
Abstract
The unfolded protein response (UPR) has evolved to counter the stresses that occur in the endoplasmic reticulum (ER) as a result of misfolded proteins. This sophisticated quality control system attempts to restore homeostasis through the action of a number of different pathways that are coordinated in the first instance by the ER stress-senor proteins IRE1, ATF6 and PERK. However, prolonged ER-stress-related UPR can have detrimental effects on cell function and, in the longer term, may induce apoptosis. Connective tissue cells such as fibroblasts, osteoblasts and chondrocytes synthesise and secrete large quantities of proteins and mutations in many of these gene products give rise to heritable disorders of connective tissues. Until recently, these mutant gene products were thought to exert their effect through the assembly of a defective extracellular matrix that ultimately disrupted tissue structure and function. However, it is now becoming clear that ER stress and UPR, because of the expression of a mutant gene product, is not only a feature of, but may be a key mediator in the initiation and progression of a whole range of different connective tissue diseases. This review focuses on ER stress and the UPR that characterises an increasing number of connective tissue diseases and highlights novel therapeutic opportunities that may arise.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The endoplasmic reticulum (ER) is a distinct compartment of eukaryotic cells and plays major roles in the synthesis, folding and trafficking of proteins entering the secretory pathway. In addition, the ER is involved in Ca2+ storage and controlling various aspects of lipid and sterol synthesis (see Schröder 2008). The general area of molecular chaperones, ER stress and the unfolded protein response (UPR) has been extensively reviewed recently (Bukau et al. 2006; Szegezdi et al. 2006; Bernales et al. 2006; Malhotra and Kaufman 2007; Ron and Walter 2007; Lin et al. 2008). In this review, we will therefore give a brief description of these areas and focus upon the impact that misfolding mutant forms of extracellular matrix (ECM) proteins have on ER homeostasis in human or mammalian cells and the possible downstream cellular consequences as they are related to disease pathogenesis.
Protein folding in the ER
Proteins destined for the secretory pathway of the cell are synthesised in the cytoplasm on ribosomes bound to the ER (forming the rough ER) and co-translationally translocated into the lumen of the ER via the Sec61 translocon. Within the lumen of the ER, newly synthesised proteins undergo a series of post-translational modifications such as signal peptide cleavage, asparagine (N)-linked glycosylation, disulphide-bond formation and collagen-specific modifications such as hydroxylation of prolyl and lysyl residues and glycosylation of hydroxylysyl residues (Bateman et al. 2009). These post-translational modifications occur as the protein is folding to attain its mature form and many of the modifications are a prerequisite for correct folding. Folding is assisted by a host of chaperones, co-chaperones, foldases and oxidoreductases. Chaperones such as BiP/Grp78 and Grp98 prevent unfolded proteins from aggregating, by means of their exposed hydrophobic sequences, and provide an environment conducive to protein folding. Enzymes such as protein disulphide isomerases (PDI) catalyse disulphide-bond formation and exchange. Many ancillary proteins are involved in securing and delivering the energy input required to drive the energy-consuming process of protein folding and in maintaining the correct redox state within the ER lumen to permit the reversible formation of disulphide bonds.
The ER has a sophisticated quality control system for ensuring that misfolded proteins do not accumulate or pass further long the secretory pathway but are instead retained within the ER and targeted for degradation (Anelli and Sitia 2008). For proteins that undergo N-linked glycosylation, the addition of a high mannose oligosaccharide from a dolicolpyrophosphate donor presages a series of trimming and reglucosylation reactions that govern the interaction of the folding glycoproteins with the lectin chaperones calnexin and calreticulin. The extent of carbohydrate trimming serves as a molecular clock and misfolded or slowly folding proteins are targeted for degradation (Molinari 2007). Non-glycosylated proteins that misfold or fold too slowly are recognised by their sequential interactions with chaperones such as BiP, Grp98 and PDI, and are targeted for degradation (Schröder 2008). The degradation of misfolded ER proteins can take place by two mechanisms. First, the protein can be retrotranslocated back into the cytoplasm where it is ubiquinated and degraded by the proteasome in a process known as ER-associated degradation (ERAD; Brodsky 2007). For proteins that cannot be retrotranslocated (perhaps because they have aggregated or assemble into multimers), autophagy appears to be activated. Portions of ER engorged with misfolded protein are budded off and become incorporated within a double-membrane structure known as an autophagosome that subsequently fuses with a lysosome in which the contents are degraded (Bernales et al. 2007; Schröder 2008; Ishida et al. 2009).
ER stress and the UPR
Mammalian cells have evolved a complex homeostatic mechanism for maintaining protein folding equilibrium within the ER known as the UPR.
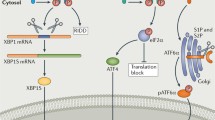
The ER responds to increases in the luminal levels of unfolded protein via three pathways activated respectively by the transmembrane ER-stress-sensing proteins inositol-requiring enzyme-1 (IRE1), pancreatic ER eukaryotic translation initiation factor (eIF)-2a kinase (PERK) and activating transcription factor-6 (ATF6). All three of these proteins are bound in their inactive states by the chaperone BiP and become dissociated from BiP during activation (see Fig. 1), although the exact mechanism by which each protein senses increases in the luminal concentration of unfolded protein is not known.

The unfolded protein response (UPR). Unfolded protein (UP) in the lumen of the endoplasmic reticulum (ER) is bound by the chaperone BiP. BiP also binds the ER luminal domains of the three ER stress sensors: pancreatic ER eukaryotic translation initiation factor (eIF)-2a kinase (PERK), inositol-requiring enzyme-1 (IRE1) and activating transcription factor-6 (ATF6). If the concentration of unfolded protein in the ER lumen increases, e.g. because of the expression of a mutant extracellular matrix gene, BiP preferentially binds the unfolded protein thereby freeing the stress sensors. PERK and IRE1 are activated by dimerisation and autophosphorylation. PERK then phosphorylates eIF2α, which suppresses general protein synthesis thus decreasing the entry of newly synthesised protein into the ER. ATF4 mRNA is more efficiently translated when eIF2α is phosphorylated and ATF4 up-regulates a set of ER-stress-sensitive genes including that for CCAAT/enhancer-binding protein homologous protein (CHOP). Activated IRE1 produces an unconventional splice in cytoplasmic X-box binding protein 1 (XBP1) mRNA and this spliced XBP1 up-regulates genes encoding chaperones to increase the protein folding capacity of the ER and genes controlling endoplasmic-reticulum-associated degradation (ERAD), a mechanism by which misfolded protein is retrotranslocated into the cytoplasm and proteasomally degraded. When BiP releases ATF690, the protein is translocated to the Golgi where it is cleaved by site-1 and site-2 proteases (S1P, S2P) releasing the cytoplasmic 50-kDa domain (ATF650) which is an active transcription factor up-regulating genes involved in the UPR, including chaperones, XBP1 and CHOP (rER rough endoplasmic reticulum)
The IRE1 branch of UPR is evolutionarily the oldest found in lower eukaryotes such as yeast and throughout the animal kingdom, whereas PERK and ATF6 are only found in higher animals such as nematodes, insects and mammals (Shen et al. 2005). The 110-kDa type 1 transmembrane IRE1 protein exists in two closely related forms: IRE1α, which is expressed ubiquitously, and IRE1β whose expression is restricted to gut epithelium (Bertolotti et al. 2001). The IRE1 protein has a cytoplasmic domain that possesses serine/threonine kinase and endoribonuclease activities, a transmembrane domain and an ER luminal domain. In its inactive state, the ER luminal domain of IRE1 is bound by the ER chaperone BiP. Upon sensing an increase in the luminal concentration of unfolded protein (Credle et al. 2005), IRE1 dissociates from BiP and oligomerises in the plane of the ER membrane; this leads to an autocatalytic phosphorylation of the cytoplasmic domain and activation of its ribonuclease activity. The endoribonuclease domain catalyses an unconventional splice within X-box binding protein 1 (XBP1) mRNA, which, in mammalian cells, produces an alternative carboxyl terminal domain in the translated protein, termed XBP1s (s for spliced), which is a basic leucine zipper (bZIP) family transcription factor. XBP1s promotes the transcription of a number of genes involved in the UPR including chaperones that increase the protein-folding capacity of the ER and proteins involved in ERAD (Todd et al. 2008). There is increasing evidence that, in addition to processing XBP1 mRNA, IRE1(α and β) directly or indirectly promotes the rapid cleavage and destruction of a subset of mRNAs associated with the rough ER thereby reducing the entry of new proteins into the ER lumen (Hollien and Weissman 2006; Iqbal et al. 2008). IRE1 also plays a direct role in signalling. The active phosphorylated form of IRE1 interacts with tumour necrosis factor receptor associated factor 2, which subsequently can promote activation of JUN N-terminal kinase (JNK). Activated JNK can then determine downstream consequences such as apoptosis or autophagy (Ogata et al. 2006; Szegezdi et al. 2006; Oh and Lim 2009). IRE1 activity is modulated by its interaction with BAX inhibitor 1 and members of the BCL-2 family of proteins (Hetz and Glimcher 2008; Lisbona et al. 2009)
PERK is also a type 1 transmembrane protein of ~120 kDa with a C-terminal cytoplasmic domain that possesses serine/threonine kinase activity and an ER luminal domain that is similar to that of IRE1 and binds BiP (Bertolotti et al. 2000). As the levels of unfolded protein in the lumen of the ER increase, PERK dissociates from BiP leading to autophosphorylation, oligomerisation and activation of the kinase domain (Fig. 1). Activated PERK phosphorylates and inactivates eukaryotic translation initiation factor (eIF) 2α, which is required for 80S ribosome assembly. Consequently, general protein synthesis is down-regulated decreasing the flow of newly synthesised unfolded protein into the ER. Paradoxically, some mRNAs such as that encoding the 39-kDa transcription factor ATF4, are translated more efficiently when eIF2α is phosphorylated. ATF4 mRNA has a series of 5′ open reading frames that result in its inefficient translation under non-stressed conditions. However, the phosphorylation of eIF2α alters the dynamics by with which these upstream open reading frames are read and results in increased synthesis of ATF4 (Lu et al. 2004). ATF4 is a transcription factor in the cAMP-response element binding (CREB) family and activates many genes involved in controlling the UPR including chaperones such as BiP and GRP94, genes involved in suppressing oxidative stress, and genes involved in amino acid metabolism and transport (Harding et al. 2003). ATF4 also induces the expression of CCAAT/enhancer-binding protein homologous protein (CHOP also known as GADD153 or DDIT3), a gene that has been associated with ER-stress-induced apoptosis (Zinszner et al. 1998).
