
Abstract
Dissolved organic matter (DOM) plays a fundamental role in nutrient cycling dynamics in riverine systems. Recent research has confirmed that the concentration of riverine DOM is not the only factor regulating its functional significance; the need to define the chemical composition of DOM is a priority. Past studies of riverine DOM rested on bulk quantification, however technological advancements have meant there has been a shift towards analytical methods which allow the characterisation of DOM either at compound class or more recently molecular level. However, it is important to consider that all analytical methods only consider a defined analytical window. Thus, herein, we explore the use of a hierarchy of methods which can be used in combination for the investigation of a wide range of DOM chemistries. By using these methods to investigate the DOM composition of a range of streams draining catchments of contrasting environmental character, a wide range of compounds were identified across a range of polarities and molecular weight, thereby extending the analytical window. Through the elucidation of the DOM character in stream samples, information can be collected about likely the sources of DOM. The identification of individual key compounds within the DOM pool is a key step in the design of robust and informative bioassay experiments, used to understand in-stream ecosystem responses. This is critical if we are to assess the role of DOM as a bioavailable nutrient resource and/or ecotoxicological factor in freshwater.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dissolved organic matter (DOM) plays a fundamental role in nutrient cycling dynamics in riverine systems, accounting for the majority of the reactive carbon (C) present in aquatic ecosystems (Evans et al. 2017) and a bioavailable source of nitrogen (N) and phosphorus (P) for both stream autotrophs and heterotrophs (Brailsford et al. 2019, 2021; Mackay et al. 2020). As a result, DOM plays an important role in stream elemental nutrient cycling and the ecological functioning of aquatic systems (Cole et al. 2007; Prairie 2008; Battin et al. 2009; von Schiller et al. 2017). In addition, DOM also plays a vital role in the transport and bioavailability of trace metals (Christensen et al. 1996; Temminghoff et al. 1997) and other pollutants (Kalbitz et al. 1997; Aiken et al. 2011) within river systems. It is also a substrate for the formation of disinfection by-products in waters abstracted and treated for drinking (Lavonen et al. 2013; Ike et al. 2019), which are potentially carcinogenic. Recent research has confirmed that the concentration of DOM in a waterbody is not the only factor regulating its functional significance; the need to define the chemical composition of DOM is increasingly being recognised as an important research priority for scientists, water companies and regulators (Riedel et al. 2016; Kaiser et al. 2017; Worms et al. 2019) as different compound classes play differing roles and are metabolised in different ways by the stream biota. Therefore, in order to understand the distribution and chemical processing of organic matter in the environment, elucidating the origin of the uncharacterised portion at molecular level is crucial (Hedges et al. 2000).
Some studies of riverine DOM character (particularly early studies) largely rest on bulk nutrient quantification (DOC, DON, DOP) (e.g. Aud et al. 2003; Worrall et al. 2004; Halliday et al. 2014; Volta et al. 2020). More recent technological advancements have supported a shift towards more specific analytical methods which allow the characterisation of the DOM pool either at the compound class level or, most recently, the molecular level. The optical properties of DOM have been used widely to define composition at a compound class level, specifically UV–Vis spectrophotometry (Helms et al. 2008; Werner et al. 2019; Lee et al. 2021; Pang et al. 2021; Yates et al. 2019, 2016) and fluorescence spectrophotometry (Baker 2002; Hudson et al. 2007; Spencer et al. 2008, 2019; Wang et al. 2019; Yates et al. 2019; Ma and Li 2020; Pisani et al. 2020). The development of in-situ optical sensors capable of monitoring at specific wavelengths have even allowed highly temporally resolved datasets to be collected, such that the dynamic changes in DOM composition under diurnal or storm event conditions can start to be determined (Werner et al. 2019; Croghan et al. 2021). In addition to techniques which consider the optical properties of the DOM pool, there has also been an increase in the number of studies characterising riverine DOM using size-exclusion chromatography coupled with organic carbon, organic nitrogen or UV detectors (Dulaquais et al. 2018; Wu et al. 2019; Hawkes et al. 2019; Trubetskaya et al. 2020). These techniques allow the dynamic flux of different compound classes to be assessed, but they lack the analytical power to determine the individual organic compounds in a complex DOM mixture. Achieving molecular level resolution is vital as the concentrations of specific compounds will vary in relation to source character, temporally and spatially within and along the length of a river lake, estuary or wetland.
Many studies have used targeted analysis of organic compounds in rivers, for example pesticides (Tan & Mustafa 2004; Stone et al. 2014; Lundqvist et al. 2019), pharmaceutical and personal care products (PPCPs) (Ashton et al. 2004; Joshua et al. 2020; Ngo et al. 2021) and other industrial pollutants, such as flame retardants (Andresen et al. 2004; Cristale et al. 2013). These tend to be carried out exclusively on compounds which have been anthropogenically introduced and are of known concern. However, the latter are generally trace components leaving the majority of DOM present in aquatic systems (potentially tens of thousands of compounds representing the majority of the overall DOM load), meaning a wide range of both natural and anthropogenic compounds remain uncharacterised. Hence, there is a need for updated molecular-scale methodologies which provide a wider analytical window and allow untargeted characterisation of DOM pool composition across the all the major molecular classes, and not merely target compounds, such as priority pollutants.
Technological advances in chromatography and mass spectrometry have begun to allow more in-depth probing of DOM compounds from complex environmental mixtures, and in some cases have provided unequivocal identification and quantification of compounds. Some studies have used chemical degradation methods followed by GC/MS analysis to examine specific compound classes in more detail, for example for dissolved black carbon (Dittmar 2008; Stubbins et al. 2012, 2015) and dissolved lignin (Kaiser & Benner 2012). In addition, there has been an increase in the number of studies using high-resolution mass spectrometry techniques, such as Orbitrap MS (Pemberton et al. 2020; Prasert et al. 2021) or Fourier-Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR MS) (Kamjunke et al. 2019; Simon et al. 2019; Spencer et al. 2019; Chen et al. 2021) as an analytical tool to investigate riverine DOM composition. A recent review by Kim et al. (2021), for example, discusses the most recent advances in ultrahigh-resolution MS for terrestrial organic matter. These novel molecular-scale approaches are now enabling significant advances in the elucidation of riverine DOM composition. However, it is important to appreciate that no analytical method is capable of achieving comprehensive molecular characterisation of DOM, with all analytical methods only considering a defined analytical window. Due to the low concentrations of individual organic compounds and other confounding factors such as the presence of inorganic salts, analytical protocols involving extraction and pre-concentration steps are always necessary prior to molecular level analysis. DOM pre-concentration is often undertaken using solid-phase extraction (SPE) and it that case the analytical window is limited by the chosen solid-phase or eluting solvent (Li & Minor 2015; Li et al. 2017). In addition, the type of MS (FTICR, TOF, Quadrupole), ionisation mode and efficiency (Patriarca et al. 2020), i.e. electron ionisation (EI) versus electrospray ionisation (ESI), ionisation polarity, i.e. positive versus negative (Hawkes et al. 2020), and mass range will affect the range of compounds detected. Therefore, a range of extraction and mass spectrometric methods is essential for obtaining a more complete characterisation of the composition of DOM at the molecular level.
The most common protocol for DOM extraction in environmental research utilises SPE cartridges packed with a styrene–divinylbenzene polymer-based sorbent and using methanol as the eluting solvent, followed by ESI and MS analysis in the negative ion mode (e.g.Stubbins et al. 2010; Spencer et al. 2019; Prasert et al. 2021). This provides detailed information regarding the polar components of DOM which are of intermediate molecular weight (m/z 150–2000). Meanwhile, pyrolysis–gas chromatography/MS (Py-GC/MS) has been employed specifically to investigate the presence of lignin phenols in river water, providing information about plant-derived DOM components (e.g. Williams et al. 2016; Kaal et al. 2020). In another example Lynch et al. (2019) used EI GC/MS analysis after a SPE preconcentration step, to characterise DOM components which could be chromatographically separated using a low polarity phase (TG-5MS) column. Each approach tackles a particular size fraction and class of compounds. In order to achieve this, it is necessary to not only optimise the compositional information gained from an individual technique, but to use it in combination with other molecular scale analyses to maximise the analytical window, such that potential impact of DOM in the environment is to be understood in relation to the molecular composition key compound classes.
Herein, we report the assembly of a suite of complementary chromatographic and mass spectrometric methodologies which can be used to intensively examine riverine DOM chemistry over a wide molecular weight range, encompassing low molecular weight chemical species to macromolecular components. The utility of these methods is then tested in a selection of streams draining catchments of contrasting environmental character, displaying marked differences in both their DOC, DON and DOP and inorganic nutrient loading rates, and the optical properties of their DOM pool (Lloyd et al. 2019; Yates et al. 2019) (Fig. 1).

Hierarchy of analytical methods for non-targeted analysis of riverine DOM
Methodology
Site descriptions
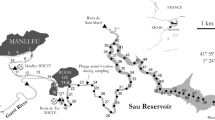
To demonstrate the applicability of this complementary approach across a selection of DOM chemistries, three well-researched contrasting UK exemplar field sites were chosen. Figure 2 shows the location of the field sites, which include a ‘natural’ peatland site in the headwaters of the River Conwy in North Wales (Nant y Brwyn hereafter), and two anthropogenically impacted catchments: a clay site impacted by agriculture in the headwaters of the River Sem, a tributary of the Hampshire Avon (Priors Farm hereafter) and a clay site impacted by a sewage treatment works on the River Chew at Chew Stoke, Somerset (Chew Stoke hereafter). The headwaters of the River Conwy drain blanket peatland supporting extensive sheep grazing, as it flows north before eventually reaching the Irish Sea. The River Sem predominately drains clay soils supporting intensive cattle production, as it flows west to join the River Nadder before flowing south into the Hampshire Avon which drains through Poole Harbour to the English Channel. The River Chew has its source in Chewton Mendip, draining an intensively farmed Carboniferous Limestone catchment, from where it drains over heavy clay soils supporting cattle production running northwest to Chew Stoke, then northeast to join the Bristol Avon, ultimately discharging to the Severn Estuary. Table 1 summarises the catchment characteristics and summary nutrient data from our prior research at each of these field sites. Of note, the human population density and discharge of treated effluent and untreated human waste from septic tanks varies markedly between the three catchments.

