
Abstract
The phyllosphere of bamboo is rich in microorganisms that can disrupt the intestinal microbiota of the giant pandas that consume them, potentially leading to their death. In the present study, the abundance, diversity, biological functions (e.g., KEGG and CAZyme), and antibiotic resistance genes (ARGs) of bacteria and fungi in two bamboo species phyllosphere (Chimonobambusa szechuanensis, CS; Bashania fangiana, BF) in Daxiangling Nature Reserve (an important part of the Giant Panda National Park) were investigated respectively by amplicon sequencing of the whole 16S rRNA and ITS1-ITS2 genes on PacBio Sequel and whole-metagenome shotgun sequencing on Illumina NovaSeq 6000 platform. The results suggested that there were respectively 18 bacterial and 34 fungi biomarkers between the phyllosphere of the two species of bamboo. Beta diversity of bacteria and fungi communities exited between the two bamboos according to the (un)weighted UniFrac distance matrix. Moreover, the functional analysis showed that the largest relative abundance was found in the genes related to metabolism and global and overview maps. Glycoside hydrolases (GHs) and glycosyl transferases (GTs) have a higher abundance in two bamboo phyllospheres. Co-occurrence network modeling suggested that bacteria and fungi communities in CS phyllosphere employed a much more complex metabolic network than that in BF, and the abundance of multidrug, tetracycline, and glycopeptide resistance genes was higher and closely correlated with other ARGs. This study references the basis for protecting bamboo resources foraged by wild giant pandas and predicts the risk of antibiotic resistance in bamboo phyllosphere bacterial and fungal microbiota in the Giant Panda National Park, China.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Giant pandas have survived on bamboo diets for more than 2 My according to fossil records (Pei 1974; Jin et al. 2007). However, over 99% of their diet now consists of bamboo (Schaller et al. 1985), and only a small proportion of which is digested by giant pandas: 75–90% of the protein, 27% of the hemicellulose, and 8% of the cellulose (Dierenfeld et al. 1982). A forepaw with a pseudothumb, expanded zygomatic arches and well-developed mandible structure are facilitating feeding on bamboo (Wei et al. 2015). Seasonal vertically migrating and changing in feeding bamboo species and parts increase their nutritional intake (Wei et al. 1999; Hong et al. 2015, 2016; Hu et al. 2017). Except for the bamboo shoot period, however, bamboo leaves are still their predominant food throughout the year (Wu et al. 2017).
In the absence of genes that encode enzymes involved in breaking down cellulose and hemicellulose (Li et al. 2010), symbiosis microbes (such as Clostridium groups) in giant pandas’ gut can promote cellulose breakdown and maximize nutritional intake (Zhu et al. 2011). Different bamboo phyllosphere-origin microorganisms may cause changes in the gut microbiota of giant pandas, even an imbalance of gut microbial homeostasis. Specially, giant pandas typically die from gastrointestinal tract illness (GIT) in a state of low immunity (Zhao et al. 2021). Escherichia coli, Klebsiella pneumoniae, and Clostridium welchii in giant pandas’ gut have been also shown to cause GIT (Wang et al. 2013; Zou et al. 2018). In addition, the widespread use of antibiotics has made antibiotic resistance genes widely present in humans, animals, and complex environments, which has become one of the world’s most significant public health threats (Chen et al. 2019; Pei et al. 2019). And bamboo leaf microbes also form a reservoir of antibiotic resistance genes (ARGs) (Sun et al. 2021; Kang et al. 2022), which may cause the risk of drug resistance for giant pandas (Mustafa et al. 2021; Kang et al. 2022).
Different bamboo species have different microbial communities, which may differentially influence on the gut microbiota of wild giant pandas (Jin et al. 2020). By traditional culture methods, Xu et al. (2014) found the different microbial compositions in the phyllosphere of the bamboo species evaluated. Those differences also were found in endophytic bacteria and fungi in bamboo leaves (Helander et al. 2013) and in endophytic fungi in Phyllostachys heteroclada between leaf and branch (Zhou et al. 2017). Using high-throughput amplicon sequencing (V4 region of 16S rRNA and ITS1 region for bacteria and fungi respectively), differences about the richness and diversity of bacteria and fungi between bamboo species also were found (Jin et al. 2020; Long et al. 2021; Kang et al. 2022). Whole-metagenome shotgun sequencing also was used to find differences in biological functions (e.g., KEGG, CAZyme) and ARGs of fungi and bacteria in Arundinaria spanostachya phyllosphere between in spring and autumn in Xiaoxiangling mountains (Long et al. 2021; Kang et al. 2022). Until now, there was no study on amplicon sequencing of the whole 16S rRNA and ITS1-ITS2 genes of phyllosphere bacteria and fungi communities in giant panda staple food bamboos and comparing of biological functions (e.g., KEGG, CAZyme) and ARGs among different bamboo species. Full-length amplicon (whole 16S rRNA and ITS1-ITS2 genes) by single molecule sequencing technology was thought to provide better resolution for species identification (Singer et al. 2016; Ihara et al. 2019). Therefore, we carry out those researches in Daxiangling Nature Reserve (Daxiangling NR) by third generation single molecule sequencing, which is an important part of the Giant Panda National Park.
In the present study, we used third-generation amplicon sequencing of the whole 16S rRNA and ITS1-ITS2 genes on PacBio Sequel and whole-metagenome shotgun sequencing on Illumina NovaSeq 6000 platform to investigate the diversity, abundance, and functions (e.g., KEGG, CAZyme) and ARGs of bacteria and fungi between in CS and BF phyllosphere in Daxiangling NR, China. The goals were to (1) expose differences of bacterial and fungal community composition between in the phyllosphere of CS and BF; (2) compare the diversity, richness, and community structure of phyllosphere bacteria and fungi communities between CS and BF; (3) investigate the interactions between fungi and bacteria in CS and BF phyllosphere using co-occurrence network analysis; (4) reveal the differences in KEGG, CAZyme of fungi and bacteria between in CS and BF phyllosphere; and (5) obtain interactive relationship of ARGs of the bacteria and fungi communities in CS and BF phyllosphere and their differences between the two bamboo species. This study would serve as a guide for managing bamboo resources forged by giant pandas (such as the bamboo forests restoration and protection) in the Giant Panda National Park, China.
