
Abstract
Despite an increase in the rates of survival in patients suffering myocardial infarction, as yet there is no therapy specifically targeting ischaemia and reperfusion injury of the myocardium. With a greater understanding of immune activation during infarction, more potential treatment targets are now being identified. The innate immune system is believed to play an important role in the myocardium after ischaemia-driven cardiomyocyte death. The release of intracellular contents including DNA into the extracellular space during necrosis and cell rupture is now believed to create a pro-inflammatory milieu which propagates the inflammatory process. DNA and DNA fragments have been shown to activate the innate immune system by acting as Danger-Associated Molecular Patterns (DAMPs), which act as ligands on toll-like receptors (TLRs). Stimulation of TLRs, in turn, can activate intracellular cell death pathways such as pyroptosis. Here, we review the role of DNA fragments during ischaemia and reperfusion, and assess their potential as a target in the quest to preserve cardiomyocyte viability following myocardial infarction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Targeting Cell-Free DNA in Myocardial Infarction
Despite our increasing understanding of the pathogenesis of myocardial infarction, it remains a leading cause of premature death in the Western world [1, 2]. The widespread adoption of percutaneous coronary intervention has resulted in a significant reduction in the duration of coronary ischaemia once the clinical diagnosis of coronary artery occlusion and ST elevation myocardial infarction (STEMI) has been made. Restoring blood flow to the myocardium promptly can prevent excessive cardiomyocyte death. Conversely, delaying treatment is associated with worse outcome and death. As a consequence of the success in instigating rapid reperfusion therapy, there has been an increase in the survival of STEMI patients, but this has led to a greater incidence of subsequent heart failure [3]. Paradoxically, both ischaemia and the subsequent reperfusion lead to excessive cardiomyocyte death which results in profound “remodelling” of the heart associated with fibrotic replacement of the myocardial cytoskeleton, altering the geometry of the ventricle and resulting in impaired pump function and heart failure—also called ventricular remodelling [4].
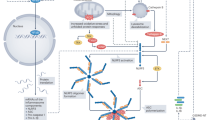
In order to help preserve cardiomyocyte viability after ischaemia-reperfusion injury, attention has focussed for many years on trying to understanding the process of cardiomyocyte death and inflammation after myocardial reperfusion [5,6,7,8]. There is increasing evidence that inflammation induced by ischaemia-reperfusion may actually contribute to cardiomyocyte death, excessive scar formation, and poor ventricular remodelling [9, 10]. Unfortunately, the results of the majority of clinical trials into the use of anti-inflammatory therapies for treating MI have been disappointing, illustrating our lack of understanding of ischaemia-reperfusion-induced inflammation in the myocardium. One important target is the process by which cardiomyocytes, which are viable at the point of reperfusion, die during reperfusion. This type of cell death differs from phagocytosis or apoptosis in that it is more uncontrolled and results in the rupture of the sarcolemma and release of the intracellular contents into the extracellular space. Current well-established cardioprotective strategies such as ischaemic preconditioning preserve cardiomyocytes during ischaemia-reperfusion injury, thereby limiting the release of intracellular debris [10]. During this process, the dying cells propagate the inflammatory response throughout the reperfusion zone, as the intracellular debris act as Danger-Associated Molecular Patterns (DAMPs) which are ligands for activation of the innate immune system [11] (Fig. 1). This type of cell death is called “necrosis”. Examples of DAMPs include mobility group box-1 protein (HMGB1), heat shock proteins, adenosine, extracellular RNA, mitochondrial DNA, and interleukin (IL)-1α all of which may stimulate the innate immune response. Recently, it has been shown that cells can also undergo a type of programmed necrosis, referred to as “necroptosis” or “pyroptosis” [12,13,14]. Identifying and targeting these DAMPs has provided varying results in attempts to save myocytes from the deleterious effects of reperfusion. This is often difficult because many identified DAMPs such as HMGB1 have complex, multifaceted roles, and inhibiting their function may instead be detrimental as inflammation is important in the process of cardiac repair after an insult. One potential DAMP that may be a promising target is DNA itself, which has the benefit of having no such multifaceted effect once outside the cell. There is now some evidence to suggest that cell death may be propagated by intracellular material such as DNA in an extracellular environment, contributing to excessive myocyte death in the myocardium after ischaemia-reperfusion injury [15,16,17,18]. One potential DAMP that has been highlighted is DNA and its components. This review aims to highlight this potentially promising target for future cardioprotective therapies.