Map showing the location of the three contrasting UK field sites
Complementary analytical chemical methods
There were a number of criteria which needed to be fulfilled when designing a robust methodology for untargeted characterisation of riverine DOM. The methods needed to: (a) allow compound identification; (b) encompass a range of compound polarities, molecular weights and functional groups; (c) allow the analysis of natural and anthropogenic DOM, and (d) encompass a wide range of concentrations from trace biomarker components to macronutrients.
With these criteria in mind, complementary chromatographic and MS-based protocols were developed and tested as shown in Fig. 1. These were optimised for the analysis of riverine DOM, and crucially the number of laboratory processing steps was kept to a minimum to avoid the potential loss of volatile or low abundance compounds. Details of the methodologies and their optimisation are outlined below. In all cases samples of river water were collected using a pre-combusted (450 °C for 4 h) amber glass bottle, attached to a solvent-washed stainless-steel pole if access to the river was difficult. The bottles were filled to the rim to avoid any headspace and topped with a piece of pre-combusted (450 °C for 4 h) aluminium foil before the lid was applied to avoid any sources of plastic, and therefore contamination of the sample. The samples were transported back to the laboratory within 24 h of collection at 4 °C in the dark and filtered using glass filter kits (Merck Millipore, Feltham, UK) and glass fibre (GF) filter papers with a nominal pore size of 0.5 µm (Advantec, Cole‐Palmer, Hanwell, UK). All glassware and filter papers were pre-combusted before use as above. The use of pre-combusted GF filters was found to be important due to contamination of molecular-scale analyses, most notably via py-GC/MS, by cellulose nitrate filters at the ultra-low concentrations for individual DOM compounds addressed in this study (Lloyd et al. In press). Cellulose nitrate filters are the most commonly used filter for the filtration of river water samples for DON and DOP analysis. A pore size of 0.5 µm was chosen to match as closely as possible the standard 0.45 µm pore size used to define the ‘dissolved fraction’ in freshwater research. Water samples were stored at 4 °C, then filtered within 24 h of collection and were either processed immediately or stored at − 20 °C before being freeze-dried depending on the method requirements. For a full discussion of all aspects of sampling, pre-processing and storage of samples for molecular-scale analysis of DOM see Lloyd et al. (In press). HPLC grade water was used for procedural blanks which were processed via all the methods described below to check for any laboratory-derived contamination. This is critical when analysing compounds at trace concentrations.
Non-polar compounds
River water is rarely analysed for non-polar compounds as they are hydrophobic and often associated with particulates and therefore would not be expected to be present or their concentrations negligible in the filtered fraction. However, the notion of the purely ‘dissolved’ fraction is flawed as in reality particulate and dissolved organic matter should be viewed as a continuum, spanning from full solution phase through colloidal matter to particulate phase. As a result, the < 0.45 µm fraction will contain colloidal material, including non-polar compounds which may be in colloidal or fine particulate phase. These compounds are diagnostically useful as they can include a variety of both natural and anthropogenically produced ‘biomarker’ compounds (van Bergen et al. 1997; Bull et al. 1998) which can help in defining the character of the riverine DOM and distinguish between different contributing sources.
The analysis of the non-polar fraction in this programme was carried out using liquid–liquid extraction of ‘raw’ filtered river water. Filtered river water (150 mL) was placed into a glass separatory funnel along with 5α-androstane as an internal standard. The mixture was extracted with n-hexane (3 × 20 mL), chosen due to its low polarity and immiscibility in water. This meant that it was possible to obtain a water-free fraction without the need for a drying step (using a sodium sulphate column for example) which could introduce potential contaminants and risk compounds of interest being lost by adsorption onto the drying column. A number of other organic solvents were trialled including dichloromethane, diethyl ether and cyclohexane. Dichloromethane and n-hexane extracts produced very similar chromatograms, and the widest range of compounds compared with the other solvents and therefore n-hexane was chosen as it had better immiscibility. The three aliquots of n-hexane were combined and rotary evaporated until 1–2 mL of solvent remained, and samples were stored at − 20 °C until analysis. Prior to analysis via gas chromatography/mass spectrometry (GC/MS), the samples were blown down to dryness under a gentle stream of N2, then 70 µL of N-methyl-N-(trimethylsilyl)trifluoroacetamide with 1% trimethylchlorosilane (MSTFA) (Sigma–Aldrich, Gillingham, UK) was added and the mixture heated at 37 °C for 40 min to produce TMS-derivatives. MSTFA was chosen over N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) as during method testing it was found that lower molecular weight compounds such as urea were not effectively derivatised using BSTFA, yet an optimal signal was observed when the MSTFA reagent was used. No other differences were observed between the range of compounds derivatised between the two reagents. The derivatised samples were then blown to dryness under N2 and re-dissolved in ethyl acetate for analysis. They were then analysed using a Thermo Scientific TRACE 1300 gas chromatograph coupled with a Thermo Scientific ISQ LT Single Quadrupole MS. The GC was fitted with a HP-1 (50 m × 0.32, film thickness 0.17 µm) column and used helium as the carrier gas. The GC temperature programme was: 50 °C (1 min) to 300 °C (held for 10 min) at 10 °C min−1. The MS was operated using electron ionisation at 70 eV with the quadrupole mass analyser scanning a range of m/z 50–650 (scan time 0.2 s). Compounds were assigned based on library mass spectra, known elution orders and comparisons of retention times with reference compounds.
Polar building block compounds
Amino acids and carbohydrates are polar building block compounds which are fundamental in all ecosystems, they are the base constituents of biomacromolecules. The ability to detect their presence in the stream DOM pool would provide an insight into nutrient processing within streams of differing environmental character. Here we refine and test methods previously developed for the determination of amino acids and carbohydrates in soils, adapting them for freshwater samples. The methods extract the total hydrolysable pools (from proteinaceous material) as it is generally assumed that the free pools will be extremely small in river water (Robarts et al. 1990) or undetectable (Lytle & Perdue 1981) as the compounds are turned over so rapidly by the stream biota.
Amino acids
Total hydrolysable amino acids were extracted using the method previously reported by Charteris et al. (2016). Approximately 10 mg of freeze-dried residue (from filtered river water) along with 10 µL of norleucine (1000 µg mL−1) as an internal standard was subjected to hydrolysis using 5 mL of HCl (6 M) for 24 h at 100 °C under N2. Centrifugation was then used to separate the hydrolysates, which were then dried at 60 °C under a stream of N2 and stored at − 20 °C in 0.1 M HCl. A Dowex® 50WX8 cation exchange resin column was used to extract the amino acids which were subsequently eluted using 2 M NH4OH. The amino acids were then derivatised to produce their N-acetyl, O-isopropyl derivatives. Acetyl chloride mixed with isopropanol (4:1 v/v) was added to the hydrolysed amino acids (0.25 mL) and heated at 100 °C for 1 h. After quenching the reaction at − 25 °C, the excess reagent was removed under a gentle stream of N2. To ensure all the solvent was removed, the sample was redissolved in 0.25 mL of dichloromethane and the solvent evaporated again under N2. Following this, 1 mL of acetone, triethylamine and acetic anhydride (5:2:1 v/v/v) was added and the sample heated at 60 °C for 10 min and once cooled the reagent were evaporated under N2. Then the sample was redissolved in 2 mL ethyl acetate and 1 mL of saturated sodium chloride solution was added. After mixing a phase separation was performed by extracting the organic top layer, this step was done three times and the organic phases combined and the solvent again evaporated under N2. These derivatives were then extracted twice with 3 mL of dichloromethane to remove any remaining aqueous reagents, then dried under a gentle stream of N2 at room temperature then redissolved in ethyl acetate and stored at − 20 °C until analysis.
The individual amino acid compounds were then separated using GC and detected using a flame ionization detector (GC-FID) (Agilent Technologies 7890B, CA, USA). The GC was fitted with a DB-35 column (35%-phenyl-methylpolysiloxane, mid-polarity, 30 m × 0.32 mm inner diameter, film 0.5 µm). Helium was used as a carrier gas (2 mL min−1), operating under constant flow conditions. The GC oven was programmed as follows: 70 °C (held for 2 min) to 150 °C at 15 °C min−1, then increasing to 210 °C at 2 °C min−1, then to 270 °C (held for 10 min) at 8 °C min−1. The compounds were identified by their retention times compared with a mixed amino acid standard. The samples were also screened using a GC-Quadrupole-Time-of-Flight mass spectrometer (GC-Q-TOF MS) (Agilent Technologies 7200 Accurate Mass, CA, USA) to confirm the identity of the compounds. The instrument was fitted with the same column as described above for the GC-FID and the GC was operated at constant flow, using helium as the carrier gas at 1.5 mL min−1. The GC oven was set to the same temperature programme described above. The MS was operated in electron ionization (EI) mode at 70 eV, the source temperature was 230 °C and the emission current was 3.1 µA. Data acquisition was between 50 and 1050 amu at 5 spectra s−1.
Neutral carbohydrates
Neutral carbohydrates were extracted from approximately 10 mg of freeze-dried residue from the filtered river water. The samples, along with an internal standard (20 µL of myo-inositol, 450 µg mL−1), were subjected to strong acid hydrolysis using H2SO4 (12 M) at room temperature for 1 h under N2, followed by mild acid hydrolysis (H2SO4 1.2 M, 100 °C, 3 h), also under N2. The two-step hydrolysis is designed to breakdown any α-cellulose which may have been introduced from vascular plant material into glucose. The hydrolysates were separated by centrifugation, then the solution was neutralised to pH ~ 6 with CaCO3. NaBH4 0.5 mg mL−1 was added to the supernatant, and the mixture heated to 40 °C for 90 min to reduce the neutral sugars. Any remaining NaBH4 was then removed by adding glacial acetic acid (200 µL) and methanol (4 × 4 mL) and then dried under a gentle stream of N2. Acetic anhydride (2 mL) was added, and the mixture heated to 100 °C for 16–18 h to acetylate the alditols. Any excess reagent was quenched by the addition of 5 mL of double distilled water, then the acetylated alditols were extracted using dichloromethane (4 × 2 mL), dried under N2, redissolved in ethyl acetate, and stored at -20 °C until analysis.