Methods
Sample collection
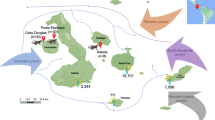
Daxiangling NR is situated in Yingjing county, Sichuan province, China (102° 29′ 36″–102° 52′ 24″ E, 29° 28′ 33″–29° 43′ 54″ N) (Fu et al. 2008; Li et al. 2020), which had 7 giant pandas according to the Fourth National Giant Panda Survey (Sichuan Forestry Bureau 2015). The habitat of giant pandas in the reserve covers 26,670 ha, accounting for 92% of the total area of the reserve, and Chimonobambusa szechuanensis (CS) is the staple bamboo foraged by giant pandas, accounting for 48.86% of the bamboo distribution area within the giant panda habitat (Yang et al. 2016). Besides, Bashania fangiana (BF) is also a staple bamboo species foraged by the giant panda, which is mainly distributed in the south of the Daxiangling NR at higher altitudes. Because CS is consumed by the most of giant pandas in Daxiangling NR, followed by BF, these two species were selected in our study.
We conducted wild sampling during May 5th to 15th in 2021. First, we set up three transects in CS and BF forests in Daxiangling NR, respectively. The transects were set from low altitude to high altitude, and the distance between them was no less than 200 m. Secondly, we collected three to six samples in each transect, with the altitude distance of adjacent collecting sits on the same transect no less than 50 m. The mixed 200 g bamboo leaves were collected with sterile gloves, packed into sterile zip-lock bags, immediately transported to the laboratory (less than 2 h), and stored at − 80 °C for further DNA extraction within 48 h. Overall, 15 samples were collected from CS and BF forests, respectively.
DNA extraction and PCR amplification
Five healthy leaves were randomly selected from each sample from 15 CS and 15 BF bamboo for further DNA extraction in accordance with the OMEGA E.Z.N.A.® Soil DNA kit’s instructions (Long et al. 2021; Kang et al. 2022). The DNA samples were shipped to Shanghai Majorbio Bio-pharm Technology Co. Ltd on dry ice. For bacterial communities, the universal bacterial primers 27F (AGRGTTYGATYMTGGCTCAG-3′) and 1492R (5′-RGYTACCTTGTTACGACTT-3′) (Weisburg et al. 1991) were used to amplify the whole 16S rRNA genes. The ITS1-ITS2 sequences were amplified for fungi communities using the primers ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS4R (5′-TCCTCCGCTTATTGATATGC-3′). Then, PCR amplification was used to carry out: initial denaturation at 95 °C for 3 min, then 27 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s, followed by a single extension at 72 °C for 10 min, and ending at 10 °C. Purified products were pooled in equimolar and the DNA library was constructed using the SMRTbell® Express Template Prep Kit 2.0 (Pacific Biosciences, CA, USA) according to PacBio’s instructions. Purified SMRTbell libraries were sequenced on the Pacbio Sequel System (Pacific Biosciences, CA, USA) by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China).
Bioinformatics analysis
Sequences with < 1000 or > 1800 bp and < 300 bp or > 900 bp were removed for bacteria and fungi, respectively. UPARSE (V7.1) (Stackebrandt and Goebel 1994) was used to cluster the optimized CCS reads into operational taxonomic units (OTUs) with a 97% degree of sequence similarity. As a representative sequence, the most abundant sequence for each OTU was selected. All chloroplast and bamboo sequences from all samples were removed by manually filtering the OTU table. The number of 16S rRNA and ITS gene sequences from each sample was filtered to 9331 and 9343, respectively, to minimize the effects of sequencing depth on alpha and beta diversity measures while still yielding an average. RDP Classifier (V2.2) was used to analyze the taxonomy of each OTU representative sequence against the ITS gene database (Unite v8.0) and 16S rRNA gene database (Silva v138) using a confidence threshold of 0.7.
Using the OTU data, Mothur v1.30.1 calculated the Shannon index and Sobs index. To determine whether the index values were substantially different between the two species, the Wilcoxon rank-sum test was utilized (P < 0.05). The similarities and differences between the phyllosphere microorganism community structures in CS and BF were compared using principal coordinate analysis (PCoA) based on the (un)weighted UniFrac distance and ANOSIM analysis at the OTU level. The significantly abundant taxa (phyla and genera) of bacteria and fungi between the two groups were determined using the linear discriminant analysis (LDA) (LDA score > 4, P < 0.05) effect size calculation.
Whole-genome shotgun sequencing and analysis
Eight samples (4 from CS and 4 from BF) were used for whole-genome shotgun sequencing in order to further investigate KEGG and CAZyme functions and ARGs on the Illumina NovaSeq 6000 platform. Fastp was used to clean up the raw reads from the metagenome sequencing by eliminating adaptor sequences, trimming, and removing low-quality reads (criteria for inclusion: reads with N bases, a minimum length threshold of 50 bp, and a minimum quality threshold of 20). These high-quality reads were then assembled into contigs using MEGAHIT. Contigs of ≥ 300 bp were selected as the final assembling result. Using MetaGene, open reading frames (ORFs) in contigs were identified (Noguchi et al. 2006). Using the NCBI translation table, the predicted ORFs of > 100 bp were retrieved and translated into amino acid sequences.
Using CD-HIT (V4.6.1), a non-redundant gene catalog was constructed with 90% sequence identity and 90% coverage. Using SOAPaligner (V2.21), high-quality reads were aligned to non-redundant gene catalogs to calculate gene abundance with 95% identity. Diamond (Buchfink et al. 2015) was used to align representative sequences from the non-redundant gene catalog to the NR database using an e-value limit of 1e−5 for taxonomy categorization. Diamond was used to annotate the Kyoto Encyclopedia of Genes and Genomes (KEGG) database using an e-value cutoff of 1e−5. Carbohydrate-active enzyme (CAZyme) annotation was conducted using hmmscan against the CAZyme database with an e-value cutoff of 1e−5. The non-redundant gene sets were mapped into CARD using DIAMOND (V0.8.35), and the results of the annotation for bacterial and fungal antibiotic resistance genes (ARGs) in CS and BF phyllospheres were obtained. The most abundant different KEGG Level 2 (LDA score ≥ 2), CAZyme features (LDA scores > 3), and CARD databases (LDA score > 2) were determined using the linear discriminant analysis (LDA) effect size (LEfSe).