During necrosis, the cell membrane breaks down and the fragmented intracellular contents enter the extracellular space. Here, certain components such as DNA, heat shock proteins and histones can act as danger-associated molecular patterns (DAMPs), further activating intracellular cell death pathways via toll-like receptor (TLR). TLRs trigger an intracellular signaling cascade that culminates in the translocation of NF-κB to the nucleus where it stimulates the synthesis of proteins including the components of the inflammasome complex, pro-IL-1β and pro-caspase-1. Inflammasome activation is dependent on a secondary signal. Extracellular DAMPs such as ATP can trigger K+ efflux, triggering the formation and activation of the inflammasome complex. This facilitates autocatalytic activation of pro-caspase-1 into caspase-1 and cleavage of the pro-IL-1β into IL-1β. The active caspases contribute to pyroptosis and cell membrane rupture. The subsequent release of intracellular contents including DNA into the extracellular space results in this debris functioning as additional DAMPs, thereby propagating a wave of cellular injury and death
Inflammation and Ischaemia-Reperfusion Injury
Ischaemia causes the cardiomyocytes to switch to anaerobic glycolysis to generate ATP, but this increases lactate production which causes a rapid drop in intracellular pH, driving ion exchangers to extrude protons at the expensive of accumulating intracellular Na and subsequently Ca ions. While the acidic conditions inhibit the opening of the MPTP (mitochondrial permeability transition pore), the cytosolic calcium overload results in cardiomyocyte hypercontracture. With reperfusion, the arrival of oxygen permits the re-activation of the electron transport chain, but also results in a burst of ROS (reactive oxygen species) production. In combination with Ca overload, ROS induce the opening of the MPTP, depleting ATP and damaging intracellular structures. This can either directly cause cellular death via necrosis or can cause extensive damage to cell membranes by lipid peroxidation and enzyme denaturation as well as direct oxidative damage to DNA, such that the resultant non-viable cell activates intracellular cell death signalling pathways including pyroptosis. As a consequence, the necrotic or pyroptotic cell releases its intracellular contents into the extracellular milieu. At a later time-point, several hours after reperfusion, the pro-inflammatory milieu attracts neutrophils which invade the necrotic region [10, 14, 19, 20].
During the process of reperfusion, there is an intense inflammatory process activated within the ischaemic area, coordinated by the huge influx of immune cells that circulating blood brings with it. A crucial aspect of the activated innate immune response is leukocyte infiltration into the necrotic myocardium. Leukocytes, the vast majority of which reside in the circulation or lymphatic tissue, play an important role during the process of myocardial infarction and repair. Neutrophils are the most abundant leukocyte and they infiltrate the injured myocardium early during ischaemia [21, 22]. Within the milieu of the infarcted myocardium, they exert direct effects such as tissue infiltration [23, 24], proteolysis, generation of oxygen free radicals [25], the release of pro-inflammatory cytokines, and stimulation of the complement cascade [26]. The resulting immune cascade is beyond the scope of this review; however, its complexity and the presence of multifunctioning mediators are undoubtedly why an effective anti-inflammatory therapy is yet to be developed.
Targeting Inflammation to Reduce MI Injury
There have been attempts to target the immune response during and after an infarction; however, the results of the majority of clinical trials have been disappointing. Earlier attempts were focussed on using corticosteroids, an idea borne out of cell-based studies showing that they conferred protection upon cardiomyocytes during ischaemia [27]. However, despite this promising theory, clinical trials gave conflicting results and in some cases raised serious concerns into the use of corticosteroid therapy post-infarction [28, 29]. Indeed, there is growing evidence that corticosteroids may impede fibroblast function and thus prevent healthy repair [30].