The acetylated alditols were separated and detected using GC-FID (Hewlett-Packard 5890 Series II, TX, USA). A VF23-ms column (cyanopropyl, high polarity, 60 m × 0.32 mm inner diameter, film 0.15 µm) was used for the separation. Helium was used as the carrier gas (2 mL min−1), operating under constant pressure. The GC oven was programmed as follows: 50 °C (held for 1 min) to 200 °C at 20 °C min−1, then to 230 °C (held for 22 min) at 4 °C min−1. Compounds were identified by their retention times compared with individual standard solutions. The samples were also screened using the GC-Q-TOF MS to confirm their identification. The GC was set up the same as for the GC-FID, using the same column and oven conditions. The MS was set up as described for the AA analysis above.
Polar intermediate molecular weight compounds
The analysis of the polar, intermediate molecular weight fraction follows that of Pemberton et al. (2020). The compounds in this fraction can be described as polar but have lower polarity compared with amino acids and carbohydrates described above. One litre of filtered river water was acidified to pH 2 using HCl and then the DOM was pre-concentrated using Oasis Hydrophilic–Lipophilic Balance (HLB) solid phase extraction cartridges (SPE, 400 mg bed mass, 60 μm particle size, Waters Ltd, Elstree, UK). Extracts were then eluted from the cartridges using methanol and dried under N2. The samples were then redissolved in methanol water (1:1, v/v) before being analysed. Direct-infusion High Resolution MS (DI-HRMS) spectra were recorded in positive ion mode using an Orbitrap™ Elite Hybrid Ion Trap‐Orbitrap™ Mass Spectrometer (Thermo Scientific, Hemel Hempstead, UK) with a heated electrospray ionisation (HESI) source. Electrospray ionisation (ESI) is a soft ionisation method that yields almost no fragmentation so each ion is a molecular species corresponding to either protonated adducts [M + H]+, or Na+ or K+ adducts. Thus, ESI mass spectra constitute distributions of molecular species, complicated by adductions. These can be recognised by the mass defects of 22 and 38 between the [M + Na]+ and [M + K]+ adducts and [M + H]+ adducts, respectively. Data handling protocols are as described by Pemberton et al. (2020).
Macro-molecular material
The analysis of macro-molecular material provides insight into higher molecular weight fraction which will likely include proteinaceous, polysaccharide and lignin-derived components. Previous studies have investigated the presence of lignin-phenols in water by isolating the compounds using solid phase extraction, followed by analysis via GC/MS or pyrolysis-GC/MS (Louchouarn et al. 2000; Williams et al. 2016). This method is very effective for specifically targeting lignin derived compounds but if a more comprehensive, untargeted approach is desired then a different pre-concentration procedure is necessary. Therefore, in this study ‘raw’ filtered river water was freeze-dried to dryness and ~ 1 mg of the residue was weighed into pre-combusted (1000 °C for 4 h) quartz tubes. An internal standard, 2 µL of 5α-androstane (0.01 µg mL−1), was added. Samples were then pyrolysed at 610 °C for 20 s at 20 °C ms−1, using a pyroprobe (CDS Analytical 6200, PA, USA). The pyrolysis products were transferred to a temperature programmable vaporisation inlet (transfer line temperature was maintained at 310 °C) which was operated at a temperature of 300 °C with a split ratio of 10:1. The products were separated using a Trace 1310 GC (Thermo Scientific, Bremen, Germany), which was fitted with a Rtx-1 column (dimethyl polysiloxane, non-polar, 60 m × 0.32 mm ID, film 0.25 µm). The GC was operated under constant flow (He, 2 mL min−1) using a temperature programme as follows: 40 °C (held for 4 min) to 300 °C at 4 °C min−1 and held for 15 min. The separated compounds were then analysed using an ISQ7000 mass spectrometer (Thermo Scientific, Bremen, Germany) using electron ionisation at 70 eV; the source temperature was 300 °C and the emission current was 50 µA. Data were acquired between 50 and 650 amu at 0.2 scan s−1). Samples were analysed both with and without thermochemolysis (when 5 µL of tetramethylammonium hydroxide (TMAH) (25% in methanol, Sigma Aldrich) was added to the samples prior to analysis).
Results and discussion
Non-polar fraction
Figure 3 shows total ion chromatograms gained from the analysis of the non-polar fractions from each of the three contrasting field sites. The three sites show some common compounds in the form of fatty acids (generic structure is shown in Fig. 4a i), particularly palmitic and stearic acid (C16 and C18) most likely derived from plant material surrounding the sample locations as these make up the largest proportion of the fatty acids in higher plants (Tjellström et al. 2012; Meï et al. 2015), or from in-stream aquatic productivity (Sushchik et al. 2010). The chromatogram from Nant y Brwyn displays the widest range of straight-chain fatty acids where a series of 9 homologues can be observed ranging from C9 to C24. Also, the C9 dicarboxylic acid (azelaic acid) is present (generic structure shown in Fig. 4a ii). These compounds have previously been identified in earlier research examining the lipidic oxidation products of peat (Hanninen 1986). Recent molecular characterisation of peat carried out by Vinci et al. (2020) showed that the majority of the organo-soluble fraction of peat was comprised of saturated long-chain fatty acids, with C16 and C18 being the most prevalent. This finding is reflected in the river water draining through the peat landscape in this study. In addition to these fatty acids, two steroidal compounds which elute in the last 10 min of the GC analysis, were identified as cholesterol (C27) (Fig a iii) and sitosterol (C29) (Fig a iv). Sitosterol is a common plant sterol and cholesterol could have algal origins (Patterson 1971). These compounds along with the organic acids are indicative of plant material which will comprise most of the organic matter input to this peat headwater stream. This distribution of compounds can be contrasted with those seen in the chromatograms from the two intensively farmed and populated catchments.

Total ion chromatograms of the non-polar fraction of DOM from the a River Conwy at Nant y Brwyn, b River Sem at Priors Farm and c River Chew at Chew Stoke

Chemical structures identified in a the non-polar, polar intermediate molecular weight and macro-molecular, b amino acid and c neutral carbohydate fractions
At Priors Farm, a range of saturated long-chain fatty acids are observed (C8, C9, C16, C18) along with the C26 alcohol (Fig. 4a v) which are indicative of the grassland character of the field location. While the C26 alcohol was the only identifiable alcohol at this site it is well-established in the literature that the C26 is the dominant fatty acid observed in grassland and grazed sites (van Bergen et al. 1997; Otto et al. 2005; Trendel et al. 2010) and therefore likely the only compound which was abundant enough to be detected in the river water draining through this intensively grazed grassland catchment. In addition, there are some key diagnostic compounds which reflect cattle grazing, such as urea which is abundant in cattle urine and slurries and the steroidal compound 5ẞ-stigmastanol (Fig. 4a vi), which is only produced via biohydrogenation of plant matter in the gut during rumination (Bull et al. 2002; Lloyd et al. 2012). Also visible on the chromatogram is a slight hump of yet unresolved material between 19 and 24 min, this highlights the complexity of the DOM signal, and this would be a target for future research.
At Chew Stoke, the fatty acids are less dominant, with only C9, C16 and C18 detectable. Instead, the some of the most abundant compounds are the steroidal biomarkers eluting between 25 and 28 min. The most abundant of these is cholesterol, followed by coprostanol (Fig. 4a vii), a C27 stanol produced by the biohydrogenation of cholesterol in higher animals and therefore an important biomarker of human sewage (Leeming et al. 1996; Bull et al. 2002). Unlike the other two sites, the sampling site on the River Chew is immediately downstream from a major sewage treatment works (STW) at Chew Stoke. The presence of stearamide (Fig. 4a viii), a fatty amide of stearic acid, is also indicative of human activity with potential origins from industrial products such as adhesives or additives from plastic packaging (Lau and Wong 2000; Hahladakis et al. 2018). Fatty acid amides are the most commonly used slipping agents in plastic films and stearamide has been shown to readily migrate (up to 95%) from low density polyethylene film packaging into food (Cooper and Tice 1995). Further evidence of anthropogenic chemicals in the river is provided by the presence of tris(1-chloro-2-propyl)phosphate (TCIPP) (Fig. 4a ix), which may originate from industrial processing as it is a flame retardant commonly used in polymers such polyurethane foams (Van den Eede et al. 2016). Its use has been increasing since the ban on brominated flame retardants and as a result it has been reported as an emerging contaminant in the natural environment (Van der Veen and de Boer 2012). Phosphate-based flame retardants are not normally chemically bonded into the manufactured materials and therefore are easily leached into the environment (Rodriguez et al. 2006). These industrially produced compounds are not observed at either of the other sites further supporting the argument that these anthropogenic contaminants are being introduced to the river via the final effluent discharged from the Chew Stoke STW.
Polar building block compounds
Amino acids
The distribution of amino acids measured for the three sites is provided in Fig. 5a (the chemical structures are shown in Fig. 4b). A total of 14 hydrolysed amino acids were identified (including the internal standard norleucine) from two of the freeze-dried river water samples, from Conwy and Priors Farm. Compound identification was confirmed using the GC-Q-TOF/MS. The total hydrolysable amino acid pool was an order of magnitude higher in the Conwy compared with Priors Farm, measuring 0.46 mg L−1 (58 µg N L−1, 0.17 mg C L−1, ~ 5% DOC) and 0.04 mg L−1 (5.1 µg N L−1, 5 µg C L−1, ~ 0.3% DOC), respectively, suggesting that that the DOM at Priors Farm is more degraded and perhaps proteins are more rapidly utilised by an active and/or larger microbial community. This is supported by further work conducted by Brailsford et al. (2021) where water was collected from both Nant y Brwyn and Priors Farm and 14C-labelled amino acids were added in an incubation experiment. The results showed after 24 h double the amount of labelled amino acids had been degraded in the Priors Farm water compared with Nant y Brwyn, suggesting that turnover rates of these compounds were greater in the agricultural catchment. However, further experiments would be needed to confirm this holds for complex proteins. Figure 4a shows the distribution of the amino acids as a percentage of the total amino acid pool at the two field sites. While the distributions are not wholly dissimilar there are some key differences, with threonine and serine and lysine notably more prominent in the Nant y Brwyn, whereas alanine, glycine and proline are more prominent at Priors Farm. In general the distribution of amino acids at Nant y Brwyn are in agreement with reported compositions for peatland sites. Silvester et al. (2021) show the most abundant amino acids in a peatland river as glycine, aspartic acid, glutamic acid, alanine, serine, and threonine, which is also true at Nant y Brwyn. The Nant y Brwyn sample did show an increased relative abundance of lysine compared with the literature, although this isan important amino acid in sphagnum moss which is the dominant vegetation at the field site, along with sedges. Glutamic acid, glycine, alanine, and aspartic acid have been shown previously as the most dominant amino acids in DOC (Hedges et al. 1994; Dittmar et al. 2001), however, at Priors Farm the relative abundance of leucine, valine and proline were also increased compared with Nant y Brwyn. These three amino acids are important components (along with glutamic acid, glycine, alanine and aspartic acid) in cow manure (Chen et al. 2003) suggesting that their prominence in the DOM profile reflects the intensive cattle production in this catchment, compared to low intensity sheep grazing in the Nant y Brwyn catchment.