Co-occurrence network analysis
OTUs with relative abundances greater than 0.1% were chosen using the OTU data for bacteria and fungi that were obtained via sequencing. Network analysis was carried out. The networks of bacterial and fungal co-occurrence among CS and BF were built using R.Gephi (Version 0.9.2). Network analyses were also carried out, and ARGs with relative abundances greater than 0.1% were chosen R. Gephi (Version 0.9.2) was also used to build the network of microbial co-occurrence. The statistical robustness is implied if Spearman’s correlation coefficient |ρ| is not less than 0.8 and the P values are less than 0.01.
Results
Microbial community composition in CS and BF phyllosphere
In this study, 310,714 and 368,599 high-quality 16S rRNA gene sequences were recovered from 15 CS and 15 BF leaf samples, respectively. At a 97% similarity level, there were 157 OTUs from all sequences. The overlap OTU number in CS and BF was 139 (Fig. S1). After annotating the representative sequences, 9 phyla, 10 classes, 15 orders, 18 families, and 29 genera were got. The relative abundance of Proteobacteria accounted for more than 75% of all bacteria observed in the 30 samples and the rest of the bacterial phyla were distributed in Acidobacteria, Bacteroidetes, Armatimonadota, etc. (Fig. 1A). At the genus level, 1174–901-12, Acidiphilium, Granulicella, and Methylocella were the dominant phyllosphere bacterial genera in both bamboo species (Fig. 1C). The LEfSe shows a significant difference in bacterial communities between CS and BF, and there were 18 biomarkers, including one phylum, three classes, four orders, three families, and seven genera. The relative abundance of Acidobacteriota in BF was much higher than that in CS, while that of Rhizobiales, Beijerinckiaceae, and 1174–901-12 in CS was higher than in BF (Fig. 2A).

Relative abundance of bacteria (A) and fungi (B) at the phylum and bacteria (C) and fungi (D) at genus level in CS and BF phyllosphere

Linear discriminant analysis (LDA) and effect size (LEfSe) analysis of bacteria (A) and fungi (B) in CS and BF (LDA scores > 4)
For fungi, a total of 349,908 and 363,595 ITS1-ITS2 sequences were obtained from 15 CS and 15 BF samples, respectively. At 97% similarity, all samples had 511 OTUs. The overlap OTU number of CS and BF was 345 (Fig. S1). All of the OTUs belonged to four phyla, 17 classes, 39 orders, 81 families, and 106 genera. Ascomycota had a maximum relative abundance (over 70%) (Fig. 1B). At the genus level, unclassified_o_Chaetothyriales, Zeloasperisporium, unclassified_c_Leotiomycetes, and Multiclavula were the dominant genera in BF and CS phyllosphere (Fig. 1D). There were 34 biomarkers, including three phylum, three classes, eight orders, nine families, and eleven genera. The relative abundance of Eurotiomycetes in BF was much higher than that in CS, while the relative abundance of Onygenales, Ascomycota, and Zeloasperisporium in BF was higher than that in CS (Fig. 2B).
Microbial diversity in CS and BF phyllosphere
The Sobs and Shannon indices for bacteria and fungi did not significantly differ between CS and BF (Fig. S2). The PCoA analysis showed significant differences in bacterial and fungal community structures between CS and BF (Fig. 3; ANOSIM analysis, P < 0.001). The PCoA based on weighted UniFrac distance was superior at distinguishing samples for bacteria community (Fig. 3A and B). For the fungal community, however, PCoA based on unweighted UniFrac distance distinguished samples more accurately than the PCoA based on weighted UniFrac distance (Fig. 3C and D).

PCoA analysis plots of bacterial communities using the weighted (A) and unweighted (B) UniFrac distance matrixes and fungal communities using the weighted (C) and unweighted (D) UniFrac distance matrixes in CS and BF phyllosphere at the OTU level
Co-occurrence patterns of bacteria and fungi communities between CS and BF phyllosphere
The number of nodes in BF (276) is more than that in CS (250) (Table S3). And the network density of CS is greater than that of BF (Table S3). The number of edges of Bac-Bac and Bac-Fun in CS phyllosphere is greater than that in BF phyllosphere, but the number of edges of Fun-Fun in BF phyllosphere is greater than that in CS phyllosphere (Table S3).
And the results of the co-occurrence network analysis showed that the network complexity, node connectivity, and average degree of microbe community in the CS bamboo bacteria and fungi communities were higher than those in the BF bamboo phyllosphere (Fig. 4). Fungi and fungi interaction accounted for the majority of the two bamboo species, and the degree of interaction between Bac-Bac and Bac-Fun was similar (Fig. 4; Table S3). Positive correlations between the two bamboos were more predominant in the co-occurrence network analysis (97.46% and 96.38%, respectively, Table S3).

Co-occurrence network analysis showing the bacteria interactions with fungi between CS (A) and BF (B) phyllosphere
Functions (e.g., KEGG, CAZy) of bacteria and fungi communities between CS and BF phyllosphere
Eight bamboo leaf samples (4 from CS, 4 from BF) were used for whole-genome shotgun sequencing. After filtering out low-quality and host genomic reads, 64.85 Gb of high-quality reads were obtained, with an average of 8.11 Gb for each sample. A total of 5.77 million contigs and 3.78 Gb of assembly sequence were obtained, and the average contig N50 was 692 bp (Table S4). The metagenomic analysis found 46 KEGG Level 2 categories in bacteria and fungi, respectively.