ATF6 is a 90-kDa transmembrane protein that consists of a cytoplasmic domain constituting a transcription factor with a bZIP motif and a luminal domain that binds BiP (Fig. 1). ATF6 is maintained in the ER through its binding to BiP. As unfolded protein levels rise in the ER and BiP dissociates, Golgi-localisation sequences in the ER luminal domain of ATF6 are revealed and the protein is translocated to the Golgi (Shen et al. 2002) in which it is cleaved by site-1 and site-2 proteases (Ye et al. 2000). These cleavages release the cytosolic 50-kDa domain as an active transcription factor (ATF650), which moves to the nucleus and activates the transcription of a range of genes involved in ER quality control including chaperones such as BiP, XBP1 and ERAD components and the apoptosis-inducing CHOP (Adachi et al. 2008).
Downstream consequences of elevated ER stress and the UPR
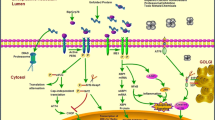
ER stress can be caused by a number of different insults including heat shock, energy deprivation, hypoxia, metabolic dysfunction and drugs such as tunicamycin, which inhibits N-linked glycosylation, and thapsigargin, which disrupts ER calcium ion balance, in addition to the expression of a mutant protein. Whatever the stimulus, elevated ER stress is characterised by increased concentrations of unfolded or misfolding protein within the ER; this triggers the UPR in an attempt to restore protein folding homeostasis. The most rapid consequences of the UPR are PERK phosphorylation of eIF2α and IRE1-dependant destabilisation of mRNA associated with the ER thereby reducing protein synthesis and the entry of newly synthesised protein into the ER lumen (Fig. 2). The transcriptional programme orchestrated by XBP1s, ATF4 and ATF650 results in increased expression of a whole range of chaperones and other proteins involved in maintaining ER protein folding quality control and of proteins that catalyse the pathways by which terminally misfolded proteins are degraded. The level of ER stress experienced by a cell is clearly going to be related to the load of protein passing through its ER, with “professional secretory cells” such as hepatocytes, insulin-producing beta cells of the pancreas and antibody-producing lymphocytes being particularly susceptible. Connective tissue cells such as chondrocytes, osteoblasts and fibroblasts also fall into this category of strongly secretory cells and are therefore likely to be particularly sensitive to ER stress induction. In the face of persistent exposure to ER stress, cells may reduce this stress to acceptable levels through the UPR, although this is frequently not without consequences and, as described below, can result in a pathogenic outcome. Alternatively, if the levels of ER stress are unacceptable, a signalling cascade triggered by IRE1-mediated JNK phosphorylation and through the activity of CHOP may result in the cell undergoing apoptosis (Szegezdi et al. 2006), which again can have pathogenic consequences (Fig. 2).

ER stress and disease
The UPR is crucial for cell homeostasis and normal development. However, it is increasingly clear that the UPR also plays a key pathogenic role in a wide range of diseases (Lin et al. 2008). In type II diabetes, metabolically induced ER stress appears to be crucial in insulin resistance (Ozcan et al. 2004), which in turn increases demands on the insulin-producing beta cells of the pancreas. The increased load of insulin passing through the beta cell ER causes elevated and persistent ER stress and can result in beta cell apoptosis and the subsequent requirement for insulin therapy (Eizirik et al. 2008). The UPR also takes a central part in the pathogenesis of inflammatory diseases (Zhang and Kaufman 2008) and many neurodegenerative conditions (Lin et al. 2008).
ER stress, skeletal development and associated diseases
The importance of certain components of the UPR for normal skeletal development is apparent from gene ablation studies. The Ire1α knockout is embryonic lethal (Urano et al. 2000) and thus its potential impact upon connective tissues has not been reported. PERK mutations in man and mice cause severe developmental defects including diabetes, growth retardation and skeletal dysplasia (Zhang et al. 2002). In the absence of Perk, mice are severely osteopenic because of a deficiency in osteoblast differentiation and maturation. The trafficking and secretion of collagen I is compromised and collagen I is abnormally retained in the ER. The mice have an osteogenesis-imperfecta-like phenotype (Wei et al. 2008). ATF4 whose translation is enhanced by PERK-activated eIF2α phosphorylation and mediates key aspects of UPR (Fig. 1) is also required for normal osteoblast differentiation and growth (Yang et al. 2004). Indeed, mild ER stress, which induces limited PERK phosphorylation and increased translation of ATF4, has been shown to enhance osteoblast function and the potential cross-talk between pathways controlling responses to ER stress and osteoblast function is being explored further (Hamamura et al. 2008). Atf6 knockout mice are viable and fertile and have no overt phenotype unless challenged by ER stress-inducing agents (Rutkowski et al. 2008). However, ablation of site-1 protease, one of the two proteases required to release and activate not only ATF6, but other related membrane-localised latent transcription factors from the Golgi, has profound affects in chondrocytes. Endochondral ossification is severely affected and the chondrocytes exhibit ultrastructural evidence of severe ER stress (Patra et al. 2007). ER stress is also a component of the disease process in inflammatory conditions such as rheumatoid arthritis (Todd et al. 2008) and recent studies have implicated elevated ER stress in the pathology associated with osteoarthritis (Ruiz-Romero et al. 2008; Hamamura et al. 2009).
ER stress induced by mutations in ECM genes
Until recently, mutations in genes encoding ECM components were largely thought to exert their effects in terms of disease pathogenesis by way of the synthesis of an ECM that was defective either because of a deficiency of the wild-type version of the mutated protein or because of the presence of the mutated protein (or a combination of both). It is becoming increasingly clear that, in addition to these two pathogenic mechanisms, the cellular consequences of increased levels of ER stress induced by misfolding mutant protein within the ER, known as the UPR, can contribute significantly to disease pathogenesis (Fig. 2).
