
Abstract
Recent evidence has demonstrated that endothelial cells can have a remarkable plasticity. By a process called Endothelial-to-Mesenchymal Transition (EndMT) endothelial cells convert to a more mesenchymal cell type that can give rise to cells such as fibroblasts, but also bone cells. EndMT is essential during embryonic development and tissue regeneration. Interestingly, it also plays a role in pathological conditions like fibrosis of organs such as the heart and kidney. In addition, EndMT contributes to the generation of cancer associated fibroblasts that are known to influence the tumor-microenvironment favorable for the tumor cells. EndMT is a form of the more widely known and studied Epithelial-to-Mesenchymal Transition (EMT). Like EMT, EndMT can be induced by transforming growth factor (TGF)-β. Indeed many studies have pointed to the important role of TGF-β receptor/Smad signaling and downstream targets, such as Snail transcriptional repressor in EndMT. By selective targeting of TGF-β receptor signaling pathological EndMT may be inhibited for the therapeutic benefit of patients with cancer and fibrosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Complex body architecture is reliant on efficient supply of oxygen and nutrients to all tissues. The vascular system achieves this task efficiently via a highly branched network of blood vessels. The inner layer of blood vessels is lined with a thin layer of endothelial cells, stabilized by a basal lamina around this endothelium (Adams and Alitalo 2007). Endothelial cells however do perform additional functions besides lining the vessel wall. One very interesting example that is emerging from recent studies is the transition of endothelial cells into mesenchymal cells (Goumans et al. 2008). This form of endothelial cells plasticity is called endothelial-to-mesenchymal transition (EndMT) and is the subject of this review.
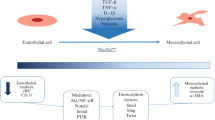
During EndMT, endothelial cells delaminate from the organized layer of cells in the vessel-lining and migrate away possibly invading the underlying tissue. The mesenchymal phenotype is characterized by the acquisition of mesenchymal markers such as smooth muscle cell actin (SMA) and Neural (N)-Cadherin and the complementary loss of endothelial marker such as CD31/Pecam-1 and Vascular-Endothelial (VE)-Cadherin. In addition, the endothelial cells lose their cell-cell junctions and gain migratory and invasive capacity. Although discovered in the process of heart-development (Markwald et al. 1975), the mechanism of EndMT has now been implicated in a wide variety of pathological conditions like fibrosis of several organs as well as in cancer (Potenta et al. 2008).
EndMT is related to the more widely known mechanism of epithelial-to-mesenchymal transition (EMT). EMT allows a polarized epithelial cell, which normally interacts with basement membrane via its basal surface, to undergo multiple changes that enable it to assume a mesenchymal cell phenotype, which includes enhanced migratory capacity, invasiveness and elevated resistance to apoptosis. One of the molecular hallmarks of EMT is the loss of Epithelial (E)-cadherin expression that is frequently mediated by the upregulation of Snail transcription factor family members. EMT can occur in many epithelial cells and is, as EndMT, a critical process in embryonic development (Thiery and Sleeman 2006). In addition EMT is thought to be very important in cancer development since an epithelial cell needs to lose cell-cell junctions before it would be able to invade the surrounding tissue and migrate away ultimately leading to metastasis.
As shown for epithelial plasticity (Xu et al. 2009; Miyazono 2009) TGF-β signaling also plays an important role in regulating endothelial plasticity. Therefore we shall first discuss TGF-β receptor signaling in controlling endothelial cell function. Subsequently, we will focus on the role of endothelial cell plasticity in health and disease.
TGF-β signaling
TGF-β is one of the best known members of a large family of secreted pleiotrophic growth factors. Other members of the family include the bone morphogenetic proteins (BMPs) and activins. Many of these cytokines have vital roles in numerous processes during development, but also in maintenance of tissue homeostasis and tissue repair in the adult (Massague 1998). Not unexpectedly, they also have roles in pathological conditions like cancer, vascular diseases and fibrosis (ten Dijke and Arthur 2007; Blobe et al. 2000).
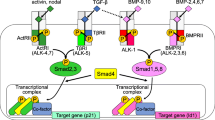
Members of the TGF-β family mediate their effects by binding specific transmembrane receptors at the cell-membrane (Feng and Derynck 2005). They bind a complex of two type I and two type II serine/threonine kinase receptors. Upon ligand induced heteromeric complex formation, the type I receptor is phosphorylated by the type II receptor. There are seven different type I receptors (ALK1 to ALK7) (also known as activin receptor-like kinases (ALK)) and five type II receptors (activin receptor type IIA (ActRIIA), activin receptor type IIB (ActRIIB), BMP type II receptor (BMPRII), TGF-β type II receptor (TGFβRII) and AMH type II receptor (AMHRII)). Different ligands can bind different combinations of type I and type II receptor thereby creating specificity of signaling. TGF-β signals mostly via ALK5 and TGFβRII, activins via ALK4 with ActRIIA and ActRIIB, and BMPs signal via ALK1, 2, 3 and 6 together with BMPRII, ActRIIA and ActRIIB. For regulation of endothelial function by TGF-β, ALK1 and ALK5 signaling are most important (van Meeteren et al. 2011).
After binding of the ligand the type I receptor phosphorylates specific transcription factors called receptor regulated (R)-Smads. Upon activation by type I receptors, R-Smads form heteromeric complexes with the common mediator (Co)-Smad (Smad4) and these heteromeric complexes accumulate into the nucleus, where they regulate the transcription of specific target genes (Moustakas and Heldin 2009).