In the KEGG level 2 categories, for bacteria, global and overview maps had a maximum relative abundance (over 38%), followed by carbohydrate metabolism, amino acid metabolism, energy metabolism, etc. (Fig. S3). The relative abundance of genes related to global and overview maps in CS is higher than that in BF (Fig. S3). Additionally, the relative abundance of genes related to carbohydrate metabolism in CS was higher than in BF (Fig. S3). For fungi, global and overview maps also had a maximum relative abundance (over 19%), followed by neurodegenerative disease, signal transduction, and cell growth and death (Fig. S4). And the relative abundance of genes related to global and overview maps in BF is higher than that in CS (Fig. S4). About the LEfse results for the KEGG level 2 categories, the relative abundance of global and overview maps had the highest differences between CS and BF in bacteria (Fig. 5A). And the relative abundance of global and overview map also had the highest differences between CS and BF in fungi (Fig. 5B).

Linear discriminant analysis (LDA) and effect size (LEfSe) analysis at KEGG level 2 in bacterial (A) and fungal (B) community between CS and BF (LDA scores > 2)
According to the bacterial CAZyme relative abundance table, the relative abundance of glycoside hydrolases in CS (41.84%) is higher than in BF (36.31%) (Table S1). The relative abundance of glycosyl transferases in BF (37.90%) is higher than in CS (34.15%) (Table S1). According to the fungal CAZyme relative abundance table, the relative abundance of glycoside hydrolases in BF (44.86%) is higher than in CS (36.48%) (Table S2). The relative abundance of glycosyl transferases in CS (30.17%) is higher than in BF(22.19%) (Table S2). About the LEfse results for the CAZyme, for bacteria, four family CAZymes have a higher relative abundance in BF than that in CS: auxiliary activities (AA3, AA3_2), carbohydrate esterases (CE10, CE9, CE1), glycoside hydrolases (GH23, GH13_11), and glycosyl transferases (GT21, GT35). Interestingly, the highest differences in CAZyme between CS and BF were found to be glycoside hydrolases (GHs; Fig. 6A). For fungi, the relative abundance of most auxiliary activities and glycoside hydrolases was significantly higher in BF than in CS (Fig. 6B). However, CS has a considerably higher relative abundance of glycosyl transferases than BF (Fig. 6B).

Linear discriminant analysis (LDA) and effect size (LEfSe) of bacterial (A) and fungal (B) CAZymes for CS and BF. GT, glycosyl transferase; CE, carbohydrate esterase; GH, glycoside hydrolase; AA, auxiliary activities (LDA scores > 3)
ARGs of bacteria and fungi communities between CS and BF phyllosphere
Using metagenomic sequencing, 136 and 77 antibiotic resistance genes were obtained in 8 bamboo samples from the phyllosphere bacteria and fungi communities, respectively. Multidrug, tetracycline, and glycopeptide resistance genes of bacteria and fungi communities were the most common types of ARGs in CS and BF phyllosphere, which closely correlated with other ARGs (Fig. 7). The relative abundance of tetracycline in bacterial is higher than fungal that in phyllosphere, but the abundance of glycopetide in fungal is higher than that in bacterial phyllosphere (Fig. 7). The fungal has rifamycin and elfamycin genes, but bacterial does not have (Fig. 7B). Other resistance genes were peptide, MLS, beta-lactam, phenicol, fosfomycin, mupirocin, etc. The LEfse results indicated that the relative abundance of glycopeptide, multidrug, mupirocin, and rifamycin of bacteria in CS phyllosphere is higher than that in BF (Fig. 8A). The relative abundance of tetracycline, MLS, phenicol, and elfamycin of bacteria in BF phyllosphere is higher than that in CS (Fig. 8A). For fungi, the relative abundance of multidrug and mupirocin in CS phyllosphere is higher than that in BF. And the relative abundance of glycopeptide and phenicol of fungi in BF phyllosphere is higher than that in CS (Fig. 8B).

ARGs co-occurrence network of bacterial (A) and fungal (B) resistance genes between CS and BF. Nodes represent genes and are colored to indicate antimicrobial classes of ARGs

LEfSe analysis of bacterial (A) and fungal (B) antibiotic resistance genes (ARGs) between CS and BF (LDA score > 2)
Discussion
Giant panda conservation and microbial community composition in bamboo phyllosphere
For bacteria, although the relative abundance of Proteobacteria accounted for more than 75% of all observed bacteria from all samples, there were also 9 phyla found after annotating by amplicon sequencing of the whole 16S rRNA genes on PacBio Sequel. The Proteobacteria of heterotrophic organisms is well-known to predominate in marine environments, as well as dominant in the gut microbiota of humans and giant pandas (Jin et al. 2020; Shin et al. 2015; Stevens et al. 2005). Acidobacteria and Bacteroidetes also were found to be the dominant bacterial phyla in the bamboo phyllosphere, which is similar to several previous studies (Jin et al. 2021; Long et al. 2021; Tian et al. 2021). The phyllosphere bacterial community of CS and BF had a similar structure to other species of staple bamboo that the giant panda foraged, including a relatively high abundance of Proteobacteria, Acidobacteria, and Bacteroidetes (Jin et al. 2021; Long et al. 2021; Tian et al. 2021). The gut microbiome of giant pandas has also been discovered to contain Acidobacteria and Bacteroidetes (Jin et al. 2020). And these microbiota have various biological roles, such as balancing plant hormones, regulating root growth, promoting nutrient absorption, and preventing disease invasion (Xu et al. 2018). Additionally, Proteobacteria, Acidobacteria, and Bacteroides are frequently found in a variety of forests, demonstrating the extensive ecological range and adaptability of these organisms (Feng et al. 2019). At the genus level, 1174–901-12, Acidiphilium, Granulicella, and Methylocella were also found to be the dominant bacterial genus in both bamboo phyllosphere; these results were consistent with previous studies (Long et al. 2021; Tian et al. 2021). Sphingomonas also was found the dominant bacterial genus in both bamboo phyllosphere in our study, which would be obtained for giant pandas from those bamboo foods to cellulose-digesting (Long et al. 2022).