As understanding of the immune process has improved, more specific immunomodulatory therapies have been trialled in the setting of ACS. Crucial to neutrophils entering the ischaemic zone is the interaction of circulating neutrophils and the vascular endothelium. One way to block this step is through inhibiting the binding of leukocyte surface adhesion molecules such as P-selectin [31,32,33]. Inclacumab, a P-selectin inhibitor, was used in a randomised trial of 544 NSTEMI patients, but the inconclusive results only showed a trend towards a reduction in troponin levels in the treatment arm [34]. The HALT-MI study looked at antibody-mediated inhibition of the CD11/CD18 integrin receptor on leukocytes in 420 patients with an acute MI; however, treatment with the antibody to the integrin did not reduce infarct size [35]. Tocilizumab, the antibody to IL-6 receptor, reduced levels of troponin during in-hospital stay in 117 NSTEMI patients [36]. The APEX-AMI trial looked at using Pexelizumab, a humanised monoclonal antibody that binds to C5 component of the complement cascade, during PCI after myocardial infarction. A total of 5745 patients were recruited but there was no difference in all-cause mortality after 90 days between the treatment and non-treatment group [37].
Despite these disappointing findings, the search for an effective anti-inflammatory therapy for acute MI continues. The exciting results of the recent CANTOS trial in the setting of atherosclerosis have reignited the hope of developing an equivalent effective immunomodulatory therapy for ischaemia-reperfusion injury. In the CANTOS trial, Canakinumab, a human monoclonal antibody targeting interleukin 1 beta (IL-1β), was administered to patients who had previously suffered a myocardial infarct and raised circulating CRP levels. Treatment demonstrated a 15% reduction in relative risk for the composite primary endpoint of non-fatal MI, non-fatal stroke, and death from cardiovascular disease [38].
A huge body of animal studies offer further tantalising hope of identifying new treatment targets in preventing the inflammatory damage in ischaemia-reperfusion injuries. Modulating pro-inflammatory cytokines in animal models have yielded exciting results; IL-1β blockade in a mouse ischaemic cardiomyopathy model significantly improved LV function [39]. Similar studies targeting IL-2 or IL-18 in animal models have demonstrated significant reduction in cardiomyocyte death and improvement in LV function [40, 41]. Chemokines [42], inflammasomes, and TGF-β [43] are some of the other targets that have demonstrated potential benefit in animal models of ischaemia-reperfusion injury.
However, one avenue that has received limited coverage as a potential new target is the role of extracellular DNA and the innate immune system. Innate immunity is the first line of defence to tissue injury and represents an evolutionary older branch in comparison with the more specific and targeted adaptive immunity. The innate immune system first appeared 750 million years ago and has been remarkably conserved throughout the evolutionary tree of life [44, 45]. A key initiator of an innate immune response is immune stimulation via a DAMP [11]. Therefore, identification of DAMPs released during ischaemia-reperfusion injury may lead to the identification of a valuable drug target. The appearance of excessive amounts of intact, high-molecular-weight, extracellular DNA is one of many differences between controlled cell death pathways such as apoptosis and uncontrolled necrotic cell death [46, 47]. When cells undergo apoptosis, intracellular DNA is methodically degraded and shielded from the immune system by retention within plasma membrane vesicles (apoptotic bodies). However, during ischaemia, cells die primarily via a process of necrosis during which DNA is released into the extracellular space and blood. The nucleus of every eukaryotic cell (except red blood cells, which do not have a nucleus) contains approximately 6 pg of DNA. Therefore, since the human myocardium contains approximately 5 billion cells, a large left ventricular myocardial infarct could cause the death of ~ 1 billion cardiomyocytes which can potentially release ~ 1 mg of DNA and DNA fragments into the extracellular space [48, 49]. This huge quantity of DNA is then free to diffuse within the necrotic milieu of the infarct zone.
Another large source of extracellular DNA during ischaemia and reperfusion comes from infiltrating leukocytes. In response to a TLR-dependent process, neutrophils, the most abundant leukocytes in the myocardium during reperfusion, discharge their nuclear DNA forming an extracellular net of DNA rich in histones [50]. This process, termed NETosis, ultimately kills the neutrophil whilst it lays down a histone-rich mechanical mesh which traps debris. NETs (neutrophil extracellular traps) can break down and release histones causing further damage to tissue remote from the initial necrotic site [51]. The NETs also play a crucial role in thrombosis, platelet aggregation, and occluding blood vessel further exaggerating coronary ischaemia [52]. Thrombi aspirated from the coronary arteries of patients who suffered STEMI demonstrate that the burden of NETosis positively correlates with infarct size and negatively correlates with ST segment resolution [53].