Inhibitory Smads (I-Smads) inhibit the activation of R-Smads by competing for type I receptor interaction and by recruiting phosphatases and ubiquitin ligases to the activated receptor complex leading to dephosphorylation or proteosomal degradation of the receptor complex (Itoh and ten Dijke 2007). R-Smads can be divided in 2 groups based on the type I receptor that is activating them. The first group consists of Smad1, 5 and 8 and these are activated by ALK1, 2, 3 and 6. The second group contains Smad2 and 3 and is activated by ALK4, 5 and 7. In addition to Smad signaling TGF-β and BMP signaling can result in activation of pathways where Smads are not involved. Non-Smad pathways include various branches of MAP kinase pathways, Rho-like GTPase signaling pathways, and PI3K/AKT pathways (Zhang 2009; Moustakas and Heldin 2005).
Co-receptors are receptors that do not signal by themselves since they lack intracellular enzymatic domains such as kinase-domains. For TGF-β signaling co-receptors endoglin and betaglycan (also called TGF-β receptor III) have been identified. Both receptors are structurally related and have a small intracellular tail and a large extracellular domain. Endoglin is highly expressed in proliferating endothelial cells, hence its name endoglin (ten Dijke et al. 2008).
To regulate the activation state of endothelial cells TGF-β can differentially activate two type I receptors, ALK1 and ALK5 (Fig. 1) (Oh et al. 2000; Goumans et al. 2002). ALK5 is ubiquitously expressed in most tissues, but ALK1 expression is typically restricted to endothelial cells. TGF-β induced ALK5 signaling leads to Smad2 and Smad3 phosphorylation resulting in inhibition of angiogenesis by inhibiting endothelial cell proliferation, migration and organization (Goumans et al. 2002, 2003). The ALK5 kinase inhibitor SB-431542 facilitated proliferation and sheet formation of embryonic stem cell derived endothelial cells and in fetal mouse metatarsal assays (Watabe et al. 2003; Liu et al. 2009). In addition, SB-431542 upregulated the expression of claudin-5, an endothelial specific component of tight junctions, suggesting a role of ALK5 signaling in regulating vascular permeability (Watabe et al. 2003). Indeed, ALK5 has been reported before to be important for TGF-β induced permeability and cytoskeletal remodeling of endothelial cells (Birukova et al. 2005). In summary, ALK5 signaling results in keeping endothelial cells in a quiescent state.

TGF-β signaling in endothelial cells. TGF-β can bind to two distinct TGF-β type II/type I receptor complexes in which TGF-β type II receptor (TβRII) is a common component and two different type I receptors, i.e. activin receptor-like (ALK)5 and ALK1 determine signaling specificity. Whereas activation of ALK5 will inhibit endothelial cell proliferation, ALK1 will elicit opposite responses. Endoglin is an auxiliary receptor that modulates TGF-β signaling responses, i.e. it stimulates TGF-β/ALK1 but inhibits TGF-β /ALK5 signaling
In contrast, TGF-β induced ALK1 signaling activates Smad1 and Smad5 leading to endothelial cell proliferation, migration and organization (Goumans et al. 2003). An important intracellular effector of ALK1 is Inhibitor of Differentiation/DNA binding (Id)1; its upregulation was shown to be required for TGF-β/ALK1-induced endothelial cell migration and tube formation (Goumans et al. 2002). However, inhibitory effects of ALK1 signaling on endothelial cells have also been reported (David et al. 2007; Lamouille et al. 2002; Mallet et al. 2006). The effect of ALK1 is likely dependent on cellular context.
Although ALK1 and ALK5 have divergent effects on endothelial cells they do interact with each other physically. ALK5-deficient endothelial cells are not only defective in ALK5 signaling but also show impaired ALK1 responses; ALK5 was found to be essential for recruitment of ALK1 into a TGF-β receptor complex, and the kinase activity of ALK5 is essential for full ALK1 activation (Goumans et al. 2003). On the other hand, ALK1 can directly antagonize ALK5 signaling at the level of Smads (Oh et al. 2000; Goumans et al. 2002). In conclusion, the cross-talk between ALK1 and ALK5 signaling provides endothelial cells with a sophisticated mechanism to fine tune endothelial function (Fig. 1).
The TGF-β co-receptors endoglin and betaglycan can both be expressed by endothelial cells. Endoglin positive but betaglycan negative endothelial cells are only responsive to TGF-β isoforms 1 and 3 but not to isoform 2 (Cheifetz et al. 1990). In endothelial cells that express both co-receptors it was shown that endoglin can form a complex with betaglycan and the TGF-β receptor complex simultaneously (Wong et al. 2000).
Signals controlling EndMT
Many endothelial cells can be induced to undergo EndMT in cell culture experiments. This has opened the opportunity to study the signaling mechanism in EndMT. To induce EndMT of endothelial cells, they are often stimulated with TGF-β or Notch ligands (Frid et al. 2002; Ishisaki et al. 2003; Timmerman et al. 2004; Noseda et al. 2004; Zeisberg et al. 2007b; Zeisberg et al. 2007a).
The molecular mechanism behind TGF-β induced EndMT has been found to involve the Snail family of transcription repressors. In mouse embryonic stem cell derived endothelial cells TGF-β induced EndMT and expression of Snail. This upregulation of Snail by TGF-β was shown to be dependent on the activation of Smad, MEK, PI3K and p38 MAPK by TGF-β (Medici et al. 2011). Subsequent knockdown of Snail blocked the TGF-β induced EndMT (Kokudo et al. 2008). Although overexpression of Snail was sufficient to induce EMT (Cano et al. 2000) for EndMT Snail expression alone is insufficient. The inhibitor of Snail, GSK-3β, needs to be inhibited by phosphorylation by kinases such as AKT to induce EndMT (Medici et al. 2011).