Plots showing the distribution of a total hydrolysable amino acids and b total neutral carbohydrates at the three field sites (Note for Chew Stoke amino acids were undetectable using the current method)
Overall, the total amino acid concentrations and the relative contributions of each compound at each site agrees with other field sites reported in the literature and reflect the environments from which the samples were collected, demonstrating the sensitivity of this approach to characterising DOM composition from catchments of contrasting environmental character and anthropogenic disturbance. By contrast, despite repeated attempts, at Chew Stoke only a small number of the amino acids were visible from the GC analysis and importantly the internal standard added was lower than would be expected and therefore suggesting interference with the derivatisation process. While the exact cause is not yet known, possible reasons for this could be the high concentrations of inorganic components in the sample compared with the other field sites, which may cause interference with the extraction or derivatisation steps in the protocol. The sample when freeze-dried, yielded a mass of residue an order of magnitude greater than the other field sites, while the DOC concentration at the site is the lowest (5 ± 0.53 mg L−1), suggesting a large quantity of inorganic material was present. In addition, the wide range of anthropogenically-derived compounds in the river close to the sewage outfall could have hitherto unknown impacts of the derivatisation chemistry, for example metals or pharmaceuticals (Pemberton et al. 2020). Further method testing is needed here to allow the method to be used at such field sites which have more complex, anthropogenically-modified background chemistries.
Neutral carbohydrates
A total of 9 neutral carbohydrates (including myo-inositol as an internal standard) were detected at all three field sites (Figs. 4c and 5b), the identity of each compound being confirmed by comparing the chromatographic retention time with standards run of each individual compound. Nant y Brwyn shows the highest concentrations, with a total carbohydrate concentration of 1.58 mg L−1 (0.6 mg C L−1, ~ 16% DOC) of river water, compared with 0.53 mg L−1 (0.2 mg C L−1, ~ 10% DOC) and 0.15 mg L−1 (0.06 mg C L−1, ~ 1% DOC) for Chew Stoke and Priors Farm, respectively. The high concentrations at the peatland site are likely due to the continual leaching of carbohydrates from the organic soils surrounding the river (Jia et al. 2008). At the anthropogenically disturbed field sites, as with the amino acids, the lower concentrations might be because of the presence of a more active or larger microbial pool which rapidly utilises the organic pool instream. Alternatively, the residence time of the inputs might be playing a role: if the material has travelled further before being sampled there will have been more time for degradation processes to have occurred. Figure 4b shows the distribution of the neutral carbohydrates as a percentage of the total pool. Like the amino acid pool, the distribution is broadly similar across the sites but with some compounds showing variations. Previous literature has shown that the source of individual neutral sugars is non-specific (Cowie & Hedges 1984; Guggenberger et al. 1994a, b; Dungait et al. 2009), but when used in combination they do have some predictive power. For example, arabinose and xylose are particularly enhanced in the Conwy site and these compounds are important constituents of plant material (Oades 1984; Guggenberger et al. 1994a), and less readily synthesised by microbes. There is evidence that deoxysugars, fucose and rhamnose can be microbially derived (Guggenberger et al. 1994b; Biersmith & Benner 1998), and these compounds are relatively more abundant at both Priors Farm and Chew Stoke where the microbial communities are hypothesised to be particularly active because of the contribution of farm and human wastes. This is supported by the microbial response to 14C-labelled tracers introduced to water and sediments in the earlier work of Brailsford and colleagues in both the Conwy catchment, including Nant y Brwyn (Brailsford et al. 2019, 2021) and Hampshire Avon (including Priors Farm) catchment (Brailsford et al. 2021).
It has been suggested that the ratio between the neutral carbohydrate components (FUC + RHA/ARA + XYL) can be used to assess the contribution of plant and microbial sources in any environmental medium, with a ratio of 0.5 or lower indicative of plant-derived carbohydrates and values around 2 being microbially-derived (Oades 1984; Guggenberger et al. 1994a). The ratio for the sites studied here are 0.47, 1.17 and 2.76 for Nant y Brwyn, Priors Farm and Chew Stoke respectively, confirming the shift from plant-derived carbohydrates at the peatland site towards an increasingly microbially-derived composition in the agricultural and sewage impacted sites. It has also been suggested that the ratio of hexoses to pentoses can be used to distinguish between terrestrial plant (higher hexose) and phytoplankton (higher pentose) contributions (Cowie and Hedges 1984; Ittekkot and Arain 1986). The hexose/pentose ratios for the sites are 1.69, 3.0 and 6.55 for the Nant y Brwyn, Priors Farm and Chew Stoke respectively, further supporting the hypothesis that there is a decrease in the relative proportion of plant-derived compounds in the DOM pool in the two anthropogenically disturbed catchments.
Polar intermediate molecular weight compounds
DI-HRMS was carried out at each of the three field sites, but the extract from the Nant y Brwyn showed contamination of the sample during collection most likely caused by a rope used to lower the sample container into the river, which was necessary because of high water levels making access to the water difficult (for a full discussion of this issue see (Lloyd et al. In press). As a result, an alternative example is shown here from another well-researched comparative headwater peatland site in mid-Wales draining into Llyn Brianne at Nant Esgair Garn (hereafter Nant Esgair Garn). Yates (2021) (this volume) provide full details of the field site, its catchment and background nutrient chemistry. Figure 6 shows the DI-HRMS mass spectra recorded for the DOM extracts from a) Nant Esgair Garn, b) Priors Farm and c) Chew Stoke. The extraction efficiency was calculated by measuring the DOC concentration of the extracts following SPE and comparing it to the concentration measured in the raw filtered river water (as NPOC using a TOC-L analyser (Shimadzu, Milton Keynes, UK)). The extraction efficiency of the samples from Nant Esgair Garn and Priors Farm were both 58%, whereas it was lower for Chew Stoke, at 40.5%, meaning that roughly half of the total C in the samples never reaches the mass spectrometer, and some compounds again might not be detected due to inefficiencies in the electrospray ionisation (Patriarca et al. 2020). The ions were extracted from each mass spectrum and yielded a similar number of ions, with 3601, 3201 and 3158 ions for Nant Esgair Garn, Priors Farm and Chew Stoke, respectively. Each of these peaks have the potential to be individual compounds in the sample, although a number could represent isotope peaks of the same compound. The sample from Nant Esgair Garn shows almost a bimodal distribution of ions with a cluster of compounds between m/z 150 and 250 and another cluster between m/z 250 and 500, with major ions (defined as > 50% relative intensity) occurring at m/z values of 267.1699, 156.1019, 376.2629, 170.1174 and 163.0697. In contrast, the sample from Priors Farm shows one Gaussian-like distribution of ions from m/z 150 to 500. There were three major ions present with m/z values of 165.0705, 431.2258 and 288.1961. The total ion current for Priors Farm was the smallest of all the samples (3.2E7), despite the DOC concentration of the water and the extraction efficiency being the highest, suggesting that the compounds present were more difficult to ionise using electrospray. The sample from Chew Stoke displayed a higher total ion current (6.6E7), suggesting that it contained compounds which were easily ionisable via electrospray. The samples exhibited a gaussian-like distribution of ions from m/z 250–750, with a modal value of m/z 447.2921, however, the ions extend beyond m/z 1000. At first glance it appears like there may be significantly fewer ions present in the sample compared with the others, however, this is not the case. The main difference is that the mass spectrum is dominated by overlapping series of prominent ions with a spacing of m/z 58 units, indicative of a mixture of oligomeric components. These oligomeric series were investigated in more detail by Pemberton et al. (2020), where they were identified as 18 separate overlapping series of polypropylene glycol (PPG) (Fig. 4a x). Pemberton et al. (2020), through further interrogation of this sample using HPLC/HRMS/MS, were able to identify 22 other compounds including pharmaceuticals such as codeine, propranolol and cocaine (Fig. 4a xi, xii, xiii). The presence of the pharmaceutical and the PPG, commonly used as an additive in food and personal care products amongst other uses, reflects the influence input of DOM the sewage treatment works is having on the DOM character in the adjacent river.

DI-HRMS spectra recorded for a Llyn Brianne at Nant Esgair Garn, b River Sem at Priors Farm and c River Chew at Chew Stoke catchments
While it was beyond the scope of this paper to identify individual compounds from all the HRMS samples, this was carried out for the Chew Stoke site in our previous paper (Pemberton et al. 2020) and full details can be found therein. It was possible here to compare the number of common masses between the sites to assess the level of similarity between the DOM constituents. Figure 7 presents a Venn diagram of the three sites which shows that the three samples share 2147 ions, suggesting that these represent ubiquitous, and potentially natural, organic matter. Priors Farm only exhibited 3 unique ions in this analytical fraction but shared 114 ions with the sample from Nant Esgair Garn which were absent from Chew Stoke. Nant Esgair Garn showed 25 unique ions which did not appear in either of the other sampling sites, suggesting that these represent peat-derived DOM compounds. This site shared 148 ions with the sample collected from Chew Stoke, which were absent in the Priors Farm sample. The identification of these compounds and their likely provenance in each catchment, using source fingerprinting approaches would be interesting avenue of further study. As could be expected the largest number of unique ions was recorded for Chew Stoke (68), this is logical given the input of DOM from the sewage works which is not present at either of the other two sites. Further analysis of these samples to isolate and identify the ions unique to each of the sites would be beneficial to better understand the specific origins of DOM compounds in each stream system.

Venn diagram showing the number of unique and shared ions detected via DI-HRMS at the three field sites
Macro-molecular material
Figure 8 shows pyrograms recorded for each of the three field sites, which also highlight differences in the character of the DOM in each catchment. While py-GC/MS is a semi-quantitative technique, by comparing the size of the 5α-androstane (IS) peak in each sample the relative abundance of DOM visible by the technique at different sites can be compared. The highest concentration of macro-molecular DOM can be seen in Nant y Brwyn, followed by Priors Farm then Chew Stoke, which reflects the DOC measurements at the field sites (See Table 1). Thus, both the quantity of DOM and the composition of the pool varies greatly between sites.