For fungi, Ascomycota and Basidiomycota had a maximum relative abundance (above 70%). At the genus level, unclassified_o_Chaetothyriales, Zeloasperisporium, unclassified_c_Leotiomycetes, and Multiclavula were the dominant genera in the two bamboo phyllospheres. This result is similar to prior investigations; the phyllosphere fungal community was structured with a high relative abundance of Ascomycota and Basidiomycota in bamboo foraging by giant panda (Jin et al. 2020; Kang et al. 2022). In addition, the presence of fungi from the phyla Ascomycota and Basidiomycota in the giant panda’s intestinal microbiome was also demonstrated (Tun et al. 2014).
LEfSe analysis showed that the relative abundance of Acidobacteriota and Ascomycota in BF was much higher than that in CS (Fig. 2). It may be related to the BF which has higher nutritional content and its leaves which contain more moisture (Jin et al. 2020). Its phyllosphere environment with sufficient nutrients promotes the growth of microbial communities (Shao et al. 2021). These microbiota have various biological roles, such as balancing plant hormones, regulating root growth, promoting nutrient absorption, and preventing disease invasion (Xu et al. 2018). So BF is most consumed by giant pandas and red pandas, followed by CS, in Daxiangling NR.
Giant panda conservation and microbial diversities, co-occurrence network patterns in bamboo phyllosphere
Here, we compared the Sobs and Shannon indices of bacteria and fungi in the CS and BF bamboo phyllosphere and found no significant difference of these two indices between the CS and BF bamboo (Fig. S2). But PCoA and ANOSIM analysis showed significant differences in bacterial and fungal community structures between CS and BF based on (un)weighted UniFrac distance at the OTU level, suggesting that the bamboo species had the greater effects on the community structure of phyllosphere fungi and bacteria. This result was similar to the findings of Isabelle (2016); they found that host species identity was a more important determinant than site or time in temperate tree phyllosphere bacterial communities. For instance, a study about lianas (Vitis vinifera) phyllosphere microbes showed that the bacterial community structure of the microbes in the phyllosphere was significantly affected by species (cultivar) difference (Singh et al. 2019).
To explore the interaction between fungi and bacteria in bamboo phyllosphere, we used a co-occurrence network analysis. The result showed that the CS phyllosphere bacterial and fungal communities had higher network complexity, node connectivity, and average microbe community degrees than the BF (Fig. 4 and Table S3). The network structure of plant fungi with different health statuses showed different network complexity and node connectivity (Lin et al. 2022). The complex interaction of bacteria and fungi on the leaf surface also has a certain impact on bamboo leaf health (Zhou et al. 2022). Physical interactions include endophytic bacteria in fungi, epiphytes on fungi surface, and their movements. These also partly explain why giant pandas mainly feed on CS in our study area (Sichuan Forestry Bureau 2015). Giant pandas may be more preferred to bamboo species with high network complexity, node connectivity, and average microbe community degrees of bacteria and fungi. Some studies have shown that bamboo phyllosphere with high microbial community diversity may be beneficial to improve the diversity of intestinal microbial communities in giant pandas and increase their adaptability (Larsen et al. 2018; Jin et al. 2020).Chemical interactions in which different small molecules produced by bacteria or fungi (antibiotics, volatile organic compounds, quorum sensing molecules, etc.) interact with each other to influence a number of physiological processes including morphology, growth, reproduction, transport/movement, nutrition, stress resistance, and pathogenicity (Zhou et al. 2022).
Giant panda conservation and KEGG and CAZyme functions of microbial community in bamboo phyllosphere
Metagenomic analysis revealed the significant differences in biological functions (KEGG and CAZymes functions) of CS and BF phyllosphere bacteria and fungi. Global and overview maps, carbohydrate metabolism, and amino acid metabolism of bacteria and fungi in all CS and BF phyllosphere were being more abundant at KEGG level 2 categories (Fig. S3 and Fig. S4). For bacterial communities, the metabolic pathways and biosynthesis of secondary metabolites gene functions were the most expressed in global and overview maps. Bacteria are capable of digesting the majority of organic matter and synthesizing a wide variety of potent small molecules thanks to their remarkable array of metabolic pathways, some of which are sometimes encoded by gene clusters (Wisecaver et al. 2014). For fungal communities, the relative abundance of metabolism functions in BF is higher than in CS. It is possible that its environment BF distributing is sunny and warm in higher elevation (Oliveira et al. 2001; Sichuan Forestry Bureau 2015).
Glycoside hydrolases (GHs) and glycosyl transferases (GTs) were being more abundant CAZymes of bacteria and fungi in all CS and BF phyllosphere. The LEfse results of the CAZyme revealed that the relative abundance of CEs (carbohydrate esterase) for BF was higher than for CS in bacteria and fungi (Fig. 6). This may also have been because higher CE contents are positively correlated with higher elevation (Dai et al. 2021). Interestingly, the relative abundance of GHs in CS may responsible for differences from BF in bacteria. As the GH family breaks down complex carbohydrates, this result indicated that CS requires more hydrolysis and synthesis of biological sugars and glycoconjugates (Lee et al. 2014).
Giant panda conservation and ARGs of microbial community in bamboo phyllosphere
Multidrug, tetracycline, and glycopeptide resistance genes of bacteria and fungi communities were the dominant types of ARGs in CS and BF phyllosphere by co-occurrence network analysis (Fig. 7; Fig. S5). Microorganisms contain a large number of resistance genes (Hulbert et al. 2001). Antibiotic resistance genes are considered a new type of environmental pollutant, which have been detected in water, soil, sediment, air, and other media. It could also pose a risk to giant pandas (Kang et al. 2022). Multidrug and tetracycline resistance genes were been identified in mice and plants (Croop et al. 1989). In view of the abundance and diversity of ARGs in bamboo phyllosphere bacteria and fungi communities, it is particularly important to investigate drug resistance genes in bamboo phyllosphere in the whole Giant Panda National Park. The relative abundance of multidrug and mupirocon genes of bacteria and fungi communities in BF phyllosphere was both higher than that in CS (Fig. 8). In bacteria, bicyclomycin resistance genes were rarely correlated with other ARGs. In fungi, diaminopyrimidine resistance genes were rarely correlated with other ARGs.