As mentioned above Notch can, as TGF-β, used in vitro to induce EndMT in endothelial cells in vitro. In this Notch induced EndMT the Snail family member Slug has been shown to be important (Leong et al. 2007). Snail and Slug are known to repress the expression of VE-cadherin (Lopez et al. 2009). Since VE-cadherin is essential for endothelial cell-cell junctions this obviously could provide a link to a mechanism by which EndMT occurs. A different factor involved in TGF-β induced EndMT was shown to be plasminogen activator inhibitor-1 (PAI-1). Although elevated levels of PAI-1 are implicated in tissue fibrosis (Ghosh and Vaughan 2011), lack of PAI-1 in the heart is associated with the development of cardiac fibrosis in aged mice (Ghosh et al. 2010). It was shown that in the PAI-deficient endothelial cells of these mice both Smad and non-Smad TGF-β signaling is spontaneously activated. This spontaneous activation leads to EndMT and subsequently the fibrosis observed in these animals (Ghosh et al. 2010). In addition, it was recently shown that c-Abelson tyrosine kinase (c-Abl) and Protein Kinase C (PKC)-δ are crucial for TGF-β-induced EndMT and therefore that imatinib mesylate and rottlerin (inhibitors of c-Abl and PKC-δ, respectively) might be effective therapeutic agents for fibroproliferative pathologies in which EndMT plays a role (Li and Jimenez 2011).
It has also been reported that pathways other than TGF-β can lead to EndMT. In myocardial infarction (MI) for example, canonical (β-catenin-dependent) Wnt signaling is induced 4 days after experimental MI in the subepicardial endothelial cells and perivascular cells. Coincidently with canonical Wnt activation EndMT was also triggered after the infarction. In addition, canonical Wnt signaling induced mesenchymal characteristics in cultured endothelial cells, suggesting a direct role of canonical Wnt signaling in EndMT (Aisagbonhi et al. 2011).
EndMT in development
EndMT was originally discovered in the embryonic heart during studies on heart development (Markwald et al. 1975). In heart development, endothelial cells lining the endocardial cushions undergo EndMT (Fig. 2). These cells subsequently invade the underlying tissue and participate in the formation of the septa and valves of the heart (Eisenberg and Markwald 1995). Studies from knockout mice have shown subsequently that signals directing EndMT in heart development include multiple TGF-β isoforms and receptors (Goumans and Mummery 2000). For example it was shown that endoglin, a TGF-β co-receptor, is required cell autonomously for EndMT during formation of the endocardial cushions (Nomura-Kitabayashi et al. 2009). In the embryonic heart the endothelial cells that undergo TGF-β-induced EndMT are exposed to high blood flow and are devoid of primary cilia (Van der Heiden et al. 2008). Egorova et al recently showed that TGF-β/ALK5 signaling is activated by blood flow in these endothelial cells (Egorova et al. 2011b), and lack of primary cilia primes endothelial cells for EndMT, thereby linking primary cilia with flow-related endothelial differentiation (Egorova et al. 2011a).

TGF-β in endothelial cell plasticity. TGF-β can stimulate the transition of endothelial into mesenchymal cells. EndMT is a complex process that involves disruption of polarized endothelial morphology into cells with spindle-shaped morphology with formation of actin stress fibers. Furthermore EndMT results in reduced cell–cell junctions through delocalization and downregulation of VE-cadherin and PECAM, increased cellular motility and expression of mesenchymal makers, such a-SMC actin, N-Cadherin and FSP-1. EndMT plays an important role in development, regeneration and pathological processes, such as fibrosis and tumor stroma formation
Role of EndMT in cancer
Fibroblasts are one the most abundant cell type in the microenvironment of tumors, being particularly prominent in carcinomas of colon, breast, pancreas, and prostate. There is substantial evidence that cancer-associated fibroblasts (CAFs) contribute to tumor growth and metastasis. This is mediated by the release of classical growth factors such as TGF-β, epidermal growth factor, hepatocyte growth factor, as well as a range of chemokines that influence diverse aspects of tumor cell behavior (Allen and Louise Jones 2011).
CAFs form a heterogeneous population, most likely related to their diverse origin. Whereas activation of local stromal fibroblasts has traditionally been considered the major source of CAFs (Kalluri and Zeisberg 2006), recently it was shown that EndMT is another unique source of CAFs (Fig. 2) (Zeisberg et al. 2007a). In this study EndMT in tumors was reported in two different mouse models of cancer and demonstrated that a substantial proportion of CAFs in these models arise through EndMT. The CAFs were shown to co-express the endothelial marker CD31 along with one of the mesenchymal markers, fibroblast specific protein (FSP)1, or α-smooth muscle actin (αSMA). Approximately, 40% of FSP1-positive CAFs were also found to be CD31 positive. To study the origin of the CAFs in more detail, tumors were grown in Tie2-Cre;R26R-lox-STOP-lox-lacZ transgenic mice, a reporter strain in which all cells of endothelial origin can be irreversibly labeled with lacZ. In 30% of the CAFs in tumors of these transgenic mice LacZ was detected making the authors conclude that the CAFs originated from endothelial cells (Zeisberg et al. 2007a). Since Tie2 is however also expressed in the hematopoietic lineage it is technically also possible that the lacZ positive CAFs originate from these cells (Tang et al. 2010; Gitler et al. 2004).