ER stress induced by mutations in fibrillar collagens
Mutations in the two genes encoding type I collagen, COL1A1 and COL1A2, result in osteogenesis imperfecta (OI) and some variants of Ehlers-Danlos syndrome. Two types of mutation cause OI. Mutations that produce premature stop codons directly or via frame shifts can cause nonsense-mediated mRNA decay and milder forms of OI that are thought to result directly from the reduced synthesis of functional collagen I trimers. The majority of mutations in the type I collagen genes cause the synthesis of mutant misfolding proα chains. Chessler and Byers (1993) first reported that BiP appeared to associate specifically with a subset of OI-causing proα1(I) chains, namely those containing mutations localised to the non-collagenous C-propeptide. Furthermore, they reported that BiP levels in the patients’ fibroblasts expressing these mutations were elevated, classic evidence of what we would nowadays consider to be UPR. Indeed, in fibroblasts cultured from a recently characterised OI mouse model (Aga2) resulting from a frameshift that introduces an extension of 90 amino acids to the C-propeptide of the proα1(I) chain, clear signs of elevated ER stress and a UPR (increased levels of BiP and other chaperones and increased expression of CHOP) together with increased rates of apoptosis have been described (Lisse et al. 2008). Intriguingly, collagen I chains harbouring OI-causing mutations in the collagenous domain do not bind BiP, and BiP levels in these fibroblasts are not elevated (Chessler and Byers 1993). The lack of a classical UPR (for example elevated BiP) in bone from an OI mouse model resulting from a collagenous domain mutation (BrtlIV carrying a Gly349Cys substitution in the Col1a1 gene) has been confirmed by proteomic analysis, although CHOP has been found to be elevated (Forlino et al. 2007). Only in one case reported to date has a mutation in the collagenous domain of a fibrillar collagen (Gly904Cys mutation in the Col2a1 gene) been reported to elevate BiP accompanied by a fragmented ER in chondrocytes of the chondrodysplastic mouse growth plate (Tsang et al. 2007). Therefore, of the collagenous domain mutations studied, most do not appear to induce a BiP-mediated classical UPR. The stress pathways triggered by misfolding collagenous domain mutations remains to be defined. Misfolded mutant fibrillar collagen chains are targeted for degradation by, as yet, poorly defined pathways. Originally described as protein suicide (Prockop 1984), a proteasomal route nowadays known as ERAD was subsequently described by Lamandé et al. (1995) and Fitzgerald et al. (1999). More recently, autophagic routes of protein degradation have been described for mutant forms of type I collagen that aggregate within the ER (Ishida et al. 2009).
ER stress induced by mutations in collagen IV
Mutations in the Col4a1 gene have been shown to cause perinatal cerebral haemorrhage, porencephaly (Gould et al. 2005) and, on a defined genetic background, ocular anterior segment dysgenesis (Gould et al. 2007). The mouse mutation studied, which resulted from the skipping of exon 40 of the Col4a1 gene because of a splice site defect, caused a 17-amino-acid deletion in the collagenous domain and prevented secretion of the mutant proα1(IV) chain. In lens epithelium, this retention of mutant collagen IV resulted in the up-regulation of the collagen-specific chaperone Hsp47, together with the up-regulation of other proteins carrying the ER-retention sequence KDEL indicative of ER stress (Gould et al. 2007).
Mutations in COMP, matrilin 3 and type XI collagen cause pseudoachondroplasia and multiple epiphyseal dysplasia
The earliest data suggesting that a mutation in a gene encoding an ECM protein (or at least a secreted protein) causes a pathology attributable to an ER-related problem comes from studies on pseudoachondroplasia (PSACH) and multiple epiphyseal dysplasia (MED). PSACH and MED are members of the same bone dysplasia family and are characterised by joint pain and stiffness, a varying degree of short-limbed dwarfism and early onset osteoarthritis (OA). The phenotypic spectrum is defined by severe PSACH and the mild Ribbing form of MED and includes mild PSACH and the severe (Fairbank) form of MED.
Morphology of PSACH and MED chondrocytes: a historical perspective
The first morphological and biochemical characterisation of cartilage from a patient with PSACH was published in 1972 and described a pathological feature that would become a hallmark of this disease for the next 35 years (Maynard et al. 1972). In this study by Maynard and colleagues, electron microscopy performed on chondrocytes isolated from fibula and iliac crest growth plates showed enlarged cisternae of ER caused by the accumulation of protein. These ER “inclusions” had a lamellar appearance with alternating electron-dense and electron-lucent layers and the authors hypothesised that they may represent abnormal proteins that were synthesised as a result of a genetic defect and that could not be transported from the ER. Several years later, a more comprehensive biochemical study was performed, which led to the hypothesis that the dilated cisternae of ER arose because of the accumulation of abnormal core protein(s) of proteoglycans (Stanescu et al. 1982). Furthermore, cell death apparently occurred more frequently when the large intracytoplasmic inclusions became “huge”.
Some two decades after the initial observation by Maynard et al. (1972), Stanescu and colleagues (1993) showed that chondrocytes from a patient with the “Fairbank type” of MED also demonstrated enlarged ER. In this study, the authors determined that the retained material could be stained with an antibody raised against the hyaluronic-acid-binding region of aggrecan, but not to type II collagen; furthermore, the retained material was resistant to collagenase. These observations led the authors to suggest that PSACH and MED were members of the same bone dysplasia family and that the abnormal cell phenotype was attributable to the accumulation of mutated aggrecan core protein (Stanescu et al. 1993).
Identification of the genetic defects in PSACH and MED
The suggestion that the retained abnormal material in the ER of PSACH and MED chondrocytes might be aggrecan provided the rationale for several linkage studies involving the use of genetic markers specific for aggrecan (Wordsworth et al. 1988; Finkelstein et al. 1991; Hecht et al. 1992; Weaver et al. 1993; Rimoin et al. 1994). However, these studies all excluded aggrecan as the causative gene for PSACH and MED in several large families. Eventually genome-wide linkage screens and positional candidate cloning approaches were successful in determining that mutations in the gene encoding cartilage oligomeric matrix protein (COMP) cause PSACH and some forms of MED (Briggs et al. 1995; Hecht et al. 1995). MED is genetically heterogeneous and the use of similar mapping approaches has determined that mutations in the genes encoding type IX collagen and matrilin-3 also cause autosomal dominant forms of MED (Briggs and Chapman 2002).
Disease mechanisms in COMP-related PSACH-MED: in vitro approaches
The confirmation that mutations in COMP cause PSACH and some forms of MED led to the first histochemical analysis of cartilage and tendon from a patient with PSACH (Maddox et al. 1997). In this study, the authors demonstrated for the first time that the retained material was indeed abnormal COMP. Type IX collagen was also present within the dilated ER, whereas type II collagen was only present in the cartilage ECM. The authors concluded that abnormally folded COMP molecules might be retained in the ER by chaperone proteins that associate with large oligomeric molecules and that this might affect normal cellular metabolism. Interestingly, the intracellular retention of mutant COMP was not observed in a tendon sample, leading the authors to suggest that it was a cell-specific phenomenon (Maddox et al. 1997). In a follow-up study on PSACH chondrocytes cultured in alginate beads, the authors confirmed the intracellular retention of mutant COMP and minimal ECM staining (Hecht et al. 1998a). Furthermore, the apparent remains of dead cells were characterised by the accumulation of “enormous cisternae” in regions of apparent cell necrosis. This led to the hypothesis that the linear polysomes observed in PSACH cells were probably the result of increased cytoplasmic protein synthesis in response to a blocked ER-Golgi system, whereas the numerous lysosomes indicated an activation of a “degenerative cycle” that allowed PSACH cells to die from defects in the secretary pathway. Whether mutant COMP is retained within the ER of tendon and ligament cells has not been fully resolved and remains controversial (Delot et al. 1998, 1999; Hecht et al. 1998b).
By 2001, the first papers were published that described the association of mutant COMP with chaperone proteins such as calreticulin (CRT), protein disulphide isomerise (PDI), Grp94, ERp72 and BiP (Grp78; Hecht et al. 2001; Vranka et al. 2001). In one such study, electron microscopy and immuno-gold labelling of a PSACH iliac crest biopsy and chondrocytes cultured in alginate beads demonstrated that five chaperones, BiP, CRT, PDI, ERp72 and Grp94 were localised to the electron-dense material in the dilated ER (Hecht et al. 2001). Furthermore, immuno-precipitation and fluorescence energy transfer experiments indicated that CRT, Grp94 and Erp72 were closely associated with mutant COMP, whereas BiP surprisingly was not. In addition, this study confirmed that type IX collagen and aggrecan also co-localised to the ER with mutant COMP; however, aggrecan was also abundant in the ECM and type II collagen was found exclusively in the ECM (Hecht et al. 2001). In a similar study, Vranka and colleagues (2001) examined an iliac crest biopsy from a young girl with PSACH and confirmed the association of chaperone proteins, including HSP47, PDI and calnexin, with mutant COMP, whereas BiP was again absent. The authors hypothesised that the UPR accounted for increased levels of these specific chaperone proteins within the dilated ER of PSACH chondrocytes. Finally, they demonstrated the presence of fibromodulin, decorin and collagen types IX, XI and XII in the rough ER, whereas aggrecan and types II and VI collagen were not retained intracellularly. The authors therefore concluded that the selective retention of mutant COMP and other molecules led to impaired cell function and a compromised ECM (Vranka et al. 2001).