Pyrograms showing the pyrolysis products from the macro-molecular DOM for a River Conwy at Nant y Brwyn, b River Sem at Priors Farm and c River Chew at Chew Stoke field sites. The superscripts on the lignin compounds denote p-hydroxyphenyl (P), guaiacyl (G) and syringyl (S) lignin units
The pyrogram from Nant y Brwyn (Fig. 8a) shows pyrolysis products after thermochemolysis, which was dominated by lignin derived compounds, which make up the majority of plant cell walls. Twenty individual compounds can be identified from the three lignin classes, determined by their phenylpropanoid units (p-hydroxyphenyl (P), guaiacyl (G) and syringyl (S) units). This illustrates the importance of plant sources contributing to the riverine DOM signal. This lignin dominated signature is typical of peat landscapes where sedges are present and has been reported previously in the literature (e.g. McClymont et al. 2011; Abbott et al. 2013). Alongside the lignin components, the two most abundant straight-chain fatty acids (C16 and C18), which were seen in the non-polar fraction are also visible. In an analysis of peat samples, Abbott et al. (2013) also reported C16 fatty acid, along with 1,2,4-trimethoxybenzene (Fig. 4a xiv), which was also seen in the present study. It should be noted that the fatty acids seen here are potentially being volatilised in the samples, rather than pyrolysis products of macro-molecular material and are likely the same source as seen in the non-polar fraction. 1,2,4-trimethoxybenzene has been shown to be produced during TMAH thermochemolysis of both carbohydrates (Fabbri & Helleur 1999) and condensed tannins (Nierop et al. 2005). A small peak assigned to methylated 4-isopropenylphenol (Fig. 4a xv) was also identified in the sample, which has been shown to be a biomarker for Sphagnum mosses (van der Heijden et al. 1997; Abbott et al. 2013). Also recorded were a small number of compounds likely of polysaccharide origin, or N-containing components, as well as toluene, phenol and 3-methyl phenol (Fig. 4a xvi, xvii, xviii), which are often reported as being of unspecific origin (Dignac et al. 2005).
In contrast to Nant y Brwyn, Priors Farm (Fig. 8b) shows a different array of pyrolysis products, the majority of which are lower molecular weight compounds. Unlike the sample from the Conwy, no additional compounds were observed when TMAH thermochemolysis was carried out. There was only one potentially lignin-derived component, 2,3-dimethoxy phenol (Fig. 4a xix) which could be gymnosperm or angiosperm monocot in origin (Ralph et al. 2004), which is likely given that Priors Farm has a grassland-dominated catchment with large areas of woodland in its headwaters. The most abundant compounds in the pyrogram are phenolic or N-containing compounds. These may be indicative of the presence of proteinaceous material in the river, as these compounds have been previous linked to polypeptides (Bracewell and Robertson 1984), which could have been introduced through intensive cattle grazing in the catchment. These compounds have also been observed in the top soil layers sampled from permanent pasture (Nierop et al. 2001) and have been attributed to amino acids, amino sugars, proteins and polypeptides (e.g. Sorge et al. 1993; Leinweber and Schulten 1998; van Bergen et al. 1998) There were also more polysaccharide derived products such as 2,5-dimethylfuran and 3-Methyl-1H-pyrrole (Fig. 4a xx, xxi), which could be of plant or animal waste origin. Overall, the DOM pool at Priors Farm contains a higher proportion of microbially-degraded, lower molecular weight compounds, and contains key compounds indicative of anthropogenic disturbance of land cover, and intensive agricultural production in its catchment.
The DOM pool sampled at the Chew Stoke yielded the lowest number of pyrolysis products suggesting the presence of less macromolecular material compared with the other two field sites. While there were a small number of compounds which were likely to be of protein (e.g. benzonitrile, phenol (Fig. 4a xxii, xvii)) or polysaccharide (e.g. 3-furaldehyde, 5-methyl 2-furancarboxaldehyde (Fig. 4a xxiii, xxiv)) origin, the pyrogram was dominated a series of dicarboxylic acids from C4 through to C8. They were the most abundant compounds aside from the straight-chain fatty acids (C16 and C18), which were ubiquitous across the three sites. This series of dicarboxylic acids is most likely anthropogenic in origin, illustrating the impact that the processed sewage effluent is having on the DOM character in the river at this site. The exact origin of the dicarboxylic acids present, namely succinic, glutaric, adipic, pimelic, suberic and azelaic acids, is currently unknown. However, these compounds are used in manufacture of synthetic polymers and additives for plastics and can be thermal and photo-oxidation degradation products of polyethylene (Hakkarainen and Albertsson 2004). It is therefore possible that these compounds have been produced simply as pyrolysis products of micro- or nano-plastics within the waterbody, or during treatment processes at the STW. Dicarboxylic acids have been shown to be abundant in aerosol samples associated the burning of plastic-enriched waste (Kumar et al. 2015) which may well occur in this populated catchment. Alternatively, the compounds may be the pyrolysis products of already partially degraded plastic which has been the subject of photo- and microbially mediated-oxidation potentially during the process of sewage treatment. This theory of the compounds being polymer-derived is also supported by data presented by Pemberton et al. (2020) who reported a number of oligomeric series in the polar DOM fraction at the same site.
Implications and future directions
The use of a range of complementary methods, as presented here, allows the analytical window to be widened to provide a more comprehensive picture of the character of DOM present in rivers. The data presented from the three contrasting case studies show that the techniques were able to identify a wide range of riverine DOM compounds, some of which would not have been identified through targeted analysis. The analysis of the non-polar fraction revealed some important diagnostic biomarker compounds, which reflected the vegetation and land cover and nature of anthropogenic disturbance, if any, in the catchment for each site. By using the distribution of sterols and stanols, faecal inputs were detected, and the specific source could be distinguished. The non-polar fraction is often overlooked when characterising DOM because compounds in this class are hydrophobic and therefore usually associated with the analysis of sediment or suspended sediment, however, they provide useful supporting information concerning DOM sources. The biomarkers identified in this fraction can provide unequivocal evidence for the contributing sources of the DOM, for example in the nature of the faecal indicator compounds as illustrated in this study.
The analysis of amino acids and carbohydrates provides information about important building block compounds, which are key ecosystem drivers. They are generally quite low in abundance in the dissolved fraction; however, this is likely because they are turned over very quickly by the stream biota, rather than an indication that they are of low importance. Both the overall quantity and the distribution of amino acids and carbohydrates were shown to vary across the range of field sites providing insights into the potential for DOM turnover by microbial communities as well as the contribution of plant and microbial sources to the carbohydrate pool.
Using DI-HRMS the complexity of the intermediate molecular weight (mostly m/z 150- 800) polar fraction was clearly shown. Thousands of ions were detected, and by comparing the distribution of ions between the three field sites many common molecular species were observed. This provides useful information about DOM components which are likely to be ubiquitous across different landscapes. In addition, ions detected which were unique to particular field sites and these ions would make interesting targets for identification and as specific source indicators in support of any source apportionment study.
The characterisation of the riverine DOM was further extended in our approach by using py-GC/MS to identify the sources of macro-molecular material within the DOM pool. Pyrolysis is a technique which is mainly used to analyse solid samples with the exception of lignin phenols in river water, particularly soils, sediments and vegetation yielded some surprising products. The advantage of using the freeze-dried residue of the whole water sample means that the pre-concentration step is not selective in the DOM recovery and therefore provides a more complete picture of the macro-molecular material within the river water samples.
While the application of this suite of methodologies has improved our ability to characterise DOM there are a number of remaining challenges in this field still to be addressed. One of the most fundamental of these is quantification. Being able to understand DOM quantitatively as well as qualitatively is the next logical step but for many of the complex mixtures described here it is non-trivial. The non-polar fraction could be quantified using a carefully chosen range of internal standards once the type of compounds present is known. The amino acid and carbohydrate fractions can be quantified using a combination of internal and external standards whereby the response factor for each individual compound in the FID can be ascertained, because each will respond differently on combustion. The greatest quantification challenges however remain when considering the DI-HRMS and pyrolysis methods. In the case of electrospray ionisation for DI-HRMS the response seen in the mass spectrometer will be controlled by the ability of each compound to ionise, therefore compounds may be present which are very abundant in the sample but difficult to ionise and, thus show a low signal when analysed and vice versa. There are several approaches which could be attempted to tackle these quantification challenges. One is using an external calibration curve run on the instrument in the same batch as the samples. However, this would have to be done for each compound of interest. Another approach would be to use standard addition, whereby samples are analysed twice with and without a spike of known concentration of target compounds and the concentration calculated based on the change in response between the mass spectra. This method also requires target compounds for quantification; however, it has the advantage that you can target multiple compounds per analytical run. The standard addition approach also has the advantage of taking account of matrix effects which may supress or enhance molecular responses. Once quantification can be robustly carried out the power of these analytical techniques will be enhanced further, and research questions based around source apportionment and ecological impact can be better addressed.
Further to quantification, there is also the challenge of defining the remaining uncharacterised DOM. There are a number of candidate compounds classes which could be added to the suite of techniques discussed herein, for example dissolved black carbon. The identity of the remaining DOM composition is a significant challenge for the scientific community to address as research questions demand. Also, more work is necessary to extend our understanding of the exact origins of the compounds seen in the analyses performed here. For example, in the non-polar fraction is the pool limited to free fatty acids? or are there ester-bound moieties present which could be captured using an additional hydrolysis step.
The other important remaining question relates to the current definition of DOM, that is, any material which passes through a 0.45 µm filter. In reality, it can be argued that organic matter should be considered a continuum, ranging from particulates of various sizes through to colloidal material and then eventually compounds that are truly dissolved. There are examples of compounds identified in this paper, for example the faecal biomarker compounds in the non-polar fraction that mostly likely exist not truly dissolved in the water but exist either as free-floating organics or associated with colloidal particles in the sub-0.45 µm fraction. With this in mind, along with the fact that the particulates are known to play an important role in nutrient cycling and ecosystem function (see for example Mena-Rivera et al. (2022) for a detailed discussion) perhaps it is most appropriate to move away from the concept of operationally defined DOM and consider all the organic matter within a particular sample window. The methodologies presented in this paper can be applied directly, or with small modifications, to particulate samples or to unfiltered river water samples containing particles along with dissolved components to extend further the analytical window.