These studies suggest that EndMT is an important mechanism for CAF recruitment to the tumour stroma. Since TGF-β signaling is a known mediator of EndMT and TGF-β is abundantly expressed in many different tumors (Zeisberg et al. 2007a), EndMT may be mediated by TGF-β produced in the tumor. Yet, the molecular mechanism of EndMT in tumors has not yet been specifically studied, but is to be expected to involve similar pathways as in fibrosis.
EndMT in tissue fibrosis
Fibrosis is an essential process of proper wound healing. In many pathological conditions however deregulated or excessive fibrosis occurs. TGF-β signaling is involved in physiological fibrosis in wound healing. Therefore it is not surprising that TGF-β signaling also plays an important role in pathological fibrosis.
The predominant cellular mediators of fibrosis are assumed to be (myo)fibroblasts, not only in heart fibrosis but also in fibrosis of organs such as lung, kidney, and the liver. Fibrosis of all these organs share similar pathways. The origin of these (myo)fibroblasts may, besides resident interstitial fibroblast, be cells derived from the bone marrow as well as fibroblastic cells that have transdifferentiated from cells of epithelial origin. More interestingly, these cells can also be derived from endothelial cells that have undergone EndMT (Fig. 2).
In the kidney for example it was shown that EndMT can generate myofibroblasts in early diabetic renal fibrosis. Using endothelial-lineage tracing with Tie2-cre; LoxP–eGFP transgenic mice a significant number of interstitial α-smooth muscle actin-positive cells (myofibroblasts) were shown to be of endothelial origin in fibrotic kidneys from mice with streptozotocin-induced diabetic nephropathy. This indicated that EndMT can contribute to the early progression of diabetic nephropathy (Li et al. 2009; Kizu et al. 2009). Earlier it was already shown that fibroblasts expressed the endothelial marker CD31 in three different mouse models of renal disease: streptozotocin-induced diabetic nephropathy, unilateral ureteral obstructive nephropathy, and a mouse model of Alport syndrome (Zeisberg et al. 2008). Approximately 30% to 50% of fibroblasts formed in the kidneys of these models co-expressed the endothelial marker CD31 and the fibroblast/myofibroblast markers FSP1 and/or αSMA (Zeisberg et al. 2008). Since TGF-β is stimulating EndMT it is not surprising that interference with TGF-β signaling has been reported to inhibit EndMT and the subsequent kidney fibrosis. First it was reported that Smad3 conditional knockout mice are resistant to streptozotocin-induced renal fibrosis and tubulointerstitial fibrosis in unilateral urethral obstruction models (Fujimoto et al. 2003; Wang et al. 2007; Sato et al. 2003). Subsequently it was shown that a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy due to a blockade of EndMT (Li et al. 2010).
Likewise in the lung, fibrosis can cause serious pathological conditions such as idiopathic pulmonary fibrosis (IPF). IPF is characterized by progressive obliteration of normal alveolar lung architecture and replacement by fibrotic tissue. The result is declining lung function, progressive dyspnea, and ultimately death within 3 to 5 years of diagnosis (Nataraj et al. 2010). Hashimoto et al have demonstrated that pulmonary capillary endothelial cells, through EndMT, can serve as a source of fibroblasts in pulmonary fibrosis (Hashimoto et al. 2010). They showed this by bleomycin-induced lung injury in mice where they found, using lineage tracing methods that the fibroblasts originated from the endothelial cells. Interestingly they also found a dependence on Ras for completion of EndMT. Only treatment with TGF-β in combination with activated Ras induced a persistent morphological change and suppression of endothelial markers consistent with EndMT (Hashimoto et al. 2010).
Endothelial-to-mesenchymal transition in cardiac fibrosis
In cardiac fibrosis the heart valves abnormally thicken due to inappropriate proliferation of cardiac fibroblasts and of disruption of normal myocardial structure through excessive deposition of extracellular matrix (Krenning et al. 2010). Several studies have given evidence for the role of EndMT in cardiac fibrosis. For example Zeisberg et al. performed lineage analysis to trace the origin of the fibroblasts in cardiac fibrosis (Zeisberg et al. 2007b). Cardiac fibrosis was induced by exposing the heart to pressure overload for 5 days via aortic banding. Analysis of the fibrotic lesions revealed the presence of fibroblasts that originated from endothelial cells. This study elegantly shows that endothelial cells can undergo EndMT and contribute to the total pool of cardiac fibroblasts, similar as in formation of the aterioventricular cushion in embryonic development.
TGF-β signaling stimulates the collagen-producing cardiac fibroblast and has therefore been implicated in the pathogenesis of cardiac fibrosis (Khan and Sheppard 2006). Progression of cardiac fibrosis is furthermore stimulated by a Smad3-dependent TGF-β signaling pathway inducing EndMT. In Smad3 heterozygous mice cardiac fibrosis was significantly reduced and associated with a decrease in the number of endothelial-derived fibroblasts (Dobaczewski et al. 2010; Bujak et al. 2007). In a different study BMP-7 inhibited EndMT and conserved the endothelial phenotype. The systemic administration of exogenous BMP-7 significantly inhibited EndMT and the progression of cardiac fibrosis in mouse models of chronic allograft rejection and pressure overload (Zeisberg et al. 2007b). Thus, although BMP cooperates with TGF-β in inducing EndMT during cushion formation, it is an antagonist in the TGF-β fibrotic pathway, preserving the endothelial phenotype and preventing fibrosis.