The crucial role of DNA breakdown in normal physiology is highlighted by the fact that mice lacking the DNA cleaving enzyme DNAse II die shortly after birth [54]. Furthermore, deficiencies in the normal biological process of DNA digestion and processing are linked to diseases with inappropriately active innate immune systems or autoimmunity [55,56,57]. Elevated levels of circulating DNA are also associated with a variety of conditions from trauma, tumour malignancy, and sepsis, all of which are themselves associated with a degree of immune activation or inflammation [58, 59]. Each DNA nucleosome core consists of superhelical DNA wound around an octamer of histones, composed of two copies of each of the core histones H2A, H2B, H3, and H4 [60]. The linker histone H1 binds to the complete nucleosome core particle and forms higher order structures [61]. After a necrotic event, cellular DNA may be released either as DNA fragments or as nucleosomes, both of which are well-recognised DAMPs [18]. The extent to which mammalian DNA components are cytotoxic was first investigated by Xu et al., who showed that intravenous injection of isolated histones into mice caused death through sepsis within minutes [62]. Curiously, administration of intact nucleosomes did not have this effect. It has now been shown that this effect is mediated through the toll-like receptors TLR2 and TLR4, two crucial receptors involved in activation of the innate immune system [63]. Unlike human DNA, foreign microbial DNA has also long been accepted as a potent stimulator of the innate immune system [64]. A heterogeneous group of pattern recognition receptors on immune cell surfaces detects foreign microbial nucleic acids, including TLR3, TLR7–TLR9 [65, 66].
A certain quantity of cell-free, circulating DNA is understood to be part of the normal physiological state in both humans and rodents; its concentration is tightly controlled by extracellular DNAases [67]. It is now believed that during normal cellular process such as cell division, the amount of extracellular DNA remains manageable through continuous degradation to maintain a level below the immuno-stimulatory threshold [68, 69]. If, however, the threshold is breached in conditions such as necrotic cell death, the DNA may act on the same pathogenic receptor pathways stimulating an innate immune response [70,71,72]. A well-documented finding in auto-inflammatory conditions is the presence of circulating cell-free DNA incorporated into immune complexes [73], confirming the ability of DNA to act as an auto-antigen. Previously, it was believed that the presence of unmethylated CpG dinucleotides in microbial DNA conferred foreign DNAs ability to interact with TLR9 and stimulate the innate immune response. Mammalian purified DNA dinucleotides are mostly methylated so in theory they should not exhibit an immuno-stimulatory response, but mammalian complexed DNA, either within histones or DNA-binding proteins, has been demonstrated to induce TLR9-mediated signalling [74, 75]. Extracellular mammalian DNA has been shown to have this effect both by interacting with TLR9 [76] to activate the innate immune system and by TLR-independent mechanism which increase the transcription of type 1 interferons a potent pro-inflammatory cytokine [77,78,79]. Unlike naked DNA, cell-free chromatin contains abundant proteins that may expose epitopes for helper T cells to identify them as foreign. Indeed, the appearance of antibodies to chromatin precedes the occurrence of anti-DNA antibodies suggesting chromatin plays a crucial role in developing an auto-inflammatory response to self-DNA [59].
The Role of Extracellular Histones in Innate Immunity
Apoptotic or necrotic cells induced by ischaemia release histones [80] either as part of nucleosome fragments or on their own. These extracellular histones have also been shown to trigger inflammation and cell death, either by stimulating pro-inflammatory cytokines resulting in the activation of cell death pathways or through the process of neutrophil extracellular traps. In human observational studies, raised histone serum levels have been demonstrated in multiple trauma patients and correlate with severity of coagulopathy, endothelial damage, and inflammation [81]. A large body of evidence demonstrates that histone-induced cell toxicity plays a crucial role in cell death during ischaemia-reperfusion injury of the myocardium. Histones are known to activate TLR2 and TLR4; furthermore, TLR knockout mice are protected from the lethal effects of histones [63]. In an ischaemic stroke model, histone infusion is correlated with large infarct size and conversely histone neutralisation via an antibody infusion results in a reduction in infarct size [82]. In a toxic liver injury model, free histones mediated cytotoxicity of liver cells via a TLR-dependent process—an effect that was abrogated by anti-histone antibodies [63]. Furthermore, in liver cells, histone-stimulated TLR activation results in activation of the intracellular NLRP3 inflammasome and subsequent pyroptosis [83]. Histones have been shown to mediate endothelial cell cytotoxicity, resulting in acute lung haemorrhage, thrombosis, and oedema [84]. Similar cytotoxic effects of histones have been demonstrated in kidney injury [85], sepsis [62], and even hair follicle death [86]. There is also increasing evidence that histones can cause cytotoxicity independent of immunostimulation, damaging endothelial cells and stimulating an influx of intracellular calcium and subsequent necrosis [84]. Thus, increasing evidence suggests free histones function as DAMPs, leading to both inflammatory and toxic responses culminating in cell death.