The host’s health is profoundly influenced by the gut microbiome, which acts as a crucial reservoir for ARGs. Food is a major influence on the gut microbes of animals (Scholz et al. 2020). Thus, bamboo phyllosphere microorganisms also may influence the giant panda gut microbiome (Jin et al. 2020). Multiple resistance genes are found in a large number of phyllosphere fungi and bacteria communities, demonstrating that phyllosphere has evolved as reservoirs of resistance genes for giant pandas. Giant pandas may be increasing their resistance to ARGs of bacteria and fungi communities in bamboo phyllosphere by coevolving with bamboo leaf-origin microorganism (Jin et al. 2020; Kang et al. 2022). In order to achieve better protection effect, ecological protection should be prioritized, and a certain amount of resource exploitation should be restricted to reduce antibiotic sources from human activities, such as grazing (Wei et al. 2020).
Conclusion
In this study, we explore the abundance, diversity, biological functions (e.g., KEGG and CAZyme), and antibiotic resistance genes (ARGs) of bacteria and fungi in two bamboo species phyllosphere (Chimonobambusa szechuanensis, CS; Bashania fangiana, BF) in Daxiangling Nature Reserve (an important part of the Giant Panda National Park). Our results suggested that although no significant alpha diversity, markedly beta diversity of bacteria and fungi communities exited between the two bamboos according to the (un)weighted UniFrac distance matrix. Moreover, the functional analysis showed that the largest relative abundance was found in the genes related to metabolism and global and overview maps. A higher relative abundance of bacterial CAZymes was discovered to be caused only by glycoside hydrolases (GHs) in CS. And the relative abundance of more fungi glycosyl transferases (GTs) in CS was also found greater than that in BF. Co-occurrence network modeling suggested that bacteria and fungi communities in CS phyllosphere employed a much more complex network than that in BF, and the abundance of multidrug, tetracycline, and glycopeptide resistance genes was higher and closely correlated with other ARGs. This integrated study improves our knowledge about microorganism of food origin and reference basis for protecting bamboo resources foraged by wild giant pandas and predicts the risk of antibiotic resistance in bamboo phyllosphere bacterial and fungal microbiota in the Giant Panda National Park, China.
Data availability
No datasets were generated or analysed during the current study.
References
Chen QL, Cui HL, Su JQ, Penuelas J, Zhu YG (2019) Antibiotic resistomes in plant microbiomes. Trends Plant Sci 24:530–541. https://doi.org/10.1016/j.tplants.2019.02.010
Croop JM, Raymond M, Haber D, Devault A, Arceci RJ, Gros P, Housman DE (1989) The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol 9(3):1346–1350. https://doi.org/10.1128/mcb.9.3.1346-1350.1989
Dai Z, Zang H, Chen J, Fu Y, Wang X, Liu H, Shen C, Wang J, Kuzyakov Y, Becker JN, Hemp A, Barberán A, Gunina A, Chen H, Luo Y, Xu J (2021) Metagenomic insights into soil microbial communities involved in carbon cycling along an elevation climosequences. Environ Microbiol 23:4631–4645. https://doi.org/10.1111/1462-2920.15655
Dierenfeld ES, Hintz HF, Robertson JB, Van Soest PJ, Oftedal OT (1982) Utilization of bamboo by the giant panda. J Nutr 112:636–641. https://doi.org/10.1093/jn/112.4.636
Feng XC, Cao XG, Ju WH, Liu KM (2019) A Study on Community Characteristics of Forest Soil Bacteria in Lushan National Nature Reserve. For Ecol Manag 6:101-107. https://doi.org/10.13466/j.cnki.lyzygl.2019.06.018
Fu JR, Sun ZY, Liu SY, Fu ZH, Liu Y, Wang X, Zhao J, Cai G (2008) A survey of bird resources in Da Xiang Ling Nature Reserve in Sichuan Province. J Sichuan For Sci Technol 29:31–37. https://doi.org/10.16779/j.cnki.1003-5508.2008.03.007
Helander M, Jia R, Huitu O, Sieber TN, Jia J, Niemelä P (2013) Endophytic fungi and silica content of different bamboo species in giant panda diet. Symbiosis 61:13–22. https://doi.org/10.1007/s13199-013-0253-z
Hong M, Yuan S, Yang Z, Yang X, Gu X, Huang F (2015) Comparison of microhabitat selection and trace abundance of giant pandas between primary and secondary forests in Liziping Nature Reserve, China: effects of selective logging. Mamm Biol 80:373–379. https://doi.org/10.1016/j.mambio.2015.05.003
Hong M, Wei W, Yang Z, Yuan S, Yang X, Gu X (2016) Effects of timber harvesting on Arundinaria spanostachya bamboo and feeding-site selection by giant pandas in Liziping Nature Reserve, China. For Ecol Manage 373:74–80. https://doi.org/10.1016/j.foreco.2016.04.039
Hu YB, Wu Q, Ma S, Ma TX, Shan L, Wang X (2017) Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas. ProcNat Acad Sci USA 114:1081–1086. https://doi.org/10.1073/pnas.1613870114
Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39:285–312. https://doi.org/10.1146/annurev.phyto.39.1.285
Ihara Y, Takeshita T, Kageyama S, Matsumi R, Asakawa M (2019) Identification of initial colonizing bacteria in dental plaques from young adults using full-length 16S rRNA gene sequencing. mSystems 4:e00360–19
Isabelle LL, Christian M, Steven WK (2016) Host species identity, site and time drive temperate tree phyllosphere bacterial community structure. Microbiome 4(1): 27. https://doi.org/10.1186/s40168-016-0174-1