Conclusions
Overall, these contrasting case studies illustrate the analytical power of a complementary suite of approaches to characterise DOM pool composition in environmental matrices, specifically here for river water samples. A similar approach has been used previously for expanding the characterisation of soil organic matter (Poirier et al. 2005). Figure 9 shows a schematic of how the theoretical analytical windows of methods used herein fit together, with some compounds being identifiable by multiple techniques. The diagram also illustrates that although using this approach has greatly extended our ability to comprehensively characterise the DOM pool, some components will remain uncharacterised. However, the data presented here clearly demonstrates the applicability of the methods to distinguish DOM source character and contributions as these vary according to environmental character and anthropogenic disturbance. The identification of unexpected compounds, for example the dicarboxylic acids and PPG oligomers at Chew Stoke clearly illustrate the advantage of adopting this type of analytical approach over targeting specific compounds known to be of importance. In addition, the suite of approaches presented here, in combination with the characterisation of the polar fraction as presented in Pemberton et al. (2020) highlights the extreme complexity of the DOM pool at molecular level and therefore how important it is to characterise this pool robustly in order to better understand its composition and likely role in stream ecosystems and their metabolic function. Furthermore, through the elucidation of the DOM character in stream and source samples using the methods presented here, information can be collected about the likely sources of DOM within a catchment, enabling the targeting of mitigation measures at key contributing sources within any catchment nutrient management plan. The identification of individual key compounds within the DOM pool at a particular site can also be used in the design of robust and informative bioassay experiments, to understand the ecosystem responses to instream DOM as this varies not only in space and time, but in terms of its molecular composition. This is critical if we are to fully understand the role of DOM as a bioavailable nutrient resource in freshwater ecosystems.

Schematic of the analytical windows of the methods applied using the hierarchical MS analytical approach (Note that the size of the circles is not proportional to the amount of DOM represented)
Data availability
Enquiries about data availability should be directed to the authors.
References
Abbott GD, Swain EY, Muhammad AB, Allton K, Belyea LR, Laing CG, Cowie GL (2013) Effect of water-table fluctuations on the degradation of Sphagnum phenols in surficial peats. Geochim Cosmochim Acta 106:177–191
Aiken GR, Hsu-Kim H, Ryan JN (2011) Influence of dissolved organic matter on the environmental fate of metals, nanoparticles, and colloids. Environ Sci Technol 45(8):3196–3201
Andresen JA, Grundmann A, Bester K (2004) Organophosphorus flame retardants and plasticisers in surface waters. Sci Total Environ 332(1):155–166
Ashton D, Hilton M, Thomas KV (2004) Investigating the environmental transport of human pharmaceuticals to streams in the United Kingdom. Sci Total Environ 333(1):167–184
Aud H, Gjertrud H, Per J (2003) Riverine Inputs of Organic Carbon and Nitrogen to Norwegian Coastal Areas. Ambio 32(6):412–417
Baker A (2002) Spectrophotometric discrimination of river dissolved organic matter. Hydrol Process 16(16):3203–3213
Battin TJ, Luyssaert S, Kaplan LA, Aufdenkampe AK, Richter A, Tranvik LJ (2009) The boundless carbon cycle. Nat Geosci 2(9):598–600
Biersmith A, Benner R (1998) Carbohydrates in phytoplankton and freshly produced dissolved organic matter. Mar Chem 63(1):131–144
Bracewell JM, Robertson GW (1984) Quantitative comparison of the nitrogen-containing pyrolysis products and amino acid composition of soil humic acids. J Anal Appl Pyrol 6(1):19–29
Brailsford FL, Glanville HC, Golyshin PN, Johnes PJ, Yates CA, Jones DL (2019) Microbial uptake kinetics of dissolved organic carbon (DOC) compound groups from river water and sediments. Sci Rep 9(1):11229
Brailsford FL, Glanville HC, Marshall MR, Yates CA, Owen AT, Golyshin PN, Johnes PJ, Jones DL (2021) Land cover and nutrient enrichment regulates low-molecular weight dissolved organic matter turnover in freshwater ecosystems. Limnol Oceanogr 66(8):2979–2987
Bull ID, Lockheart MJ, Elhmmali MM, Roberts DJ, Evershed RP (2002) The origin of faeces by means of biomarker detection. Environ Int 27(8):647–654
Bull ID, Van Bergen PF, Poulton PR, Evershed RP (1998) Organic geochemical studies of soils from the Rothamsted Classical Experiments - II, Soils from the Hoosfield Spring Barley Experiment treated with different quantities of manure. Org Geochem 28(1–2):11–26
Charteris AF, Knowles TDJ, Michaelides K, Evershed RP (2016) Compound-specific amino acid 15N stable isotope probing of nitrogen assimilation by the soil microbial biomass using gas chromatography/combustion/isotope ratio mass spectrometry. Rapid Commun Mass Spectrom 30(16):1846–1856
Chen ML, Li CL, Spencer RGM, Maie N, Hur J, McKenna AM, Yan FP (2021) Climatic, land cover, and anthropogenic controls on dissolved organic matter quantity and quality from major alpine rivers across the Himalayan-Tibetan Plateau. Sci Total Environ 754:12
Chen S, Liao W, Liu C, Wen Z, Kincaid RL, Harrison JH, Elliott DC, Brown MD, Solana AE, Stevens DJ (2003) Value-added chemicals from animal manure. In: vol PNNL-14495 5894; 820101000; TRN: US200428%%947. Pacific Northwest National Lab., Richland, WA (US), Environmental Molecular Sciences Laboratory (US), USA
Christensen JB, Jensen DL, Christensen TH (1996) Effect of dissolved organic carbon on the mobility of cadmium, nickel and zinc in leachate polluted groundwater. Water Res 30(12):3037–3049
Cole JJ, Prairie YT, Caraco NF, McDowell WH, Tranvik LJ, Striegl RG, Duarte CM, Kortelainen P, Downing JA, Middelburg JJ, Melack J (2007) Plumbing the global carbon cycle: integrating inland waters into the terrestrial carbon budget. Ecosystems 10(1):172–185
Cooper I, Tice PA (1995) Migration studies on fatty acid amide slip additives from plastics into food simulants. Food Addit Contam 12(2):235–244
Cowie GL, Hedges JI (1984) Carbohydrate sources in a coastal marine environment. Geochim Cosmochim Acta 48(10):2075–2087
Cristale J, Katsoyiannis A, Sweetman AJ, Jones KC, Lacorte S (2013) Occurrence and risk assessment of organophosphorus and brominated flame retardants in the River Aire (UK). Environ Pollut 179:194–200
Croghan D, Khamis K, Bradley C, Van Loon AF, Sadler J, Hannah DM (2021) Combining in-situ fluorometry and distributed rainfall data provides new insights into natural organic matter transport dynamics in an urban river. Sci Total Environ 755:12
Dignac MF, Houot S, Francou C, Derenne S (2005) Pyrolytic study of compost and waste organic matter. Org Geochem 36(7):1054–1071
Dittmar T (2008) The molecular level determination of black carbon in marine dissolved organic matter. Org Geochem 39(4):396–407
Dittmar T, Fitznar HP, Kattner G (2001) Origin and biogeochemical cycling of organic nitrogen in the eastern Arctic Ocean as evident from D-and L-amino acids. Geochim Cosmochim Acta 65(22):4103–4114
Dulaquais G, Breitenstein J, Waeles M, Marsac R, Riso R (2018) Measuring dissolved organic matter in estuarine and marine waters: size-exclusion chromatography with various detection methods. Environ Chem 15(7):436–449
Dungait JAJ, Bol R, Bull ID, Evershed RP (2009) Tracking the fate of dung-derived carbohydrates in a temperate grassland soil using compound-specific stable isotope analysis. Org Geochem 40(12):1210–1218
Evans CD, Futter MN, Moldan F, Valinia S, Frogbrook Z, Kothawala DN (2017) Variability in organic carbon reactivity across lake residence time and trophic gradients. Nat Geosci 10(11):832–835
Fabbri D, Helleur R (1999) Characterization of the tetramethylammonium hydroxide thermochemolysis products of carbohydrates. J Anal Appl Pyrol 49(1):277–293
Guggenberger G, Christensen BT, Zech W (1994a) Land-use effects on the composition of organic matter in particle-size separates of soil: I. Lignin and carbohydrate signature. Eur J Soil Sci 45(4):449–458
Guggenberger G, Zech W, Schulten H-R (1994b) Formation and mobilization pathways of dissolved organic matter: evidence from chemical structural studies of organic matter fractions in acid forest floor solutions. Org Geochem 21(1):51–66
Hahladakis JN, Velis CA, Weber R, Iacovidou E, Purnell P (2018) An overview of chemical additives present in plastics: migration, release, fate and environmental impact during their use, disposal and recycling. J Hazard Mater 344:179–199
Hakkarainen M, Albertsson A-C (2004) Environmental degradation of polyethylene. In: Albertsson A-C (ed) Long term properties of polyolefins. Springer, Berlin Heidelberg, Berlin, Heidelberg, pp 177–200
Halliday JS, Skeffington AR, Bowes JM, Gozzard E, Newman RJ, Loewenthal M, Palmer-Felgate JE, Jarvie PH, Wade JA (2014) The water quality of the River Enborne, UK: observations from high-frequency monitoring in a rural, Lowland River system. Water 6(1):150–180
Hanninen K (1986) Cupric oxide oxidation products of humic acids from finnish milled peat. In: Fuchsman CH (ed) Peat and Water: Aspects of water retention and dewatering in peat. Springer Netherlands