DNA and Cell Death (Inflammasome Activation and Pyroptosis)
It has been shown that extracellular DNA and histones could function as an alarmin or DAMP by causing activation of TLRs. As well as playing a crucial role in the innate immune system, activation of TLR can activate cell death pathways such as pyroptosis. It is now believed that during ischaemia and reperfusion, pyroptosis may contribute to infarct size and subsequent poor remodelling of the myocardium [12, 87, 88]. This has culminated in a great deal of interest in targeting intracellular cell death pathways to limit the degree of cell death during myocardial infarction [8, 89, 90].
During ischaemia, necrotic cell debris including DNA fragments circulate in the extracellular matrix creating a pro-inflammatory milieu. Activation of the TLRs on surviving cardiac cells in the border zone of the infarct area leads to activation of downstream intracellular signalling pathways, which convene to result in NF-κB mediated expression of the protein components that make up the NLRP3 inflammasome [91, 92]. Following a secondary trigger, the individual protein components which now exist in the cytoplasm of the cell aggregate to form a multiprotein oligomer also called the inflammasome complex [93, 94]. This complex is now able to interact with pro-caspase-1 and leads to its conversion into its active caspase-1 form [95]. Caspase-1 begins the subsequent autocatalytic activation of the pro-inflammatory cytokines IL-1β and IL-18 [96]. An additional substrate of caspase-1 is the cytosolic protein, gasdermin D (GSDMD) [97,98,99]. Following cleavage by caspase-1, the N-terminal fragments of GSDMD (GSDMD-N) oligomerise within the cell membrane to form pores. These pores result in loss of cell membrane integrity, leading pyroptotic cell death [100]. The pores also increase membrane permeability to IL-1β and IL-18 leading to their extracellular release, and these cytokines amplify the inflammatory response and mediate further injury [42, 87, 88, 101,102,103,104,105,106,107,108].
Evidence for a Role of Extracellular DNA in Ischaemia-Reperfusion Injury of the Myocardium
A number of animal studies have proven that self-DNA may be an effective target to inhibit inflammation and myocyte death during ischaemia-reperfusion injury. In a murine model, it was demonstrated that histones caused cardiomyocyte toxicity and an in vivo heart ischaemia-reperfusion model, DNAse 1 treatment, disrupted extracellular cytotoxic chromatin resulting in a reduction myocardial histone concentration [17]. This correlated with a significant improvement in left ventricular remodelling and cardiomyocyte survival. Ge et al. supported this finding in a murine model of ischaemia-reperfusion showing that DNAse with the addition of recombinant tissue-type plasminogen activator resulted in a reduction of infarct size as well as decreasing the density of neutrophil-associated NETs [109]. This also leads to an improvement in left ventricular remodelling. Curiously, this effect was not observed when DNAse or rt-PA was administered on its own [109]. Savchenko et al. administered DNAse to PAD4-/- mice which do not produce NETs as well as wild type mice [110].The study demonstrated that myocardial ischaemia-reperfusion injury caused an increase in nucleosomes, neutrophil infiltration, and histone H3 at the site of injury. Treatment with DNAse improved cardiac contractile function to a similar degree in both wild type and PAD4-/- deficient mice. This suggests that DNA fragments contribute to cardiomyocyte dysfunction during reperfusion irrespective of NETs, possibly by acting as a DAMP. Using an in vivo rat model, Downey’s group have made similar findings, demonstrating that DNAse administered after 30 min of coronary artery occlusion resulted in a significant reduction in infarct size [111]. Interestingly, the addition of mitochondrial DNA inhibitor with DNAse resulted in a greater reduction in infarct size then that which was seen with DNAse alone [111]. This would suggest that nuclear DNA could be acting through a different pathway than the well-recognised DAMP, mitochondrial DNA.