Targeting TGF-β in fibrotic diseases
With EndMT (and EMT) contributing to tissue fibrosis progression and TGF-β as pivotal mediator of this response and also inducer of extracellular matrix deposition, this opens possibilities to intervene with fibrosis by targeting TGF-β. Several clinical trials on fibrosis with anti-TGF-β agents have been done or are currently running. For example in skin fibrosis a clinical trial is testing the effect of the synthetic peptide P144 (ClinicalTrials.gov Identifier: NCT00781053). P144 encodes a part of the ligand (TGF-β) binding domain of betaglycan and can block TGF-β activity. In animal models P144 inhibited carbon tetrachloride induced liver fibrosis and more importantly it inhibited bleomycin induced skin fibrosis (Ezquerro et al. 2003).
CAT-152 (Lerdelimumab) is a fully human TGF-β2 neutralizing antibody with high affinity for TGF-β2 and lower affinity for TGF-β3 (Thompson et al. 1999). In rabbits it was capable of inhibiting scarring after glaucoma surgery (Mead et al. 2003). In the first clinical trials, CAT-152 showed possible effects in reducing scar formation in intractable glaucoma patients that received a trabeculectomy (Siriwardena et al. 2002). Disappointingly in larger phase III clinical trials these results could not be confirmed however (Khaw et al. 2007).
Systemic sclerosis is a disease in where the dermal layer of the skin undergoes fibrosis. In addition systemic sclerosis results in inflammatory responses, vascular changes and loss of function of internal organs due to scarring and extracellular matrix deposition. In a phase I/II trial CAT-192, a TGF-β1 neutralizing antibody did not show effect on early stage diffuse cutaneous systemic sclerosis, together with more adverse events occurred in the CAT-192 treated groups (ClinicalTrials.gov Identifier: NCT00043706) (Denton et al. 2007). Nevertheless this antibody is now being tested in patients with myelofibrosis (ClinicalTrials.gov Identifier: NCT01291784).
GC2008 or Fresolimumab which is the humanized version of the pan-TGF-β neutralizing antibody 1D11 is also tested in clinical trials. These trials also treat patients with diffuse systemic sclerosis (ClinicalTrials.gov Identifier: NCT01284322).
EndMT leading to loss of endothelial function
As described above the result of EndMT in pathological conditions is fibrosis because of the generation of fibroblasts and excessive extracellular matrix. However EndMT also leads to the loss of endothelium. The loss of endothelial function can lead to badly perfused tissue and subsequent tissue damage. For example following traumatic spinal cord injury, significant vascular disruption occurs at the site(s) of injury. This interruption of vascular support is thought to be a key mediator of multiple secondary injury cascades, all of which contribute to loss of functional tissue. It was demonstrated that in response to spinal ischemia/reperfusion injury TGF-β-responsive genes in the endothelial cell compartment were early and robustly transcriptional activated (Benton et al. 2009). Given the known effects of TGF-β on endothelial cells it is attractive to speculate that the loss of endothelial functions, such as increased permeability of the endothelium in spinal cord injury is due to increased TGF-β signaling and conversion of endothelial cells to a more fibroblastic phenotype.
Endothelial transdifferentiation is not limited to fibrosis
Other than differentiation towards myofibroblasts endothelial cells can also differentiate to other cell types. Interestingly TGF-β signaling was shown to play an important role also in these transdifferentiation processes.
Fibrodysplasia ossificans progressiva (FOP) is a severely debilitating disorder caused by an activating mutation in the BMP type I receptor, ALK2 (Shore et al. 2006). In FOP-patients acute inflammation causes heterotopic ossification in soft tissues at nearly any site in the body (Kaplan et al. 2008). The source of the ossifying cells was previously unknown but Medici et al. showed that bone and cartilage cells from lesions of people with FOP and mice with mutant ALK2 expressed the endothelial markers Tie2 and von Willebrand factor. This suggests an endothelial origin of the ectopic mesenchymal cells that form the heterotopic tissues (Horwitz 2010; Medici et al. 2010). Furthermore, human endothelial cells treated with BMP4 or TGF-β2, to activate endogenous ALK2, undergo EndMT resulting in mesenchymal stem cells. These mesenchymal stem cells may be of use in cell therapy to treat people in need of bone and cartilage regeneration (Medici et al. 2010).
Not only endothelial cells lining the vessel wall have been shown to be able to undergo EndMT. Also circulating endothelial progenitor cells have been shown to undergo EndMT. The conversion of these circulating endothelial progenitor cells to smooth muscle-like progeny was also shown to be stimulated by TGF-β (Moonen et al. 2010).
Endothelial plasticity also plays a role in the phenotype of the three main types of endothelial cells; arterial, venous and lymphatic. These 3 different phenotypes can be superimposed, or reverted to, by subtle alterations in the combination or in the expression levels of a few key regulators (e.g. Notch signaling, COUP-TFII and Prox1) (Johnson et al. 2008; You et al. 2005) (reviewed in (Oliver and Srinivasan 2010). Furthermore also during sprouting angiogenesis there are several different types of cells within the growing sprout. Tip cells and stalk cells are fundamentally different although they can revert to one and another quickly (Jakobsson et al. 2010). These changes in endothelial cell fates highlight again the once unappreciated plasticity of endothelial cells.