Hawkes JA, D’Andrilli J, Agar JN, Barrow MP, Berg SM, Catalán N, Chen H, Chu RK, Cole RB, Dittmar T, Gavard R, Gleixner G, Hatcher PG, He C, Hess NJ, Hutchins RHS, Ijaz A, Jones HE, Kew W, Khaksari M, Palacio Lozano DC, Lv J, Mazzoleni LR, Noriega-Ortega BE, Osterholz H, Radoman N, Remucal CK, Schmitt ND, Schum SK, Shi Q, Simon C, Singer G, Sleighter RL, Stubbins A, Thomas MJ, Tolic N, Zhang S, Zito P, Podgorski DC (2020) An international laboratory comparison of dissolved organic matter composition by high resolution mass spectrometry: are we getting the same answer? Limnol Oceanogr Methods 18(6):235–258
Hawkes JA, Sjoberg PJR, Bergquist J, Tranvik LJ (2019) Complexity of dissolved organic matter in the molecular size dimension: insights from coupled size exclusion chromatography electrospray ionisation mass spectrometry. Faraday Discuss 218:52–71
Hedges JI, Cowie GL, Richey JE, Quay PD, Benner R, Strom M, Forsberg BR (1994) Origins and processing of organic matter in the Amazon River as indicated by carbohydrates and amino acids. Limnol Oceanogr 39(4):743–761
Hedges JI, Eglinton G, Hatcher PG, Kirchman DL, Arnosti C, Derenne S, Evershed RP, Kögel-Knabner I, de Leeuw JW, Littke R, Michaelis W, Rullkötter J (2000) The molecularly-uncharacterized component of nonliving organic matter in natural environments. Org Geochem 31(10):945–958
Helms JR, Stubbins A, Ritchie JD, Minor EC, Kieber DJ, Mopper K (2008) Absorption spectral slopes and slope ratios as indicators of molecular weight, source, and photobleaching of chromophoric dissolved organic matter. Limnol Oceanogr 53(3):955–969
Hudson N, Baker A, Reynolds D (2007) Fluorescence analysis of dissolved organic matter in natural, waste and polluted waters—A review. River Res Appl 23(6):631–649
Ike IA, Lee Y, Hur J (2019) Impacts of advanced oxidation processes on disinfection byproducts from dissolved organic matter upon post-chlor(am)ination: a critical review. Chem Eng J 375:121929
Ittekkot V, Arain R (1986) Nature of particulate organic matter in the River Indus, Pakistan. Geochim Cosmochim Acta 50(8):1643–1653
Jia G, Dungait JAJ, Bingham EM, Valiranta M, Korhola A, Evershed RP (2008) Neutral monosaccharides as biomarker proxies for bog-forming plants for application to palaeovegetation reconstruction in ombrotrophic peat deposits. Org Geochem 39(12):1790–1799
Joshua DI, Praveenkumarreddy Y, Prabhasankar VP, D’Souza AP, Yamashita N, Balakrishna K (2020) First report of pharmaceuticals and personal care products in two tropical rivers of southwestern India. Environ Monit Assess 192(8):529
Kaal J, Plaza C, Nierop KGJ, Perez-Rodriguez M, Biester H (2020) Origin of dissolved organic matter in the Harz Mountains (Germany): a thermally assisted hydrolysis and methylation (THM-GC-MS) study. Geoderma 378:16
Kaiser K, Benner R (2012) Characterization of lignin by gas chromatography and mass spectrometry using a simplified CuO oxidation method. Anal Chem 84(1):459–464
Kaiser K, Canedo-Oropeza M, McMahon R, Amon RMW (2017) Origins and transformations of dissolved organic matter in large Arctic rivers. Sci Rep 7(1):13064
Kalbitz K, Popp P, Geyer W, Hanschmann G (1997) β-HCH mobilization in polluted wetland soils as influenced by dissolved organic matter. Sci Total Environ 204(1):37–48
Kamjunke N, Hertkorn N, Harir M, Schmitt-Kopplin P, Griebler C, Brauns M, von Tumpling W, Weitere M, Herzsprung P (2019) Molecular change of dissolved organic matter and patterns of bacterial activity in a stream along a land-use gradient. Water Res 164:13
Kim S, Kim D, Jung MJ (2021) Analysis of environmental organic matters by Ultrahigh-Resolution mass spectrometry-A review on the development of analytical methods. Mass Spectrom Rev 18
Kumar S, Aggarwal SG, Gupta PK, Kawamura K (2015) Investigation of the tracers for plastic-enriched waste burning aerosols. Atmos Environ 108:49–58
Lau O-W, Wong S-K (2000) Contamination in food from packaging material. J Chromatogr A 882(1–2):255–270
Lavonen EE, Gonsior M, Tranvik LJ, Schmitt-Kopplin P, Köhler SJ (2013) Selective chlorination of natural organic matter: identification of previously unknown disinfection byproducts. Environ Sci Technol 47(5):2264–2271
Lee SC, Shin Y, Jeon YJ, Lee EJ, Eom JS, Kim B, Oh NH (2021) Optical properties and C-14 ages of stream DOM from agricultural and forest watersheds during storms. Environ Pollut 272:116412
Leeming R, Ball A, Ashbolt N, Nichols P (1996) Using faecal sterols from humans and animals to distinguish faecal pollution in receiving waters. Water Res 30(12):2893–2900
Leinweber P, Schulten HR (1998) Nonhydrolyzable organic nitrogen in soil size separates from long-term agricultural experiments. Soil Sci Soc Am J 62(2):383–393
Li H, Minor EC (2015) Dissolved organic matter in Lake Superior: insights into the effects of extraction methods on chemical composition. Environ Sci Process Impacts 17(10):1829–1840
Li Y, Harir M, Uhl J, Kanawati B, Lucio M, Smirnov KS, Koch BP, Schmitt-Kopplin P, Hertkorn N (2017) How representative are dissolved organic matter (DOM) extracts? A comprehensive study of sorbent selectivity for DOM isolation. Water Res 116:316–323
Lloyd CEM, Johnes PJ, Freer JE, Carswell AM, Jones JI, Stirling MW, Hodgkinson RA, Richmond C, Collins AL (2019) Determining the sources of nutrient flux to water in headwater catchments: examining the speciation balance to inform the targeting of mitigation measures. Sci Total Environ 648:1179–1200
Lloyd CEM, Johnes PJ, Pemberton JA, Yates CA, Jones D, Evershed RP (In Press) Sampling, storage and laboratory approaches for dissolved organic matter characterisation in freshwaters: moving from nutrient fraction to molecular-scale characterisation. Sci Total Environ 2022:154105. ISSN 0048-9697. https://doi.org/10.1016/j.scitotenv.2022.154105. https://www.sciencedirect.com/science/article/pii/S0048969722011974
Lloyd CEM, Michaelides K, Chadwick DR, Dungait JAJ, Evershed RP (2012) Tracing the flow-driven vertical transport of livestock-derived organic matter through soil using biomarkers. Org Geochem 43:56–66
Louchouarn P, Opsahl S, Benner R (2000) Isolation and quantification of dissolved lignin from natural waters using solid-phase extraction and GC/MS. Anal Chem 72(13):2780–2787
Lundqvist J, von Brömssen C, Rosenmai AK, Ohlsson Å, Le Godec T, Jonsson O, Kreuger J, Oskarsson A (2019) Assessment of pesticides in surface water samples from Swedish agricultural areas by integrated bioanalysis and chemical analysis. Environ Sci Eur 31(1):53
Lynch LM, Sutfin NA, Fegel TS, Boot CM, Covino TP, Wallenstein MD (2019) River channel connectivity shifts metabolite composition and dissolved organic matter chemistry. Nat Commun 10:11
Lytle CR, Perdue EM (1981) Free, proteinaceous, and humic-bound amino acids in river water containing high concentrations of aquatic humus. Environ Sci Technol 15(2):224–228
Ma YM, Li SY (2020) Spatial and temporal comparisons of dissolved organic matter in river systems of the Three Gorges Reservoir region using fluorescence and UV-Visible spectroscopy. Environ Res 189:9
Mackay EB, Feuchtmayr H, De Ville MM, Thackeray SJ, Callaghan N, Marshall M, Rhodes G, Yates CA, Johnes PJ, Maberly SC (2020) Dissolved organic nutrient uptake by riverine phytoplankton varies along a gradient of nutrient enrichment. Sci Total Environ 722:137837
McClymont EL, Bingham EM, Nott CJ, Chambers FM, Pancost RD, Evershed RP (2011) Pyrolysis GC–MS as a rapid screening tool for determination of peat-forming plant composition in cores from ombrotrophic peat. Org Geochem 42(11):1420–1435
Meï C, Michaud M, Cussac M, Albrieux C, Gros V, Maréchal E, Block MA, Jouhet J, Rébeillé F (2015) Levels of polyunsaturated fatty acids correlate with growth rate in plant cell cultures. Sci Rep 5(1):15207
Mena-Rivera L, Lloyd CEM, Reay MK, Goodall T, Read DS, Johnes PJ, Evershed RP (2022) Biogeochemistry (This volume)
Morton RD, Rowland CS, Wood CM, Meek L, Marston CG, Smith GM (2014) Land Cover Map 2007 (1km percentage target class, GB) v1.2. NERC Environmental Information Data Centre. https://doi.org/10.5285/fdf8c8d3-5998-45a5-8431-7f5e6302fc32
Ngo TH, Van D-A, Tran HL, Nakada N, Tanaka H, Huynh TH (2021) Occurrence of pharmaceutical and personal care products in Cau River. Vietnam Environ Sci Pollut Res 28(10):12082–12091
Nierop KGJ, Preston CM, Kaal J (2005) Thermally assisted hydrolysis and methylation of purified tannins from plants. Anal Chem 77(17):5604–5614
Nierop KGJ, Pulleman MM, Marinissen JCY (2001) Management induced organic matter differentiation in grassland and arable soil: a study using pyrolysis techniques. Soil Biol Biochem 33(6):755–764
Oades JM (1984) Soil organic matter and structural stability: mechanisms and implications for management. Plant Soil 76(1):319–337
Otto A, Shunthirasingham C, Simpson MJ (2005) A comparison of plant and microbial biomarkers in grassland soils from the Prairie Ecozone of Canada. Org Geochem 36(3):425–448
Pang Y, Wang K, Sun YG, Zhou YP, Yang SY, Li YY, He C, Shi Q, He D (2021) Linking the unique molecular complexity of dissolved organic matter to flood period in the Yangtze River mainstream. Sci Total Environ 764:142803
Patriarca C, Balderrama A, Može M, Sjöberg PJR, Bergquist J, Tranvik LJ, Hawkes JA (2020) Investigating the ionization of dissolved organic matter by electrospray. Anal Chem 92(20):14210–14218
Patterson GW (1971) The distribution of sterols in algae. Lipids 6(2):120–127
Pemberton JA, Lloyd CEM, Arthur CJ, Johnes PJ, Dickinson M, Charlton AJ, Evershed RP (2020) Untargeted characterisation of dissolved organic matter contributions to rivers from anthropogenic point sources using direct-infusion and high-performance liquid chromatography/Orbitrap mass spectrometry. Rapid Commun Mass Spectrom 34(S4):e8618