In 2003, two studies, using different cell culture models, introduced the concept of increased chondrocyte apoptosis as a disease mechanism in PSACH and tried to relate this finding to growth plate defects (Hashimoto et al. 2003; Duke et al. 2003). In the first study, COS7 cells were used to recapitulate the PSACH phenotype by the overexpression of mutant COMP (Hashimoto et al. 2003). Interestingly, in addition to the retention of mutant COMP, these cells demonstrated an induction of ER stress through the phosphorylation of eIF2α, a well-characterised mechanism of down-regulating protein synthesis under ER stress conditions. The authors then proceeded to examine the levels of apoptosis in this cell culture model and were able to determine that, in cells expressing mutant COMP, the relative levels of apoptosis were significantly increased (Hashimoto et al. 2003). In a parallel study, Hecht and colleagues were able to confirm increased levels of apoptosis in PSACH chondrocytes cultured in alginate beads (Duke et al. 2003). The authors hypothesised that apoptosis in PSACH chondrocytes occurs via a modified “damaged cell” pathway. Analysis of the growth plate in an iliac crest biopsy from a PSACH patient suggested increased chondrocyte cell death in vivo, characterised by a reduced number of chondrocytes in the pre-hypertrophic and hypertrophic region and an increased number of TUNEL-positive cells. These findings led the authors to suggest that PSACH was an ER storage disease and that the retained material (presumably mutant COMP and its interacting partners) was toxic to chondrocytes (Duke et al. 2003).
MED mutations in matrilin-3 cause trafficking defects similar to COMP mutations
Missense mutations in the gene encoding matrilin-3 (MATN3) have been shown to cause some forms of MED (Chapman et al. 2001) and the related disease bilateral heredity micro-ephyseal dysplasia (BHMED; Mostert et al. 2003). Interesting, all of MED-causing mutations in MATN3 are found in the single A-domain of matrilin-3 in either the β-strand or α-helix regions (Jackson et al. 2004; Fresquet et al. 2008). The majority of these mutations have been revealed to result in the retention of mutant protein in vitro, whereas a smaller proportion of mutations allows the secretion of mutant protein (Cotterill et al. 2005; Fresquet et al. 2008). Furthermore, mutant matrilin-3 A-domains remain predominantly unfolded in the ER of cells and are associated with ERp72, a chaperone protein important for disulphide bond formation. In contrast, no association has been demonstrated between mutant matrilin-3 A-domains and the chaperone proteins PDI, Grp78/Bip and Grp94 (Cotterill et al. 2005). These data indicate a specific role for ERp72 in the recognition and retention of misfolded matrilin-3 or, alternatively, might reflect an experimental limitation in the expression of matrilin-3 A-domains alone and not the full-length molecules.
Mouse models of PSACH-MED caused by mutations in COMP and matrilin-3
In order to determine the disease mechanisms of PSACH-MED in vivo, we generated knock-in mouse models harbouring mutations in Comp (p.Thr585Met) and Matn3 (p.Val194Asp; Leighton et al. 2007; Pirog-Garcia et al. 2007). These studies are complemented by those of Schmitz and colleagues (2008) who have generated a transgenic mouse line expressing mutant Comp (p.D469del) driven by a type II collagen promoter.
Mild PSACH/MED resulting from a p.T585M missense mutation in the C-terminal domain of COMP (Comp p.T585M)
Mutant animals are normal at birth but grow more slowly than their wild-type littermates and develop mild short-limb dwarfism by adulthood. In the growth plates of mutant mice, the chondrocyte columns are sparser and poorly organised compared with wild-type littermates. Mutant COMP is secreted into the ECM but its localisation is disrupted, together with the localisation of several COMP-binding proteins, suggesting that the presence of mutant COMP disrupts ECM assembly. By 3 weeks of age, chondrocyte proliferation is significantly reduced, whereas apoptosis is both increased and spatially dysregulated in the mutant growth plates.
Although mutant COMP is not retained within the ER of chondrocytes, mild ER stress and UPR still occur that are characterised by the up-regulation of chaperone proteins BiP and calteticulin, the phosphorylation of eIF2α, cleavage of ATF6 and caspase-12 and, eventually, an increased expression of CHOP (Pirog-Garcia et al. 2007). These findings are all characteristic of ER-stress-induced UPR, finally leading to the activation of pro-apoptotic pathways (Rutkowski and Kaufman 2004); this is the first evidence of CHOP involvement in the disease mechanisms of PSACH-MED. Overall, these data suggest that the mutation in the C-terminal domain of COMP exerts a dominant-negative effect on both intra- and extracellular processes.
MED resulting from a V194D missense mutation in the A-domain of matrilin-3 (matn3:V194D)
Mice that are homozygous for this mutation develop a progressive dysplasia and have short-limbed dwarfism that is consistent in severity with the relevant human phenotype. Mutant matrilin-3 is retained within the rough ER of chondrocytes from birth and is associated with an UPR, by 5 days of age, which is characterised by the up-regulation of classical early markers of the UPR such as BiP and calreticulin. Eventually, proliferation is reduced and the apoptosis of chondrocytes is spatially dysregulated in the growth plate. However, this cell pathology does not appear to be directly related to CHOP, the expression of which is not increased in mutant chondrocytes at 3 or 5 days and 2 or 3 weeks of age in MED (Leighton et al. 2007). The precise genetic pathway(s) linking ER stress and the UPR to apoptosis therefore remain unresolved in this form of MED
Transgenic mice expressing D649del under a Col2a1 promoter
Male mice expressing a mutant Comp transgene with a BMP40 signal peptide (on a Comp-null background) show mild growth retardation. The columnar organisation of chondrocytes is disrupted and gaps are noted between columns. Mutant COMP has a peri-cellular localisation different from the inter-territorial distribution of the endogenous wild-type COMP. Some mutant COMP is also localised to intracellular compartments in chondrocytes within the proliferative zone. This intracellular retention is dependent on the Comp expression level, since a second D465 del transgenic line, in which mutant Comp expression is lower, shows no intracellular retention. Electron microscopy has confirmed that, in some cells,ER cisternae are dilated, consistent with the retention of mutant COMP. TUNEL staining shows a significant increase in apoptosis in the growth plates of mutant mice but this is restricted to the boundary with the secondary centre of ossification. The total numbers of cells within the mutant growth plate is not reduced compared with wild-type controls leading the authors to suggest that cell proliferation is not significantly altered. However, cell proliferation assays have not been performed to determine directly the relative levels of proliferation in a given time period. The role of ER stress and the UPR has not been investigated in this mouse model (Schmitz et al. 2008).
Summary of PSACH-MED disease mechanisms in vivo
A consistent finding in all three mouse models of PSACH-MED is the increased and spatially dysregulated apoptosis in the cartilage growth plate (Leighton et al. 2007; Pirog-Garcia et al. 2007; Schmitz et al. 2008). Furthermore, two of these models (Comp p.T585M and Matn3 p.V194D) also have a significant reduction in chondrocyte proliferation as measured by BrdU incorporation. These detrimental changes to chondrocyte proliferation, differentiation and survival are likely to be key factors in the development and progression of these forms of PSACH-MED. Interestingly however, the specific genetic pathways leading to apoptosis may be different depending on the specific disease gene and/or mutation. Our data confirm that the expression of Matn3 p.V194D and Comp p.T855M cause ER stress that results in an UPR characterised by the up-regulation of chaperone proteins. In the case of Comp p.T855M, the UPR appears sufficient to cause the folding and secretion of mutant COMP. However, either the UPR itself or the presence of mutant COMP in the ECM initiates a mild CHOP-mediated increase in apoptosis (Pirog-Garcia et al. 2007). In contrast, the UPR is not capable of mediating the folding and secretion of Matn3 p.V194D, which appears to accumulate in the ER of mutant chondrocytes, eventually leading to apoptosis. In this case, apoptosis is not CHOP-mediated and the specific genetic pathways remain unresolved (Leighton et al. 2007).