Endothelial dysfunction plays a crucial role in ischaemia-reperfusion injury, contributing to myocardial stunning, microvascular obstruction, and exposing the myocytes to toxic stimuli which contribute to lethal myocardial injury [112,113,114]. Heparin consists of a high volume of negatively charged sulphated proteoglycans, binds to histones, and inactivates them through high-affinity electrostatic interactions [115]. It has long been shown that heparin protects the coronary endothelium and myocardium from ischaemia-reperfusion injury [116,117,118,119]. Both heparin and chondroitin sulphate have both been shown to protect vascular endothelial cells from histone-induced cytotoxicity in vitro [120, 121]. Furthermore, heparin derivatives reduce infarct size in a rat model of ischaemia-reperfusion by inhibiting caspase-dependent, cell death pathways [122].
Summary
In the quest to protect cardiomyocytes from the deleterious effects of ischaemia-reperfusion injury, identification of pyroptosis as a contributing factor to infarct size has revealed a target for future cardioprotective therapies. Extracellular DNA fragments from dead cells and neutrophils are potent instigators of TLR-dependent inflammasome activation and subsequent pyroptosis. The use of DNAse and DNA-neutralising therapies has shown some promise in animal models of preventing the damaging effects of ischaemia-reperfusion injury. Identification and targeting the instigators of pyroptosis may provide benefit in limiting cell death post-infarction and preventing the morbidity and mortality associated with ischaemia and reperfusion injury.
References
World Health Organization. Global status report on noncommunicable diseases 2014. World Health. 2014;176–9.
Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, Teo K, et al. Reducing the global burden of cardiovascular disease, part 1: the epidemiology and risk factors. Circ Res. 2017;121:677–94.
Bansilal S, Castellano JM, Fuster V. Global burden of CVD: focus on secondary prevention of cardiovascular disease. Int J Cardiol. 2015;201:S1–7.
Briceno N, Schuster A, Lumley M, Perera D. Ischaemic cardiomyopathy: pathophysiology, assessment and the role of revascularisation. Heart. 2016;102:396–406.
Andreadou I, Cabrera-Fuentes H, Devaux Y, Frangogiannis NG, Frantz S, Guzik T, et al. Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res. 2019;115:1117–30.
Davidson SM, Arjun S, Basalay MV, Bell RM, Bromage DI, Bøtker HE, et al. The 10th biennial hatter cardiovascular institute workshop: cellular protection—evaluating new directions in the setting of myocardial infarction, ischaemic stroke, and cardio-oncology. Basic Res Cardiol. 2018;113:43.
Hausenloy DJ, Garcia-Dorado D, Bøtker HE, Davidson SM, Downey J, Engel FB, et al. Novel targets and future strategies for acute cardioprotection: position paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc Res. 2017;113:564–85.
Hausenloy DJ, Barrabes JA, Bøtker HE, Davidson SM, Di Lisa F, Downey J, et al. Ischaemic conditioning and targeting reperfusion injury: a 30 year voyage of discovery. Basic Res Cardiol. 2016;111:70.
Bell RM, Bøtker HE, Carr RD, Davidson SM, Downey JM, Dutka DP, et al. 9th Hatter Biannual Meeting: position document on ischaemia/reperfusion injury, conditioning and the ten commandments of cardioprotection. Basic Res Cardiol. 2016;111:41.
Hausenloy DJ, Yellon DM. Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol. 2016;13:193–209.
Matzinger P. The danger model: a renewed sense of self. Science (80-). 2002;296:301–5.
Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26:R543–76.
Takahashi M. NLRP3 inflammasome as a novel player in myocardial infarction. Int Heart J. 2014;55:101–5.
Rauf A, Shah M, Yellon DM, Davidson SM. Role of caspase 1 in ischemia/reperfusion injury of the myocardium. J Cardiovasc Pharmacol. 2019;74:194–200.