Pisani O, Bosch DD, Coffin AW, Endale DM, Liebert D, Strickland TC (2020) Riparian land cover and hydrology influence stream dissolved organic matter composition in an agricultural watershed. Sci Total Environ 717:137165
Poirier N, Sohi SP, Gaunt JL, Mahieu N, Randall EW, Powlson DS, Evershed RP (2005) The chemical composition of measurable soil organic matter pools. Org Geochem 36(8):1174–1189
Prairie YT (2008) Carbocentric limnology: looking back, looking forward. Can J Fish Aquat Sci 65(3):543–548
Prasert T, Ishii Y, Kurisu F, Musikavong C, Phungsai P (2021) Characterization of lower Phong river dissolved organic matters and formations of unknown chlorine dioxide and chlorine disinfection by-products by Orbitrap mass spectrometry. Chemosphere 265:128653
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH, Boerjan W (2004) Lignins: natural polymers from oxidative coupling of 4-hydroxyphenyl- propanoids. Phytochem Rev 3(1):29–60
Riedel T, Zark M, Vähätalo AV, Niggemann J, Spencer RGM, Hernes PJ, Dittmar T (2016) Molecular signatures of biogeochemical transformations in dissolved organic matter from ten world rivers. Front Earth Sci 4:85
Robarts RD, Wicks RJ, Gehr R (1990) Seasonal changes in the dissolved free amino acid and DOC concentrations in a hypertrophic African reservoir and its inflowing rivers. Hydrobiologia 199(3):201–216
Rodriguez I, Calvo F, Quintana JB, Rubi E, Rodil R, Cela R (2006) Suitability of solid-phase microextraction for the determination of organophosphate flame retardants and plasticizers in water samples. J Chromatogr A 1108(2):158–165
Silvester E, Karis T, Yusuf A, Pengelly J, Grover S, Rees GN (2021) Organic carbon and nitrogen dynamics during a peatland storm event: how dissolved combined amino acids reveal the spatial and temporal separation of organic molecules. J Hydrol 597:126191
Simon C, Osterholz H, Koschinsky A, Dittmar T (2019) Riverine mixing at the molecular scale - An ultrahigh-resolution mass spectrometry study on dissolved organic matter and selected metals in the Amazon confluence zone (Manaus, Brazil). Org Geochem 129:45–62
Sorge C, Schnitzer M, Schulten HR (1993) In-source pyrolysis-field ionization mass spectrometry and Curie-point pyrolysis-gas chromatography/mass spectrometry of amino acids in humic substances and soils. Biol Fertil Soils 16(2):100–110
Spencer RGM, Aiken GR, Wickland KP, Striegl RG, Hernes PJ (2008) Seasonal and spatial variability in dissolved organic matter quantity and composition from the Yukon River basin, Alaska. Global Biogeochem Cycles 22(4):4002
Spencer RGM, Kellerman AM, Podgorski DC, Macedo MN, Jankowski K, Nunes D, Neill C (2019) Identifying the molecular signatures of agricultural expansion in amazonian headwater streams. J Geophys Res-Biogeosci 124(6):1637–1650
Stone WW, Gilliom RJ, Ryberg KR (2014) Pesticides in U.S streams and rivers: occurrence and trends during 1992–2011. Environm Sci Technol 48(19):11025–11030
Stubbins A, Niggemann J, Dittmar T (2012) Photo-lability of deep ocean dissolved black carbon. Biogeosciences 9(5):1661–1670
Stubbins A, Spencer RGM, Chen H, Hatcher PG, Mopper K, Hernes PJ, Mwamba VL, Mangangu AM, Wabakanghanzi JN, Six J (2010) Illuminated darkness: molecular signatures of Congo River dissolved organic matter and its photochemical alteration as revealed by ultrahigh precision mass spectrometry. Limnol Oceanogr 55(4):1467–1477
Stubbins A, Spencer RGM, Mann PJ, Holmes RM, McClelland JW, Niggemann J, Dittmar T (2015) Utilizing colored dissolved organic matter to derive dissolved black carbon export by arctic rivers. Front Earth Sci 3
Sushchik NN, Gladyshev MI, Ivanova EA, Kravchuk ES (2010) Seasonal distribution and fatty acid composition of littoral microalgae in the Yenisei River. J Appl Phycol 22(1):11–24
Tan BLL, Mustafa AM (2004) The Monitoring of Pesticides and Alkylphenols in Selected Rivers in the State of Selan Jor, Malaysia. Asia Pacific Journal of Public Health 16(1):54–63
Temminghoff EJM, Van der Zee SEATM, de Haan FAM (1997) Copper mobility in a copper-contaminated sandy soil as affected by ph and solid and dissolved organic matter. Environ Sci Technol 31(4):1109–1115
Tjellström H, Yang Z, Allen DK, Ohlrogge JB (2012) Rapid kinetic labeling of arabidopsis cell suspension cultures: implications for models of lipid export from plastids. Plant Physiol 158(2):601–611
Trendel JM, Schaeffer P, Adam P, Ertlen D, Schwartz D (2010) Molecular characterisation of soil surface horizons with different vegetation in the Vosges Massif (France). Org Geochem 41(9):1036–1039
Trubetskaya OE, Richard C, Patsaeva SV, Trubetskoj OA (2020) Evaluation of aliphatic/aromatic compounds and fluorophores in dissolved organic matter of contrasting natural waters by SEC-HPLC with multi-wavelength absorbance and fluorescence detections. Spectroc Acta Pt A-Mole Biomole Spectr 238:118450
van Bergen PF, Bull ID, Poulton PR, Evershed RP (1997) Organic geochemical studies of soils from the Rothamsted classical experiments.1. Total lipid extracts, solvent insoluble residues and humic acids from Broadbalk wilderness. Org Geochem 26:117–135
van Bergen PF, Flannery MB, Poulton PR, Evershed RP (1998) Organic geochemical studies of soils from rothamsted experimental station: III nitrogen-containing organic matter in soil from Geescroft Wilderness. Nitrogen-Containing Macromolecules in the Bio- and Geosphere 707: 321-338
Van den Eede N, Tomy G, Tao F, Halldorson T, Harrad S, Neels H, Covaci A (2016) Kinetics of tris (1-chloro-2-propyl) phosphate (TCIPP) metabolism in human liver microsomes and serum. Chemosphere 144:1299–1305
van der Heijden E, Boon JJ, Rasmussen S, Rudolph H (1997) Sphagnum acid and its decarboxylation product isopropenylphenol as biomarkers for fossilised Sphagnum in peats. Anc Biomol 1:93–107
Van der Veen I, de Boer J (2012) Phosphorus flame retardants: properties, production, environmental occurrence, toxicity and analysis. Chemosphere 88(10):1119–1153
Vinci G, Mazzei P, Drosos M, Zaccone C, Piccolo A (2020) Molecular characterization of ombrotrophic peats by humeomics. Chem Biolog Technol Agric 7(1):18
Volta C, Ho DT, Maher DT, Wanninkhof R, Friederich G, Del Castillo C, Dulai H (2020) Seasonal variations in dissolved carbon inventory and fluxes in a mangrove-dominated estuary. Global Biogeochem Cycles 34(12):006515
von Schiller D, Acuña V, Aristi I, Arroita M, Basaguren A, Bellin A, Boyero L, Butturini A, Ginebreda A, Kalogianni E, Larrañaga A, Majone B, Martínez A, Monroy S, Muñoz I, Paunović M, Pereda O, Petrovic M, Pozo J, Rodríguez-Mozaz S, Rivas D, Sabater S, Sabater F, Skoulikidis N, Solagaistua L, Vardakas L, Elosegi A (2017) River ecosystem processes: a synthesis of approaches, criteria of use and sensitivity to environmental stressors. Sci Total Environ 596–597:465–480
Wang XN, Wu Y, Bao HY, Gan SC, Zhang J (2019) Sources, transport, and transformation of dissolved organic matter in a large river system: illustrated by the Changjiang River. China J Geophys Res-Biogeosci 124(12):3881–3901
Werner BJ, Musolff A, Lechtenfeld OJ, de Rooij GH, Oosterwoud MR, Fleckenstein JH (2019) High-frequency measurements explain quantity and quality of dissolved organic carbon mobilization in a headwater catchment. Biogeosciences 16(22):4497–4516
Williams JS, Dungait JAJ, Bol R, Abbott GD (2016) Contrasting temperature responses of dissolved organic carbon and phenols leached from soils. Plant Soil 399:13–27
Worms IAM, Chmiel HE, Traber J, Tofield-Pasche N, Slaveykova VI (2019) Dissolved organic matter and associated trace metal dynamics from river to lake, under ice-covered and ice-free conditions. Environ Sci Technol 53(24):14134–14143
Worrall F, Harriman R, Evans CD, Watts CD, Adamson J, Neal C, Tipping E, Burt T, Grieve I, Monteith D, Naden PS, Nisbet T, Reynolds B, Stevens P (2004) Trends in dissolved organic carbon in UK rivers and lakes. Biogeochemistry 70(3):369–402
Wu ZP, Wu WD, Lin C, Zhou SL, Xiong JF (2019) Deciphering the origins, composition and microbial fate of dissolved organic matter in agro-urban headwater streams. Sci Total Environ 659:1484–1495
Yates C (2021) Determining patterns in dissolved organic matter composition according to landscape character. Biogeochem Spec Issue
Yates CA, Johnes PJ, Owen AT, Brailsford FL, Glanville HC, Evans CD, Marshall MR, Jones DL, Lloyd CEM, Jickells T, Evershed RP (2019) Variation in dissolved organic matter (DOM) stoichiometry in UK freshwaters: assessing the influence of land cover and soil C: N ratio on DOM composition. Limnol Oceanogr 64(6):2328–2340
Yates CA, Johnes PJ, Spencer RGM (2016) Assessing the drivers of dissolved organic matter export from two contrasting lowland catchments, U.K. Sci Total Environ 569–570:1330–1340
Funding
This study was funded under the Natural Environment Research Council DOMAINE Large Grant program (NE/K010689/1) “Characterising the nature, origins and ecological significance of dissolved organic matter in freshwater ecosystems.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Responsible Editor: Stephen Maberly.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lloyd, C.E.M., Mena-Rivera, L.A., Pemberton, J.A. et al. Characterisation of riverine dissolved organic matter using a complementary suite of chromatographic and mass spectrometric methods. Biogeochemistry 164, 29–52 (2023). https://doi.org/10.1007/s10533-022-00913-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-022-00913-z