Collagen X mutations and metaphyseal chondrodysplasia type Schmid
Collagen X is a short-chain collagen that is expressed exclusively in the hypertrophic zone of the growth plate cartilage. Mutations in the COL10A1 gene cause metaphyseal chondrodysplasia type Schmid (MCDS; Warman et al. 1993; Wallis et al. 1994; Bateman et al. 2005), a relatively mild metaphyseal dysplasia. With the exception of two mutations that affect the signal peptide cleavage site of the type X collagen chain, MCDS-causing mutations are clustered in the C-terminal non-collagenous NC1 domain of the protein, the domain responsible for initiating trimerisation during collagen synthesis (Bateman et al. 2005). Mutations that introduce premature stop codons can cause a nonsense-mediated mRNA decay-like response (Bateman et al. 2005; Tan et al. 2008). Missense mutations cluster within particular subdomains of the NC1 domain (Marks et al. 1999; Bogin et al. 2000). Some mutant forms of the NC1 domain retain their ability to trimerise in an in vitro assembly system (Marks et al. 1999), whereas others are predicted to be permissive for efficient protein folding and collagen trimerisation based on structural considerations (Bogin et al. 2000). Nevertheless, the expression of a variety of these missense mutant collagen X transcripts in heterologous cell systems has demonstrated that each induces misfolding of the collagen X with impaired secretion (Chan et al. 1996, 2001; Wilson et al. 2002, 2005). Retained mutant collagen X protein is degraded by a combination of proteasomal and lysosomal routes (Chan et al. 2001; Wilson et al. 2005). In 2005, Wilson and colleagues reported that the intracellular retention of collagen X provoked a UPR based on XBP1 splicing and the up-regulation of BiP (Wilson et al. 2005).
Mouse models of MCDS
MCDS mutations cause ER stress in vivo
Transgenic mice expressing a construct driven by a collagen X promoter encoding the mouse equivalent of the first human MCDS mutation described (Warman et al. 1993), a 13-bp deletion within the COL10A1 gene that disrupts the C-terminus of the NC1 domain, exhibit a short-limb dwarfism (Tsang et al. 2007). Histological examination of the growth plate has revealed an expanded hypertrophic zone similar to that previously reported in a pig model of MCDS (Nielsen et al. 2000) and more recently in the iliac crest biopsy from a human donor (Ho et al. 2007). The hypertrophic chondrocytes expressing the mutant collagen X shows immunohistochemical evidence of impaired secretion and intracellular retention of the protein. Electron-microscopic examination has revealed that the ER of affected chondrocytes is distended and fragmented. Furthermore, the hypertrophic chondrocytes exhibit clear evidence of ER stress and a resulting UPR as assessed by Xbp1 splicing, and the up-regulation of the chaperone BiP and the transcription factor CHOP, although no evidence has been obtained for increased rates of apoptosis in the hypertrophic zone (Tsang et al. 2007). Essentially the same phenotype is exhibited by a second transgenic mouse expressing the mouse equivalent of the human MCDS p.P620fsX621 mutation from a collagen X promoter (Ho et al. 2007). In both these mouse lines, the phenotype is not affected by breeding onto a collagen X knockout background, although breeding to homozygosity for the mutant transgenic alleles produces more severe phenotypes demonstrating a clear gene-dosage effect (Tsang et al. 2007; Ho et al. 2007). Gene expression within the expanded hypertrophic zone is disrupted with the re-expression of collagen II and prehypertrophic markers such as the parathormone-related peptide receptor accompanying the down-regulation of collagen X expression and the disorganised expression of terminal hypertrophic markers such as osteopontin (Tsang et al. 2007; Ho et al. 2007). Clear evidence that hypertrophic cells reverts in phenotype to a pre-hypertrophic state including re-engagement of part of the cell cycle has been presented and the “reprogramming” of the chondrocyte differentiation process has been proposed as a strategy by which hypertrophic chondrocytes down-regulate mutant gene production in order to deal with the accumulating unfolded collagen X protein (Tsang et al. 2007).
Our own laboratory has produced a gene-targeted “knock-in” MCDS mouse expressing the Col10a1 p.N617K mutation. This mouse exhibits essentially the same growth plate phenotype as the transgenic lines described above including the intracellular retention of the mutant collagen X and a robust UPR characterised by the up-regulation of both BiP and the cleaved activated form of ATF6 (Rajpar et al. 2009). The phenotype in mice heterozygote for the mutant allele is less severe than that in homozygotes. In heterozygotes, a transient reduction in longitudinal bone growth is apparent at peak growth rates around 3 weeks of age but largely resolves compared with wild-type controls by 11 weeks of age. In contrast, the homozygote MCDS mice exhibit a 15%–20% reduction in long bone (femur) length compared with wild-type animals; this persists throughout their adult life. The expression of vascular endothelial growth factor (VEGF), which is a key factor controlling the rate of invasion and cartilage erosion at the vascular invasion front (Zelzer and Olsen 2005), in the hypertrophic zone is disrupted in the mutant mouse hypertrophic zone. In addition, the recruitment of osteoclasts to the vascular invasion front is significantly reduced in mutant mice. A reduced rate of cartilage erosion atrributable to the disruption of VEGF signalling and the resultant decrease in osteoclast recruitment to the vascular invasion front of the growth plate explains mechanistically the expansion of the hypertrophic zone in MCDS (Rajpar et al. 2009).
Direct evidence that ER stress causes the MCDS phenotype
Whereas all of the MCDS-causing mutations tested in vivo produced ER stress and a UPR, small quantities of mutant collagen X are secreted (Ho et al. 2007; Rajpar et al. 2009) and the possibility that this secreted protein plays an important role in the disease mechanism cannot be excluded. We therefore decided to test directly the role played by ER stress in the pathogenic process by targeting the expression of an exogenous ER-stress-inducing protein specifically to hypertrophic chondrocytes by using the collagen X promoter. If ER stress and the induced UPR are critical factors in the disease mechanism, mice expressing the ER-stress-inducing construct should exhibit a similar phenotype to the MCDS mouse lines described above. The ER-stress-inducing protein selected was the cog mutant form of thyroglobulin (Tgcog). Thyroglobulin is a large dimeric protein that is secreted into the lumen of the thyroid gland where it is iodinated and extensively processed to form thyroid hormones. Tgcog cannot dimerise, which is an obligate step for transport beyond the ER, and is therefore not secreted but completely retained and degraded intracellularly (Kim et al. 1996; Park and Arvan 2004). Mouse lines expressing the Tgcog transgene in hypertrophic chondrocytes exhibit increased ER stress and a UPR in the growth plate similar to that seen in the p.N617K MCDS mouse, namely, elevated levels of BiP and cleaved ATF6. Histologically, the hypertrophic zone is expanded in the Tgcog mouse lines. Particularly noteworthy, whereas the Tgcog protein is retained intracellularly, the secretion of the wild-type collagen X synthesised in these mice is not affected (Rajpar et al. 2009). Furthermore, osteoclast recruitment to the vascular invasion front is significantly reduced and gene expression patterns in the hypertrophic zone of the Tgcog mice are disrupted in a similar manner to that described above for the MCDS mouse lines. In the Tgcog line studied in most detail, mice homozygous for the mutant allele express the transgene at one-third the level of the endogenous wild-type collagen X. Reduced bone growth is apparent in rapidly growing (3–6 week old) mice expressing the Tgcog protein but is largely resolved in comparison with wild-type controls by 11 weeks of age (Rajpar et al. 2009). The finding that the Tgcog line develops a pathology strikingly similar to the MCDS phenotype directly demonstrates the central pathogenic role played by elevated ER stress and the resulting UPR.
The level of ER stress correlated with disease severity
All of the MCDS mouse models display a clear gene-dosage effect upon disease severity (Tsang et al. 2007; Ho et al. 2007; Rajpar et al. 2009); this can be attributed to the increased level of ER stress and UPR induced in mice homozygous, rather than heterozygous, for the mutant allele. Furthermore, the variation of disease severity, as judged by the extent of hypertrophic zone expansion, also appears to relate directly to the level of ER stress experienced by the hypertrophic chondrocytes. During the period of most rapid bone growth when protein synthesis rates will be at their highest (from birth to 4 weeks of age), the hypertrophic expansion in mutant animals is greatest and this expansion decreases markedly as growth rates subsequently decline (Tsang et al. 2007; Rajpar et al. 2009). In mice heterozygous for the MCDS-causing p.N617K mutation and in mice expressing the Tgcog mutant gene, bone growth rates are reduced only during the period of most rapid bone growth and, by 10–11 weeks of age, the bone lengths of these animals recover and are essentially the same as those of wild-type controls (Rajpar et al. 2009). This finding illustrates that disease severity can be influenced by the severity of the ER stress experienced and that strategies for reducing ER stress may well have a significant impact upon the clinical severity of MCDS.