Concluding remarks
Recent studies have revealed the remarkable plasticity of endothelial cells. While initially met with skepticism (Tarin et al. 2005), EMT (and therefore also EndMT) was gaining acceptance. However, the concept of EMT/EndMT is currently under intense discussion (Quaggin and Kapus 2011; Zeisberg and Duffield 2010), in particular as it relates to kidney and liver fibrosis. Whereas early lineage tracing studies demonstrated EMT/EndMT, new lineage tracing experiments failed to do so (Humphreys et al. 2010). What is clear is that epithelial/endothelial cells can undergo morphological changes, i.e. demonstrate plastic behavior, and that TGF-β drives this process. In pathological situations like cancer and fibrosis, targeting of the TGF-β/Smad signaling pathway may have therapeutic benefit and should be further explored in experimental animal models and clinical trials. Recent advances of (intravital) imaging techniques should be applied in vivo studying diseases with possible links to EMT/EndMT to see whether further evidence of these processes can be obtained (Yoshino et al. 2011). As mentioned above several clinical trials are ongoing to test if anti-TGF-β therapy can inhibit pathological conditions, like fibrosis, in which endothelial (and epithelial) plasticity plays an important role. If successful not only many fibrotic pathological conditions but also the generation of cancer associated fibroblasts might be inhibited. This would lead to a less favorable micro-environment for tumor growth and ultimately would help to fight cancer in the clinic.
Abbreviations
- ActR:
-
Activin receptor
- ALK:
-
Activin receptor-like kinase
- BMP:
-
Bone Morphogenetic Protein
- CAF:
-
Cancer-Associated Fibroblast
- E-Cadherin:
-
Epithelial-cadherin
- EMT:
-
Epithelial-to-Mesenchymal Transition
- EndMT:
-
Endothelial to Mesenchymal Transition
- FOP:
-
Fibrodysplasia Ossificans Progressiva
- Id:
-
Inhibitor of differentiation/DNA binding
- IPF:
-
Idiopathic Pulmonary Fibrosis
- MI:
-
Myocardial infarction
- N-Cadherin:
-
Neural-Cadherin
- PAI-1:
-
Plasminogen Activator Inhibitor-1
- SMA:
-
Smooth Muscle cell Actin
- Smad:
-
Small phenotype and Mothers Against Decapentaplegic related protein
- TGF-β:
-
Transforming Growth Factor-β
- TGFβR:
-
Transforming Growth Factor-β receptor
- VE-Cadherin:
-
Vascular-endothelial cadherin
References
Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8:464–478
Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK (2011) Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech. doi:10.1242/dmm.006510
Allen M, Louise Jones J (2011) Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J Pathol 223:163–177
Benton RL, Maddie MA, Dincman TA, Hagg T, Whittemore SR (2009) Transcriptional activation of endothelial cells by TGFβ coincides with acute microvascular plasticity following focal spinal cord ischaemia/reperfusion injury. ASN Neuro 1(3):181–194
Birukova AA, Adyshev D, Gorshkov B, Birukov KG, Verin AD (2005) ALK5 and Smad4 are involved in TGF-β1-induced pulmonary endothelial permeability. FEBS Lett 579:4031–4037
Blobe GC, Schiemann WP, Lodish HF (2000) Role of transforming growth factor β in human disease. N Engl J Med 342:1350–1358
Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG (2007) Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116:2127–2138
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2:76–83
Cheifetz S, Hernandez H, Laiho M, ten Dijke P, Iwata KK, Massague J (1990) Distinct transforming growth factor-β (TGF-β) receptor subsets as determinants of cellular responsiveness to three TGF-β isoforms. J Biol Chem 265:20533–20538
David L, Mallet C, Vailhe B, Lamouille S, Feige JJ, Bailly S (2007) Activin receptor-like kinase 1 inhibits human microvascular endothelial cell migration: potential roles for JNK and ERK. J Cell Physiol 213:484–489
Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, Silliman N, Streisand J, Powell J, Akesson A, Coppock J, Hoogen F, Herrick A, Mayes MD, Veale D, Haas J, Ledbetter S, Korn JH, Black CM, Seibold JR (2007) Recombinant human anti-transforming growth factor β1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum 56:323–333
Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG (2010) Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res 107:418–428
Egorova AD, Khedoe PP, Goumans MJ, Yoder BK, Nauli SM, ten Dijke P, Poelmann RE, Hierck BP (2011a) Lack of Primary Cilia Primes Shear-Induced Endothelial-to-Mesenchymal Transition. Circ Res 108(9):1093–1101
Egorova AD, Van der Heiden K, Van de Pas S, Vennemann P, Poelma C, DeRuiter MC, Goumans MJ, Gittenberger-de Groot AC, ten Dijke P, Poelmann RE, Hierck BP (2011b) Tgfβ/Alk5 signaling is required for shear stress induced klf2 expression in embryonic endothelial cells. Dev Dyn 240(7):1670–1680
Eisenberg LM, Markwald RR (1995) Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res 77:1–6
Ezquerro IJ, Lasarte JJ, Dotor J, Castilla-Cortazar I, Bustos M, Penuelas I, Blanco G, Rodriguez C, Lechuga MC, Greenwel P, Rojkind M, Prieto J, Borras-Cuesta F (2003) A synthetic peptide from transforming growth factor β type III receptor inhibits liver fibrogenesis in rats with carbon tetrachloride liver injury. Cytokine 22:12–20
Feng XH, Derynck R (2005) Specificity and versatility in tgf-β signaling through Smads. Annu Rev Cell Dev Biol 21:659–693
Frid MG, Kale VA, Stenmark KR (2002) Mature Vascular Endothelium Can Give Rise to Smooth Muscle Cells via Endothelial-Mesenchymal Transdifferentiation: In Vitro Analysis. Circ Res 90:1189–1196
