/histidine transporter. J Biol Chem 272:10205–10211
Yan HHN, Lai JCW, Ho SL, Leung WK, Law WL, Lee JFY, Chan AKW, Tsui WY, Chan ASY, Lee BCH, Yue SSK, Man AHY, Clevers H, Yuen ST, Leung SY (2017) RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 66:1645–1656
Yu L, Yin B, Qu K, Li J, Jin Q, Liu L, Liu C, Zhu Y, Wang Q, Peng X, Zhou J, Cao P, Cao K (2018) Screening for susceptibility genes in hereditary non-polyposis colorectal cancer. Oncol Lett 15:9413–9419
Zetner DB, Bisgaard ML (2017) Familial colorectal cancer type X. Curr Genomics 18:341–359
Zhang M, Chen F, Zhang D, Zhai Z, Hao F (2016) Association study between SLC15A4 polymorphisms and haplotypes and systemic lupus erythematosus in a han chinese population. Genet Test Mol Biomarkers 20:451–458
Zhang D, Li Z, Xu X, Zhou D, Tang S, Yin X, Xu F, Li H, Zhou Y, Zhu T, Deng H, Zhang S, Huang Q, Wang J, Yin W, Zhu Y, Lai M (2017) Deletions at SLC18A1 increased the risk of CRC and lower SLC18A1 expression associated with poor CRC outcome. Carcinogenesis 38:1057–1062
Zuo XB, Sheng YJ, Hu SJ, Gao JP, Li Y, Tang HY, Tang XF, Cheng H, Yin XY, Wen LL, Sun LD, Yang S, Cui Y, Zhang XJ (2014) Variants in TNFSF4, TNFAIP3, TNIP1, BLK, SLC15A4 and UBE2L3 interact to confer risk of systemic lupus erythematosus in Chinese population. Rheumatol Int 34:459–464
Acknowledgements
The authors thank the members of the families for participating in this study, Genomics and Proteomics Core Facility (GPCF) of the German Cancer Research Center (DKFZ) for providing excellent library preparation and sequencing services and the Omics IT and Data Management Core Facility (ODCF) of the DKFZ for the whole exome sequencing data management.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was funded by COST Action CA17118, supported by COST (European Cooperation in Science and Technology) and Transcan ERA-NET funding from the German Federal Ministry of Education and Research (BMBF). KH was supported by the EU Horizon 2020 program grant No. 856620.
Author information
Authors and Affiliations
Contributions
Conceptualization: KH, AF and ORB; methodology: DS, AS and BM; software: AK, NP, MS and OR; validation: DD and JL; formal analysis: DS, AK, NP, AS and ORB; investigation: DS and ORB; resources: DD and JL; data curation: DS, AK, NP, MS and ORB; writing-original draft preparation: DS and ORB; writing-review and editing: KH, AF and ORB; visualization: DS; supervision: KH, AF and ORB; project administration: KH, AF and ORB; Funding acquisition: KH. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Ethical approval
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Bioethics Committee of the Pomeranian Medical Academy in Szczecin (protocol code No: BN-001/174/05).
Informed consent
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patients to publish this paper.
Additional information
Communicated by Shuhua Xu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Skopelitou, D., Srivastava, A., Miao, B. et al. Whole exome sequencing identifies novel germline variants of SLC15A4 gene as potentially cancer predisposing in familial colorectal cancer. Mol Genet Genomics 297, 965–979 (2022). https://doi.org/10.1007/s00438-022-01896-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-022-01896-0