Unanswered questions about ER stress and MCDS
For MCDS, many questions still remain. First, what is the mechanism that causes the hypertrophic chondrocyte experiencing ER stress apparently to revert to a pre-hypertrophic stage in order to deal with the accumulated mutant protein prior to re-engaging the hypertrophic differentiation programme? An understanding of the mechanism that drives this apparent “reverse differentiation” is of particular relevance to developing novel therapeutic approaches for treating osteoarthritis in which inappropriate hypertrophic differentiation of chondrocytes is apparent (Kawaguchi 2008). Second, how do mutations in the collagen X gene that result in virtually complete nonsense-mediated decay of the mutant mRNA (see Bateman et al. 2005), and therefore essentially no synthesis of mutant protein, produce a disease phenotype that is clinically indistinguishable from that produced by the expression of mutant protein that triggers UPR? One of the mouse lines described above (Ho et al. 2007) was generated to examine a nonsense-mediated decayed allele but, in the mouse, this mutant allele, which encoded a premature stop codon, generated a relatively stable mRNA that was effectively translated into a mutant protein that elicited increased ER stress. Nevertheless, the possibility that the high levels of nonsense-mediated mRNA decay required effectively to degrade the mutant collagen X mRNA may in some way be capable of triggering a stress response involving the ER in the hypoxic, energy-depleted and autophagic environment of the hypertrophic chondrocyte (Srinivas et al. 2009) should not be ignored. Third, can we produce novel stress-alleviating treatment strategies (see below) that will significantly reduce the level of ER stress experienced in the growth plate and thus reduce clinical severity of the disease?
ER stress may contribute to a range of connective tissue pathologies
UPR as a result of increased ER stress is being described in an increasing number of connective tissue disorders, as we have attempted to collate in this review. Whereas the UPR is the cell’s attempt to restore protein-folding homeostasis to the ER and as such is a protective mechanism, the process of restoring homeostasis can interfere with normal cell function and produce a biological consequence such as decreased rates of protein synthesis and altered gene expression profiles. If homeostasis is rapidly restored, there may be little consequence but, in situations in which the ER is exposed to prolonged periods of stress, such as in a connective tissue cell expressing a mutant ECM protein, pathological consequences can ensue. First, chronic ER stress can trigger apoptosis and cell loss becomes a major factor in the pathology. This is possibly the case for PSACH in which mutant COMP expression in the proliferative zone of the growth plate causes cell loss (Pirog-Garcia et al. 2007) or for mutations of type I collagen, which can trigger osteoblast apoptosis in OI (Lisse et al. 2008). However, chronic ER stress and the UPR do not necessarily always trigger apoptosis. For example, only a minority of cells in the affected tissues of the PSACH and OI mouse models mentioned above undergo apoptosis. The surviving cells exhibit strong UPR and would therefore have reduced general protein synthesis rates whist up-regulating chaperone synthesis and the synthesis of genes involved with degrading the accumulated unfolded or misfolded protein. This general decrease in protein synthesis provoked by the expression of a mutant ECM gene means that not only is the surrounding ECM deficient in the mutant gene product, but it may also be deficient in many of the non-mutated ECM proteins. This effect has been most clearly demonstrated in the Tgcog mouse model of MCDS where expression of the mutant Tgcog protein provokes the down-regulation in the expression the wild-type collagen X gene (Rajpar et al. 2009); exactly the same phenomenon has previously been described in the other MCDS mouse models reported (Tsang et al. 2007; Ho et al. 2007). Decreased rates of protein synthesis attributable to eIF2α phosphorylation might also contribute significantly to the decreased rates of chondrocyte proliferation seen in the PSACP and MED mouse models reported above (Pirog-Garcia et al. 2007; Leighton et al. 2007).
Novel therapeutic opportunities for treating disorders caused by mutant ECM proteins
For MCDS, we have been able to demonstrate that the pathology arises largely as a result of the UPR per se (Rajpar et al. 2009). The possibility that UPR induced by the expression of mutant ECM genes may be a contributory pathogenic factor in many of the resulting diseases warrants further investigation because new strategies for alleviating or reducing ER stress are constantly being developed. Chemical or low molecular weight chaperones that have the capacity to assist proteins fold more efficiently and thus reduce ER stress (Perlmutter 2002; Papp and Csermely 2006) have proved of therapeutic benefit in experimental models of some ER-stress-associated disorders such as type II diabetes (Ozcan et al. 2006) and stroke (Qi et al. 2004). The level of ER stress can also be reduced by speeding up the rates at which misfolded proteins can be degraded either by ERAD and proteasomal routes or by autophagy, and many new treatment strategies that affect the cell’s capacity to degrade misfolded proteins are being devised and tested (e.g. Rochet 2007; Rubinsztein et al. 2007; Kudo et al. 2008; Williams et al. 2008).
In order to take advantage of recent progress made in this area, we need to study the characteristics of the cell stress induced by specific mutations in specified ECM proteins and, furthermore, to understand the way that the cell deals with the resulting misfolding protein. As is clear from studies on collagen I mutations, various stress pathways might be activated by different mutations in the same gene (Chessler and Byers 1993) and, similarly, various pathways of protein degradation might also be activated by different mutations in the same gene (Ishida et al. 2009). However, the possibility that the alleviation of ER stress may in many connective tissue disorders produce a decrease in the clinical severity of the disease is an exciting and unexpected new opportunity for treatment.
The concept that a connective tissue disease can be treated without addressing the underlying genetic defect is not new. For instance, patients with Marfan syndrome have mutations in their fibrillin 1 gene resulting in excessive transforming growth factor (TGF)-beta signalling. Marfan patients are now undergoing clinical trials with drugs that directly suppress the raised TGF-beta signalling but have no impact upon the underlying gene defect, since such a strategy has produced spectacular results in terms of treatment when tested in mouse models (Pearson et al. 2008). ER stress associated with a range of connective tissue diseases offers a similar opportunity in cases in which treatment of the downstream consequences, rather than the root-cause of the disease, might produce significant reductions in clinical severity.