Fujimoto M, Maezawa Y, Yokote K, Joh K, Kobayashi K, Kawamura H, Nishimura M, Roberts AB, Saito Y, Mori S (2003) Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun 305:1002–1007
Ghosh AK, Vaughan DE (2011) PAI-1 in Tissue Fibrosis. J Cell Physiol. doi:10.1002/jcp.22783
Ghosh AK, Bradham WS, Gleaves LA, De TB, Murphy SB, Covington JW, Vaughan DE (2010) Genetic deficiency of plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged mice: involvement of constitutive transforming growth factor-β signaling and endothelial-to-mesenchymal transition. Circulation 122:1200–1209
Gitler AD, Kong Y, Choi JK, Zhu Y, Pear WS, Epstein JA (2004) Tie2-Cre-induced inactivation of a conditional mutant Nf1 allele in mouse results in a myeloproliferative disorder that models juvenile myelomonocytic leukemia. Pediatr Res 55:581–584
Goumans MJ, Mummery C (2000) Functional analysis of the TGFβ receptor/Smad pathway through gene ablation in mice. Int J Dev Biol 44:253–265
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P (2002) Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J 21:1743–1753
Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P (2003) Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol Cell 12:817–828
Goumans MJ, van Zonneveld AJ, ten Dijke P (2008) Transforming Growth Factor [β]-Induced Endothelial-to-Mesenchymal Transition: A Switch to Cardiac Fibrosis? Trends Cardiovasc Med 18:293–298
Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, Shimokata K, Hasegawa Y (2010) Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 43:161–172
Horwitz EM (2010) Building bone from blood vessels. Nat Med 16:1373–1374
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176:85–97
Ishisaki A, Hayashi H, Li AJ, Imamura T (2003) Human umbilical vein endothelium-derived cells retain potential to differentiate into smooth muscle-like cells. J Biol Chem 278:1303–1309
Itoh S, ten Dijke P (2007) Negative regulation of TGF-β receptor/Smad signal transduction. Curr Opin Cell Biol 19:176–184
Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, Schulte-Merker S, Gerhardt H (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12:943–953
Johnson NC, Dillard ME, Baluk P, McDonald DM, Harvey NL, Frase SL, Oliver G (2008) Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev 22:3282–3291
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Kaplan FS, Shen Q, Lounev V, Seemann P, Groppe J, Katagiri T, Pignolo RJ, Shore EM (2008) Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP). J Bone Miner Metab 26:521–530
Khan R, Sheppard R (2006) Fibrosis in heart disease: understanding the role of transforming growth factor-β in cardiomyopathy, valvular disease and arrhythmia. Immunology 118:10–24
Khaw P, Grehn F, Hollo G, Overton B, Wilson R, Vogel R, Smith Z (2007) A phase III study of subconjunctival human anti-transforming growth factor β(2) monoclonal antibody (CAT-152) to prevent scarring after first-time trabeculectomy. Ophthalmology 114:1822–1830
Kizu A, Medici D, Kalluri R (2009) Endothelial-mesenchymal transition as a novel mechanism for generating myofibroblasts during diabetic nephropathy. Am J Pathol 175:1371–1373
Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K (2008) Snail is required for TGFβ-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci 121:3317–3324
Krenning G, Zeisberg EM, Kalluri R (2010) The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol 225:631–637
Lamouille S, Mallet C, Feige JJ, Bailly S (2002) Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood 100:4495–4501
Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, Karsan A (2007) Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med 204:2935–2948
Li Z, Jimenez SA (2011) Protein kinase C delta and the c-Abl kinase are required for transforming growth factor-β induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum 63(8):2473–2483
Li J, Qu X, Bertram JF (2009) Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol 175:1380–1388
Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, Yamamoto H, Bertram JF (2010) Blockade of Endothelial-Mesenchymal Transition by a Smad3 Inhibitor Delays the Early Development of Streptozotocin-Induced Diabetic Nephropathy. Diabetes 59:2612–2624
Liu Z, Kobayashi K, van Dinther M, van Heiningen SH, Valdimarsdottir G, van Laar T, Scharpfenecker M, Lowik CW, Goumans MJ, ten Dijke P, Pardali E (2009) VEGF and inhibitors of TGFβ type-I receptor kinase synergistically promote blood-vessel formation by inducing alpha5-integrin expression. J Cell Sci 122:3294–3302
Lopez D, Niu G, Huber P, Carter WB (2009) Tumor-induced upregulation of Twist, Snail, and Slug represses the activity of the human VE-cadherin promoter. Arch Biochem Biophys 482:77–82
Mallet C, Vittet D, Feige JJ, Bailly S (2006) TGFβ1 induces vasculogenesis and inhibits angiogenic sprouting in an embryonic stem cell differentiation model: respective contribution of ALK1 and ALK5. Stem Cells 24:2420–2427
Markwald RR, Fitzharris TP, Smith WN (1975) Sturctural analysis of endocardial cytodifferentiation. Dev Biol 42:160–180
Massague J (1998) TGF-β signal transduction. Annu Rev Biochem 67:753–791
Mead AL, Wong TT, Cordeiro MF, Anderson IK, Khaw PT (2003) Evaluation of anti-TGF-β2 antibody as a new postoperative anti-scarring agent in glaucoma surgery. Invest Ophthalmol Vis Sci 44:3394–3401
Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16:1400–1406
Medici D, Potenta S, Kalluri R (2011) Transforming Growth Factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signaling. Biochem J 437(3):515–520
Miyazono K (2009) Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad Ser B Phys Biol Sci 85:314–323