Jin CZ, Ciochon RL, Dong W, Hunt RM, Liu JY, Jaeger M, Zhu QZ (2007) The first skull of the earliest giant panda. Proc Natl Acad Sci USA 104:10932–10937. https://doi.org/10.1073/pnas.0704198104
Jin L, Wu D, Li C, Zhang A, Xiong Y, Wei R, Zhang G, Yang S, Deng W, Li T, Li B, Pan X, Zhang Z, Huang Y, Zhang H, He Y, Zou L (2020) Bamboo nutrients and microbiome affect gut microbiome of giant panda. Symbiosis 80:293–304. https://doi.org/10.1007/s13199-020-00673-0
Jin L, He YG, Yang XJ, Deng WW, Yang L, Jiang CY, Li B, Li CW, Zhou Y, Zeng W, Li T, Huang Y, Zhang HM, Zhou SQ, Zou LK (2021) Nutrient composition and microbial communities of bamboo at Wolong National Nature Reserve. Chin J Appl Environ Biol 27:1210–1217. https://doi.org/10.19675/j.cnki.1006-687x.2020.08039
Kang L, Luo W, Dai Q, Zhou H, Wei W, Tang J, Han H, Yuan Y, Long J, Zhang Z, Hong M (2022) Giant pandas’ staple food bamboo phyllosphere fungal community and its influencing factors. Front Microbiol 13:1009588. https://doi.org/10.3389/fmicb.2022.1009588
Larsen OFA, Claassen E (2018) The mechanistic link between health and and gut microbiota diversity. Scic Rep 8(1). https://doi.org/10.1038/s41598-018-20141-6
Lee S, Cantarel B, Henrissat B, Gevers D, Birren BW, Huttenhower C, Ko G (2014) Gene-targeted metagenomics analysis of glucan-branching enzyme gene profiles among human and animal fecal microbiota. ISME J 8:493–503. https://doi.org/10.1038/ismej.2013.167
Li RQ, Fan W, Tian G, Zhu HM, He L, Cai J, Huang QF, Cai QL, Li B, Bai YQ (2010) The sequence and de novo assembly of the giant panda genome. Nature 463:311–317. https://doi.org/10.1038/nature08696
Li P, Fu MX, Qi DW, Song XQ, Wei W, Yang WJ, Chen YX, Zhou YS, Liu JB, Ma R, Yu J, Yang H, Chen P, Hou R (2020) Camera-trapping survey of wild mammals and birds in Daxiangling nature reserve, Sichuan Province. Biodivers Sci 28:905–912. https://doi.org/10.17520/biods.2019381
Lin T, Xiao YS, Zeng WA, Gu SS, Zhai ZG, Wu SL, Li PF, Feng K, Deng Y, Hu QL (2022) Network analysis reveals the root endophytic fungi associated with fusarium root rot invasion. Appl Soil Ecol 178(104567):0929–1393. https://doi.org/10.1016/j.apsoil.2022.104567
Long JJ, Luo W, Xie JM, Yuan Y, Wang J, Kang LW, Li Y, Zhang ZJ, Hong MS (2021) Environmental factors influencing Phyllosphere bacterial communities in Giant Pandas' staple food bamboos. Front Microbiol 12:748141. https://doi.org/10.3389/fmicb.2021.748141
Long JJ (2022) Seasonal changes of intestinal microorganisms in captive giant and red pandas and their relationship with leaf-peripheral microorganisms[D]. (In Chinese)
Mustafa GR, Li CW, Zhao SY, Jin L, He XP, Shabbir MZ (2021) Metagenomic analysis revealed a wide distribution of antibiotic resistance genes and biosynthesis of antibiotics in the gut of giant pandas. BMC Microbiol 21:15. https://doi.org/10.1186/s12866-020-02078-x
Oliveira IC, Brenner E, Chiu J, Hsieh MH, Kouranov A, Lam HM, Shin MJ, Coruzzi G (2001) Metabolite and light regulation of metabolism in plants: lessons from the study of a single biochemical pathway. Brazilian Journal of Medical and Biologica Research = Revista brasileira de pesquisas medicas e biologicas 34(5):567–575. https://doi.org/10.1590/s0100-879x2001000500003
Pei WZ (1974) Evolutionary history of giant panda. Acta Zool Sin 20:188–190
Pei M, Zhang B, He Y, Su J, Gin K, Lev O (2019) State of the art of tertiary treatment technologies for controlling antibiotic resistance in wastewater treatment plants. Environ Int 131:105026. https://doi.org/10.1016/j.envint.2019.105026
Schaller GB, Hu JC, Pan WS, Zhu J (1985) The giant panda of Wolong. University of Chicago Press, Chicago. https://doi.org/10.1086/414647
Scholz M, Staudacher H, Fava F, Tuohy K, Whelan K (2020) Food & nutrition: the driving factors of our gut microbes. Proc Nutr Soc 79:699. https://doi.org/10.26226/morressier.5d5e518abedcf39b7663c72a
Shao PS, Lynch L, Xie HT, Bao XL, Liang C (2021) Tradeoffs among microbial life history strategies influence the fate of microbial residues in subtropical forest soils. Soil Boil Biochem 153:108112. https://doi.org/10.1016/j.soilbio.2020.108112
Shin NR, Whon TW, Bae JW (2015) Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 33:496–503. https://doi.org/10.1016/j.tibtech.2015.06.011
Singer E, Bushnell B, Coleman-Derr D, Bowman B, Bowers RM, Levy A, Gies EA, Cheng J-F, Copeland A, Klenk H-P (2016) High-resolution phylogenetic micobial community profiling. ISME J 10:2020–2032 https://doi.org/10.1038/ismej.2015.249
Singh P, Santoni S, Weber A, This P, Péros JP (2019) Understanding the phyllosphere microbiome assemblage in grape species (Vitaceae) with amplicon sequence data structures. Sci Rep 9:14294. https://doi.org/10.1038/s41598-019-50839-0
Stevens H, Stübner M, Simon M, Brinkhoff T (2005) Phylogeny of Proteobacteria and Bacteroidetes from oxic habitats of a tidal flat ecosystem. FEMS Microbiol Ecol 54:351–365. https://doi.org/10.1016/j.femsec.2005.04.008
Sun Y, Snow D, Walia H, Li X (2021) Transmission routes of the microbiome and resistome from manure to soil and lettuce. Environ Sci Technol 55:11102–11112. https://doi.org/10.1021/acs.est.1c02985
Sichuan Forestry Bureau (2015) The Giant panda of Sichuan: report of the fourth National Giant Panda Survey. Sichuan Science and Technology Press