References
Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K (2008) ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33:75–89
Anelli T, Sitia R (2008) Protein quality control in the early secretory pathway. EMBO J 27:315–327
Bateman JF, Wilson R, Freddi S, Lamandé SR, Savarirayan R (2005) Mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. Hum Mutat 25:525–534
Bateman JF, Boot-Handford RP, Lamandé SR (2009) Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Genet 10:173–183
Bernales S, Papa FR, Walter P (2006) Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22:487–508
Bernales S, Schuck S, Walter P (2007) ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy 3:285–287
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2:326–332
Bertolotti A, Wang X, Novoa I, Jungreis R, Schlessinger K, Cho JH, West AB, Ron D (2001) Increased sensitivity to dextran sodium sulfate colitis in IRE1 β-deficient mice. J Clin Invest 107:585–593
Bogin O, Kvansakul M, Rom E, Singer J, Yayon A, Hohenester E (2000) Insight into Schmid metaphyseal chondrodysplasia from the crystal structure of the collagen X NC1 domain trimer. Structure 10:165–173
Briggs MD, Chapman KL (2002) Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat 19:465–478
Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, Cekleniak JA, Knowlton RG, Cohn DH (1995) Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet 10:330–336
Brodsky JL (2007) The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem J 404:352–362
Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein folding. Cell 125:443–451
Chan D, Weng YM, Hocking AM, Golub S, McQuillan DJ, Bateman JF (1996) Site-directed mutagenesis of human type X collagen. Expression of alpha1(X) NC1, NC2, and helical mutations in vitro and in transfected cells. J Biol Chem 271:13566–13572
Chan D, Ho MS, Cheah KS (2001) Aberrant signal peptide cleavage of collagen X in Schmid metaphyseal chondrodysplasia. Implications for the molecular basis of the disease. J Biol Chem 276:7992–7997
Chapman KL, Mortier GR, Chapman K, Loughlin J, Grant ME, Briggs MD (2001) Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat Genet 28:393–396
Chessler SD, Byers PH (1993) BiP binds type I procollagen proα chains with mutations in the carboxyl-terminal propeptide synthesized by cells from patients with osteogenesis imperfecta. J Biol Chem 268:18226–18233
Cotterill SL, Jackson GC, Leighton MP, Wagener R, Makitie O, Cole WG, Briggs MD (2005) Multiple epiphyseal dysplasia mutations in MATN3 cause misfolding of the A-domain and prevent secretion of mutant matrilin-3. Hum Mutat 26:557–565
Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P (2005) On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci USA 102:18773–18784
Delot E, Brodie SG, King LM, Wilcox WR, Cohn DH (1998) Physiological and pathological secretion of cartilage oligomeric matrix protein by cells in culture. J Biol Chem 273:26692–26697
Delot E, King LM, Briggs MD, Wilcox WR, Cohn DH (1999) Trinucleotide expansion mutations in the cartilage oligomeric matrix protein (COMP) gene. Hum Mol Genet 8:123–128
Duke J, Montufar-Solis D, Underwood S, Lalani Z, Hecht JT (2003) Apoptosis staining in cultured pseudoachondroplasia chondrocytes. Apoptosis 8:191–197
Eizirik DL, Cardozo AK, Cnop M (2008) The role of endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Finkelstein J, Doege EK, Yamada Y, Pyeritz RE, Graham JM, Moeschler JR Jr, Pauli RM, Hecht JT, Francomano CA (1991) Analysis of the chondroitin sulfate proteoglycan core protein (CSPGCP) gene in achondroplasia and pseudoachondroplasia. Am J Hum Genet 48:97–102
Fitzgerald J, Lamandé SR, Bateman JF (1999) Proteasomal degradation of unassembled mutant type I collagen pro-alpha1(I) chains. J Biol Chem 274:27392–27398
Forlino A, Tani C, Rossi A, Lupi A, Campari E, Gualeni B, Bianchi L, Armini A, Cetta G, Bini L, Marini JC (2007) Differential expression of both extracellular and intracellular proteins is involved in the lethal or nonlethal phenotypic variation of BrtllV, a murine model for osteogenesis imperfecta. Proteomics 7:1877–1891
Fresquet MG, Jackson C, Loughlin J, Briggs MD (2008) Novel mutations in exon 2 of MATN3 affect residues within the alpha-helices of the A-domain and can result in the intracellular retention of mutant matrilin-3. Hum Mutat 29:330
Gould DB, Phalan FC, Breedveld GJ, Mil SE van, Smith RS, Schimenti JC, Aguglia U, Knaap MS van der, Heutink P, John SWM (2005) Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 308:1167–1171
Gould DB, Marchant JK, Savinova OV, Smith RS, John SWM (2007) Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet 16:797–807
Hamamura K, Liu Y, Yokota H (2008) Microarray analysis of thapsigargin—induced stress to the endoplasmic reticulum of mouse osteoblasts. J Bone Miner Metab 26:231–240
Hamamura K, Goldring MB, Yokota H (2009) Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes in response to surviving stress to endoplasmic reticulum. Arch Oral Biol 54:279–286
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633
Hashimoto Y, Tomiyama T, Yamano Y, Mori H (2003) Mutation (D472Y) in the type 3 repeat domain of cartilage oligomeric matrix protein affects its early vesicle trafficking in endoplasmic reticulum and induces apoptosis. Am J Pathol 163:101–110
Hecht JT, Blanton SH, Wang Y, Daiger SP, Horton WA, Rhodes C, Yamada Y, Francomano CA (1992) Exclusion of human proteoglycan link protein (CRTL1) and type II collagen (COL2A1) genes in pseudoachondroplasia. Am J Med Genet 44:420–424
Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FF, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J (1995) Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet 10:325–329
Hecht JT, Montufar-Solis D, Decker G, Lawler J, Daniels K, Duke PJ (1998a) Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol 17:625–633
Hecht JT, Deere M, Putnam E, Cole W, Vertel B, Chen H, Lawler J (1998b) Characterization of cartilage oligomeric matrix protein (COMP) in human normal and pseudoachondroplasia musculoskeletal tissues. Matrix Biol 17:269–278
Hecht JT, Hayes E, Snuggs M, Decker G, Montufar-Solis D, Doege K, Mwalle F, Poole R, Stevens J, Duke PJ (2001) Calreticulin, PDI, Grp94 and BiP chaperone proteins are associated with retained COMP in pseudoachondroplasia chondrocytes. Matrix Biol 20:251–262
Hetz C, Glimcher L (2008) The daily job of night killers: althernative roles of the BCL-2 family in organelle physiology. Trends Cell Biol 18:38–44
Ho MS, Tsang KY, Lo RL, Susic M, Mäkitie O, Chan TW, Ng VC, Sillence DO, Boot-Handford RP, Gibson G, Cheung KM, Cole WG, Cheah KS, Chan D (2007) COL10A1 nonsense and frame-shift mutations have a gain-of-function effect on the growth plate in human and mouse metaphyseal chondrodysplasia type Schmid. Hum Mol Genet 16:1201–1215
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum localized mRNAs during the unfolded protein response. Science 313:104–107
Iqbal J, Dai K, Seimon T, Jungreis R, Oyadomari M, Kuriakose G, Ron D, Tabas I, Hussain MM (2008) IRE1beta inhibits chylomicron production by selectively degrading MTP mRNA. Cell Metab 7:445–455
Ishida Y, Yamamoto A, Kitamura A, Lamandé SR, Yoshimori Y, Bateman JF, Kubota H, Nagata K (2009) Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Cell Biol 20:2744–2754
Jackson GC, Barker FS, Jakkula E, Czarny-Ratajczak M, Makitie O, Cole WG, Wright MJ, Smithson SF, Suri M, Rogala P, Mortier GR, Baldock C, Wallace A, Elles R, Ala-Kokko L, Briggs MD (2004) Missense mutations in the beta strands of the single A-domain of matrilin-3 result in multiple epiphyseal dysplasia. J Med Genet 41:52–59
Kawaguchi H (2008) Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol Cells 25:1–6
Kawai T, Fan J, Mazan-Mamczarz K, Gorospe M (2004) Global mRNA stabilization preferentially linked to translational repression during the endoplasmic reticulum stress response. Mol Cell Biol 24:6773–6787
Kim PS, Kwon O-Y, Arvan P (1996) An endoplasmic reticulum storage disease causing congenital goiter with hypothyroidism. J Cell Biol 133:517–527
Kudo T, Kanemoto S, Hara H, Morimoto N, Morihara T, Kimura R, Tabira T, Imaizumi K, Takeda M (2008) A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ 15:364–375
Lamandé SR, Chessler SD, Golub SB, Byers PH, Chan D, Cole WG, Sillence DO, Bateman JF (1995) Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro alpha 1 (I) chain carboxyl-terminal propeptide which impair subunit assembly. J Biol Chem 270:8642–8649
Leighton MP, Nundlall S, Starborg T, Meadows RS, Suleman F, Knowles L, Wagener R, Thornton DJ, Kadler KE, Boot-Handford RP, Briggs MD (2007) Decreased chondrocyte proliferation and dysregulated apoptosis in the cartilage growth plate are key features of a murine model of epiphyseal dysplasia caused by a matn3 mutation. Hum Mol Genet 16:1728–1741
Lin JH, Walter P, Yen TS (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 3:399–425
Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, Reed JC, Glimcher LH, Hetz C (2009) BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell 33:679–691
Lisse TS, Thiele F, Fuchs H, Hans W, Przemeck GKH, Abe K, Rathkolb B, Quintanilla-Martinez L, Hoelzlwimmer G, Helfrich M, Wolf E, Ralston SH, Angelis MH de (2008) ERstress-mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet 4:e7
Lu PD, Harding HP, Ron D (2004) Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167:27–33