Moonen JR, Krenning G, Brinker MG, Koerts JA, van Luyn MJ, Harmsen MC (2010) Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc Res 86:506–515
Moustakas A, Heldin CH (2005) Non-Smad TGF-β signals. J Cell Sci 118:3573–3584
Moustakas A, Heldin CH (2009) The regulation of TGFβ signal transduction. Development 136:3699–3714
Nataraj D, Ernst A, Kalluri R (2010) Idiopathic pulmonary fibrosis is associated with endothelial to mesenchymal transition. Am J Respir Cell Mol Biol 43:129–130
Nomura-Kitabayashi A, Anderson GA, Sleep G, Mena J, Karabegovic A, Karamath S, Letarte M, Puri MC (2009) Endoglin is dispensable for angiogenesis, but required for endocardial cushion formation in the midgestation mouse embryo. Dev Biol 335:66–77
Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R, Shahidi R, Dorovini-Zis K, Li L, Beckstead B, Durand RE, Hoodless PA, Karsan A (2004) Notch Activation Results in Phenotypic and Functional Changes Consistent With Endothelial-to-Mesenchymal Transformation. Circ Res 94:910–917
Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E (2000) Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 97:2626–2631
Oliver G, Srinivasan RS (2010) Endothelial cell plasticity: how to become and remain a lymphatic endothelial cell. Development 137:363–372
Potenta S, Zeisberg E, Kalluri R (2008) The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer 99:1375–1379
Quaggin SE, Kapus A (2011) Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int 80(1):41–50
Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A (2003) Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 112:1486–1494
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS (2006) A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 38:525–527
Siriwardena D, Khaw PT, King AJ, Donaldson ML, Overton BM, Migdal C, Cordeiro MF (2002) Human antitransforming growth factor β(2) monoclonal antibody–a new modulator of wound healing in trabeculectomy: a randomized placebo controlled clinical study. Ophthalmology 109:427–431
Tang Y, Harrington A, Yang X, Friesel RE, Liaw L (2010) The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis 48:563–567
Tarin D, Thompson EW, Newgreen DF (2005) The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res 65:5996–6000
ten Dijke P, Arthur HM (2007) Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol 8:857–869
ten Dijke P, Goumans MJ, Pardali E (2008) Endoglin in angiogenesis and vascular diseases. Angiogenesis 11:79–89
Thiery JP, Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7:131–142
Thompson JE, Vaughan TJ, Williams AJ, Wilton J, Johnson KS, Bacon L, Green JA, Field R, Ruddock S, Martins M, Pope AR, Tempest PR, Jackson RH (1999) A fully human antibody neutralising biologically active human TGFβ2 for use in therapy. J Immunol Methods 227:17–29
Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL (2004) Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 18:99–115
Van der Heiden K, Hierck BP, Krams R, de CR, Cheng C, Baiker M, Pourquie MJ, Alkemade FE, DeRuiter MC, Gittenberger-de Groot AC, Poelmann RE (2008) Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 196:542–550
van Meeteren LA, Goumans MJ, ten Dijke P (2011) TGF-β Receptor Signaling Pathways in Angiogenesis; Emerging Targets for Anti-Angiogenesis Therapy. Curr Pharm Biotechnol (in press)
Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA, Laping NJ, Chen S (2007) Interference with TGF-β signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol 293:F1657–F1665
Watabe T, Nishihara A, Mishima K, Yamashita J, Shimizu K, Miyazawa K, Nishikawa S, Miyazono K (2003) TGF-β receptor kinase inhibitor enhances growth and integrity of embryonic stem cell-derived endothelial cells. J Cell Biol 163:1303–1311
Wong SH, Hamel L, Chevalier S, Philip A (2000) Endoglin expression on human microvascular endothelial cells - Association with betaglycan and formation of higher order complexes with TGF-β signalling receptors. Eur J Biochem 267:5550–5560
Xu J, Lamouille S, Derynck R (2009) TGF-β-induced epithelial to mesenchymal transition. Cell Res 19:156–172
Yoshino T, Saito D, Tadokoro R, Takahashi Y (2011) In vivo gene manipulations of epithelial cell sheets: A novel model to study epithelial-to-mesenchymal transition. Dev Growth Differ 53:378–388
You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ, Tsai SY (2005) Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 435:98–104
Zeisberg M, Duffield JS (2010) Resolved: EMT produces fibroblasts in the kidney. J Am Soc Nephrol 21:1247–1253
Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R (2007a) Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res 67:10123–10128
Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R (2007b) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13:952–961
Zeisberg EM, Potenta SE, Sujimoto H, Zeisberg M, Kalluri R (2008) Fibroblasts in kidney fibrosis emerge via endothelial to mesenchymal transition. J Am Soc Nephrol 19(12):2282–2287
Zhang YE (2009) Non-Smad pathways in TGF-β signaling. Cell Res 19:128–139
Acknowledgements
We thank Drs. Beerend Hierck and Dorien Peters for helpful discussions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Research in our lab on TGF-β in angiogenesis is supported by the Dutch Cancer Society (to LAvM), Netherlands Institute for Regenerative Medicine (NIRM), the Le Ducq foundation and the Centre for Biomedical Genetics.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Meeteren, L.A., ten Dijke, P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res 347, 177–186 (2012). https://doi.org/10.1007/s00441-011-1222-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-011-1222-6