Tian CY, Hong MS, Long JJ, Xie JM (2021) Seasonal variation of bacterial diversity in phyllosphere of giant pandas’ staple food bamboo. Journal of Sichuan Forestry Science and Technology 42:1–7. https://doi.org/10.12172/202107050001
Tun HM, Mauroo NF, Yuen CS, Ho JC, Wong MT, Leung FC (2014) Microbial diversity and evidence of novel homoacetogens in the gut of both geriatric and adult giant pandas (Ailuropoda melanoleuca). PLoS One 9:e79902. https://doi.org/10.1371/journal.pone.0079902
Wang X, Yan Q, Xia X, Zhang Y, Li D, Wang C, Chen S, Hou R (2013) Serotypes, virulence factors, and antimicrobial susceptibilities of vaginal and fecal isolates of Escherichia coli from giant pandas. Appl Environ Microbiol 79:5146–5150. https://doi.org/10.1128/aem.01367-13
Wei F, Feng Z, Wang Z, Li M (1999) Feeding strategy and resource partitioning between giant and red pandas. Mammalia 63:417–430. https://doi.org/10.1515/mamm.1999.63.4.417
Wei FW, Hu Y, Yan L, Nie Y, Wu Q, Zhang ZJ (2015) Giant pandas are not an evolutionary cul-de-sac: evidence from multidisciplinary research. Mol Biol Evol 32(1):4–12. https://doi.org/10.1093/molbev/msu278
Wei FW, Swaisgood RR, Pilfold NW, Owen MA, Dai Q, Wei FW, Han H, Yang ZS, Yang XY, Gu XD, Zhang JD, Yuan SB, Hong MS, Tang JF, Zhou H, He K, Zhang ZJ (2020) Assessing the effectiveness of China’s panda protection system. Curr Biol 30:1–7
Wisecaver JH, Slot JC, Rokas A (2014) Correction: the evolution of fungal metabolic pathways. PLoS Genet 10:e1004816. https://doi.org/10.1371/journal.pgen.1004816
Wu Q, Wang X, Ding Y, Hu YB, Nie YG, Wei W (2017) Seasonal variation in nutrient utilization shapes gut microbiome structure and function in wild giant pandas. Proc Biol Sci 284:20170955. https://doi.org/10.1098/rspb.2017.0955
Xu XL, Bai Y, Liu H, Yang CL, Ling YG (2014) Microflora analysis on six kinds of bamboo leaves in Sichuan Province. J Huazhong Agric Univ 33:53–59 (In Chinese)
Xu J, Zhang Y, Zhang P, Trivedi P, Riera N, Wang Y, Liu X, Fan G, Tang J, Coletta-Filho HD, Cubero J, Deng X, Ancona V, Lu Z, Zhong B, Roper MC, Capote N, Catara V, Pietersen G et al (2018) The structure and function of the global citrus rhizosphere microbiome. Nat Commun 9:4894. https://doi.org/10.1038/s41467-018-07343-2
Yang LJ, Zhong X, Yan TT, Ran JH, Zhang M, Cheng Y, Tang KC (2016) Impactsof bamboo shoot-collecting on the growth of Chimonobambusa szechuanensis and Giant panda activities. Sichuan Journal of Zoology 35:321–326. https://doi.org/10.11984/j.issn.1000-7083.20150409
Zhao S, Li C, Zhu T, Jin L, Deng W, Zhao K, He Y, Li G, Xiong Y, Li T, Li B, Huang Y, Zhang H, Zou L (2021) Diversity and composition of gut bacterial community in giant panda with anorexia. Curr Microbiol 78:1358–1366. https://doi.org/10.1007/s00284-021-02424-w
Zhou ZY (2017) Adaptation of intestinal flora to high-fiber environment in giant pandas (Ailuropoda melanoleuca) during dietary transition[D]. Sichuan Agricultural University, Sichuan (In Chinese)
Zhou YK, Shen XY, Hou CL (2017) Diversity and antimicrobial activity of culturable fungi from fishscale bamboo (Phyllostachys heteroclada) in China. World J Microbiol Biotechnol 33:104. https://doi.org/10.1007/s11274-017-2267-9
Zhou YQ, Wang HK, Xu S, Liu K, Qi H, Wang MC, Chen XYL, Berg G, Ma ZH, Cernava T, Chen Y (2022) Bacterial-fungal interactions under agricultural settings: from physical to chemical interactions. Stress Biol 2:22. https://doi.org/10.1007/s44154-022-00046-1
Zhu LF, Wu Q, Dai JY, Zhang SN, Wei FW (2011) Evidence of cellulose metabolism by the giant panda gut microbiome. Proc Natl Acad Sci USA 108:17714–17719. https://doi.org/10.1073/pnas.1017956108
Zou W, Li C, Yang X, Wang Y, Cheng G, Zeng J, Zhang X, Chen Y, Cai R, Huang Q, Feng L, Wang H, Li D, Zhang G, Chen Y, Zhang Z, Zhang H (2018) Frequency of antimicrobial resistance and integron gene cassettes in Escherichia coli isolated from giant pandas (Ailuropoda melanoleuca) in China. Microb Pathog 116:173–179. https://doi.org/10.1016/j.micpath.2018.01.034
Acknowledgements
We appreciate the help of all the staff that participated in the experiments and data collection. Thanks were also given to the anonymous reviewers for their constructive comments.
Funding
This research was funded by the Second Qinghai-Tibet Plateau Comprehensive Scientific Survey (2019QZKK05010502), Giant Pandas International Cooperation Foundation of State Forestry Administration (2023), the Innovation Team Funds of China West Normal University (grant no. KCXTD2022-7), and the National Natural Science Foundation of China (grant number 31900337).
Author information
Authors and Affiliations
Contributions
Material preparation, data collection and analysis were performed by all authors. The first draft of the manuscript was written by Xinyue Wang, Yi Li and Mingsheng Hong. Mingsheng Hong planned and supervised the project. And all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Li, Y., Kang, L. et al. Diversity, functions, and antibiotic resistance genes of bacteria and fungi are examined in the bamboo plant phyllosphere that serve as food for the giant pandas. Int Microbiol (2024). https://doi.org/10.1007/s10123-024-00583-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10123-024-00583-x