Maddox BK, Keene DR, Sakai LY, Charbonneau NL, Morris NP, Ridgway CC, Boswell BA, Sussman MD, Horton WA, Bachinger HP, Hecht JT (1997) The fate of cartilage oligomeric matrix protein is determined by the cell type in the case of a novel mutation in pseudoachondroplasia. J Biol Chem 272:30993–30997
Malhotra JD, Kaufman RJ (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18:716–731
Marks DS, Gregory CA, Wallis GA, Brass A, Kadler KE, Boot-Handford RP (1999) Metaphyseal chondrodysplasia type Schmid mutations are predicted to occur in two distinct three-dimensional clusters within type X collagen NC1 domains that retain the ability to trimerize. J Biol Chem 274:3632–3641
Maynard JA, Cooper RR, Ponseti IV (1972) A unique rough surfaced endoplasmic reticulum inclusion in pseudoachondroplasia. Lab Invest 26:40–44
Molinari M (2007) N-glycan structure dictates extension of protein folding or onset of disposal. Nat Chem Biol 3:313–320
Mostert AK, Dijkstra PF, Jansen BR, Van Horn JR, De Graaf B, Heutink P, Lindhout D (2003) Familial multiple epiphyseal dysplasia due to a matrilin-3 mutation: further delineation of the phenotype including 40 years follow-up. Am J Med Genet 120A:490–497
Nielsen VH, Bendixen C, Arnbjerg J, Sorensen CM, Jensen HE, Shukri NM, Thomsen B (2000) Abnormal growth plate function in pigs carrying a dominant mutation in type X collagen. Mamm Genome 11:1087–1092
Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26:9220–9231
Oh SH, Lim SC (2009) Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by the extent of c-Jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung epithelial fibroblast cells. Pharmacol Exp Ther 329:112–122
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140
Papp E, Csermely P (2006) Chemical chaperones: mechanisms of action and potential use. Handb Exp Pharmacol 172:405–416
Park Y-N, Arvan P (2004) The acetylcholinesterase homology region is essential for normal conformational maturation and secretion of thyroglobulin. J Biol Chem 279:17085–17089
Patra D, Xing X, Davies S, Byran J, Franz C, Hunziker EB, Sandell LJ (2007) Site-1 protease is essential for endochondral bone formation in mice. J Cell Biol 179:687–700
Pearson GD, Devereux R, Loeys B, Maslen C, Milewicz D, Pyeritz R, Ramirez F, Rifkin D, Sakai L, Svensson L, Wessels A, Van Eyk J, Dietz HC, National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group (2008) Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on research in Marfan syndrome and related disorders. Circulation 118:785–791
Perlmutter DL (2002) Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatric Res 52:832–836
Pirog-Garcia KA, Meadows RS, Knowles L, Heinegard D, Thornton DJ, Kadler KE, Boot-Handford RP, Briggs MD (2007) Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum Mol Genet 16:2072–2088
Prockop DJ (1984) Osteogenesis imperfecta: phenotypic heterogeneity, protein suicide, short and long collagen. Am J Hum Genet 36:449–505
Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y (2004) Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol 66:899–908
Rajpar HM, McDermott B, Kung L, Eardley R, Knowles L, Heeran M, Thornton DJ, Wilson R, Bateman JF, Poulom R, Arvan P, Kadler KE, Briggs MD, Boot-Handford RP (2009) Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PloS Genet (in press)
Rimoin DL, Rasmussen IM, Briggs MD, Roughley PJ, Gruber HE, Warman ML, Olsen BR, Hsia YE, Yuen J, Reinker K, et al (1994) A large family with features of pseudoachondroplasia and multiple epiphyseal dysplasia: exclusion of seven candidate gene loci that encode proteins of the cartilage extracellular matrix. Hum Genet 93:236–242
Rochet JC (2007) Novel therapeutic strategies for the treatment of protein-misfolding diseases. Expert Rev Mol Med 9:1–34
Ron D, Walter P (2007) Signal transduction in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Rubinsztein DC, Gestwickl JE, Murphy LO, Klionsky D (2007) Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6:304–312
Ruiz-Romero C, Carreira V, Rego I, Remeseiro S, Lόpez-Armada MJ, Blanco FJ (2008) Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics 8:495–507
Rutkowski DT, Kaufman RJ (2004) A trip to the ER: coping with stress. Trends Cell Biol 14:20–28
Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, Katze MG, Hussain MM, Song B, Swathirajan J, Wang J, Yau GD, Kaufman RJ (2008) UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell 15:829–840
Schmitz M, Niehoff A, Miosge N, Smyth N, Paulsson M, Zaucke F (2008) Transgenic mice expressing D469Delta mutated cartilage oligomeric matrix protein (COMP) show growth plate abnormalities and sternal malformations. Matrix Biol 27:67–85
Schröder M (2008) Endoplasmic reticulum stress responses. Cell Mol Life Sci 65:862–894
Shen J, Chen X, Hendershot L, Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3:99–111
Shen X, Ellis RE, Sakaki K, Kaufman RJ (2005) Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet 1:e37
Srinivas V, Bohensky J, Zahm AM, Shapiro IM (2009) Autophagy in mineralizing tissues. Microenvironmental perspectives. Cell Cycle 8:391–393
Stanescu R, Stanescu V, Muriel MP, Maroteaux P (1993) Multiple epiphyseal dysplasia, Fairbank type: morphologic and biochemical study of cartilage. Am J Med Genet 45:501–507
Stanescu V, Maroteaux P, Stanescu R (1982) The biochemical defect of pseudoachondroplasia. Eur J Pediatr 138:221–225
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic stress-induced apoptosis. EMBO Rep 7:880–885
Tan JT, Kremer F, Freddi S, Bell KM, Baker NL, Lamandé SR, Bateman JF (2008) Competency for nonsense-mediated reduction in collagen X mRNA is specified by the 3′ UTR and corresponds to the position of mutations in Schmid metaphyseal chondrodysplasia. Am J Hum Genet 82:786–793
Todd DJ, Lee A-H, Glimcher LH (2008) The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol 8:663–674
Tsang KY, Chan D, Cheslett D, Chan WCW, So CL, Melhado IG, Chan TWY, Kwan KM, Hunziker EB, Yamada Y, Bateman JF, Cheung KMC, Cheah KSE (2007) Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biology 5:e44
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666
Vranka J, Mokashi A, Keene DR, Tufa S, Corson G, Sussman M, Horton WA, Maddox K, Sakai L, Bachinger HP (2001) Selective intracellular retention of extracellular matrix proteins and chaperones associated with pseudoachondroplasia. Matrix Biol 20:439–450
Wallis GA, Rash B, Sweetman WA, Thomas JT, Super M, Evans G, Grant ME, Boot-Handford RP (1994) Amino acid substitutions of conserved residues in the carboxyl-terminal domain of the α1 chain of type X collagen occur in two unrelated families with metaphyseal chondrodysplasia type Schmid. Am J Hum Genet 54:169–178
Warman ML, Abbott M, Apte SS, Hefferon T, McIntosh I, Cohn DH, Hecht JT, Olsen BR, Francomano CA (1993) A type X collagen mutation causes Schmid metaphyseal chondrodysplasia. Nat Genet 5:79–82
Weaver EJ, Summerville GP, Yeh G, Hervada-Page M, Oehlmann R, Rothman R, Jimenez SA, Knowlton RG (1993) Exclusion of type II and type VI procollagen gene mutations in a five-generation family with multiple epiphyseal dysplasia. Am J Med Genet 45:345–352
Wei J, Sheng X, Feng D, McGrath B, Cavener DR (2008) PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J Cell Physiol 217:693–707
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O’Kane CJ, Floto RA, Rubinsztein DC (2008) Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4:295–305
Wilson R, Freddi S, Bateman JF (2002) Collagen X chains harboring Schmid metaphyseal chondrodysplasia NC1 domain mutations are selectively retained and degraded in stably transfected cells. J Biol Chem 277:12516–12524
Wilson R, Freddi S, Chan D, Cheah KSE, Bateman JF (2005) Misfolding of collagen X chains harboring Schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation intracellular retention and activation of the unfolded protein response. J Biol Chem 280:15544–15552
Wordsworth P, Ogilvie D, Priestley L, Smith R, Wynne-Davies R, Sykes B (1988) Structural and segregation analysis of the type II collagen gene (COL2A1) in some heritable chondrodysplasias. J Med Genet 25:521–527
Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, Hanauer A, Karsenty G (2004) ATF4 is a substrate of RSK2 and essential regulator of osteoblast biology: implication for Coffin–Lowry syndrome. Cell 117:387–398
Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, Brown MS, Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6:1355–1364
Zelzer E, Olsen BR (2005) Multiple roles of vascular endothelial growth factor (VEGF) in skeletal development, growth, and repair. Curr Top Dev Biol 65:169–187
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462
Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR (2002) The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22:3864–3874
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12:982–995
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Additional information
The work included from the authors laboratories was funded by the Wellcome Trust (M.D.B. is a Wellcome Trust Senior Research Fellow: grant 084353/Z/07/Z), the EU (LSHM-CT-2007-037471), NIH (RO1 AR49547-01) and the ARC (18253).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Boot-Handford, R.P., Briggs, M.D. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res 339, 197–211 (2010). https://doi.org/10.1007/s00441-009-0877-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-009-0877-8