Kalbitz M, Grailer JJ, Fattahi F, Jajou L, Herron TJ, Campbell KF, et al. Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J. 2015;29:2185–93.
Fattahi F, Russell MW, Malan EA, Parlett M, Abe E, Zetoune FS, et al. Harmful roles of TLR3 and TLR9 in cardiac dysfunction developing during polymicrobial Sepsis. Biomed Res Int. 2018;2018:1–10.
Vogel B, Shinagawa H, Hofmann U, Ertl G, Frantz S. Acute DNase1 treatment improves left ventricular remodeling after myocardial infarction by disruption of free chromatin. Basic Res Cardiol. 2015;110:15.
Pisetsky DS. The origin and properties of extracellular DNA: from PAMP to DAMP. Clin Immunol. 2012;144:32–40.
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35.
Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003;35:339–41.
Engler RL, Dahlgren MD, Peterson MA, Dobbs A, Schmid-Schönbein GW. Accumulation of polymorphonuclear leukocytes during 3-h experimental myocardial ischemia. Am J Phys. 1986;251:H93–100.
Jolly SR, Kane WJ, Hook BG, Abrams GD, Kunkel SL, Lucchesi BR. Reduction of myocardial infarct size by neutrophil depletion: effect of duration of occlusion. Am Heart J. 1986;112:682–90.
Bienvenu K, Granger DN. Molecular determinants of shear rate-dependent leukocyte adhesion in postcapillary venules. Am J Phys. 1993;264:H1504–8.
Gasic AC, McGuire G, Krater S, Farhood AI, Goldstein MA, Smith CW, et al. Hydrogen peroxide pretreatment of perfused canine vessels induces ICAM-1 and CD18-dependent neutrophil adherence. Circulation. 1991;84:2154–66.
Duilio C, Ambrosio G, Kuppusamy P, DiPaula A, Becker LC, Zweier JL. Neutrophils are primary source of O2 radicals during reperfusion after prolonged myocardial ischemia. Am J Physiol Heart Circ Physiol. 2001;280:H2649–57.
Frangogiannis N. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–73.
Libby P, Maroko PR, Bloor CM, Sobel BE, Braunwald E. Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest. 1973;52:599–607.
Giugliano GR, Giugliano RP, Gibson CM, Kuntz RE. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am J Cardiol. 2003;91:1055–9.
Roberts R, DeMello V, Sobel BE, Koerting A, Ren G, Abou-Khamis T, et al. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 1976;53:I204–6.
Kloner RA, Fishbein MC, Lew H, Maroko PR, Braunwald E. Mummification of the infarcted myocardium by high dose corticosteroids. Circulation. 1978;57:56–63.
Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer T A. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood. 1996;88:146–57.
Burns AR, Bowden RA, Abe Y, Walker DC, Simon SI, Entman ML, et al. P-selectin mediates neutrophil adhesion to endothelial cell borders. J Leukoc Biol. 1999;65:299–306.
Jones DA, Abbassi O, McIntire LV, McEver RP, Smith CW. P-selectin mediates neutrophil rolling on histamine-stimulated endothelial cells. Biophys J. 1993;65:1560–9.
Tardif JC, Tanguay JF, Wright SS, Duchatelle V, Petroni T, Grégoire JC, et al. Effects of the P-selectin antagonist inclacumab on myocardial damage after percutaneous coronary intervention for non-st-segment elevation myocardial infarction: results of the SELECT-ACS trial. J Am Coll Cardiol. 2013;61:2048–55.
Faxon DP, Gibbons RJ, Chronos NAF, Gurbel PA, Sheehan F. The effect of blockade of the CD11/CD18 integrin receptor on infarct size in patients with acute myocardial infarction treated with direct angioplasty: the results of the HALT-MI study. J Am Coll Cardiol. 2002;40:1199–204.
Kleveland G, Bratlie M, Ueland T, Amundsen B, Aakhus S, Damaas JK, et al. The interleukin-6 receptor antagonist tocilizumab reduces inflammation and myocardial damage in non-ST elevation myocardial infarction-a randomized, double-blind, placebo controlled study. Eur Heart J. 2015;36:27.
Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D, O’Neill WW, et al. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA. 2007;297:43–51.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31.
Toldo S, Mezzaroma E, Bressi E, Marchetti C, Carbone S, Sonnino C, et al. Interleukin-1beta blockade improves left ventricular systolic/diastolic function and restores contractility reserve in severe ischemic cardiomyopathy in the mouse. J Cardiovasc Pharmacol. 2014;64:1–6.
Abbate A, Salloum FN, van Tassell BW, Vecile E, Toldo S, Seropian I, et al. Alterations in the interleukin-1/interleukin-1 receptor antagonist balance modulate cardiac remodeling following myocardial infarction in the mouse. PLoS One. 2011;6:11.
Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J Biol Chem. 2009;284:7853–65.
Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–87.
Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–38.
Kimbrell DA, Beutler B. The evolution and genetics of innate immunity. Nat Rev Genet. 2001;2:256–67.
Cooper MD, Herrin BR. How did our complex immune system evolve? Nat Rev Immunol. 2010;10:2–3.
Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517.
Kerr JFR, Wyllie A, Currie A. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. J Intern Med. 1972;258:479–517.
Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–30.
Michlewska S, McColl A, Rossi A, Megson I, Dransfield I. Clearance of dying cells and autoimmunity. Autoimmunity. 2007;40(4):267–73.
Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis. 2017;8:2812.
Pfeiler S, Stark K, Massberg S, Engelmann B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica. 2017;102:206–13.
Hoeksema M, Van Eijk M, Haagsman HP, Hartshorn KL. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol. 2016;11:441–53.
Mangold A, Alias S, Scherz T, Hofbauer T, Jakowitsch J, Panzenböck A, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. 2015;116:1182–92.
Kawane K, Fukuyama H, Yoshida H, Nagase H, Ohsawa Y, Uchiyama Y, et al. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol. 2003;4:138–44.
Nishimoto S, Kawane K, Watanabe-Fukunaga R, Fukuyama H, Ohsawa Y, Uchiyama Y, et al. Nuclear cataract caused by a lack of DNA degradation in the mouse eye lens. Nature. 2003;424:1071–4.
Crow YJ, Rehwinkel J. Aicardi-Goutie’res syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet. 2009;18:130–6.
Krieser RJ, MacLea KS, Park JP, Eastman A. The cloning, genomic structure, localization, and expression of human deoxyribonuclease IIbeta. Gene. 2001;269:205–16.
Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012;189:2689–95.
Soni C, Reizis B. DNA as a self-antigen: nature and regulation. Curr Opin Immunol. 2018;55:31–7.
Koyama M, Kurumizaka H. Structural diversity of the nucleosome. J Biochem. 2018;163:85–95.
Kirmes I, Szczurek A, Prakash K, Charapitsa I, Heiser C, Musheev M, et al. A transient ischemic environment induces reversible compaction of chromatin. Genome Biol. 2015;16:1–19.
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21.
Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–31.
Tokunaga T, Yamamoto H, Shimada S, Abe H, Fukuda T, Fujisawa Y, et al. Antitumor activity of deoxyribonucleic acid fraction from Mycobacterium bovis BCG. I. Isolation, physicochemical characterization, and antitumor activity. J Natl Cancer Inst. 1984;72:955–62.
Engel A, Barton GM. Compartment-specific control of signaling from a DNA-sensing immune receptor. Sci Signal. 2010;3:150.
Lim KH, Staudt LM. Toll-like receptor signaling. Cold Spring Harb Perspect Biol. 2013;5:1.
Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet. 2001;28:313–4.
Nagata S, Nagase H, Kawane K, Mukae N, Fukuyama H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003;10:108–16.
Viorritto ICB, Nikolov NP, Siegel RM. Autoimmunity versus tolerance: can dying cells tip the balance? Clin Immunol. 2007;122:125–34.
Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2010;51:621–32.
Chen C, Feng Y, Zou L, Wang L, Chen HH, Cai JY, et al. Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. J Am Heart Assoc 2014;3.
Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med. 2010;207:2921–30.
Chan RWY, Jiang P, Peng X, Tam L-S, Liao GJW, Li EKM, et al. Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc Natl Acad Sci. 2014;111:E5302–11.
Suzuki K, Mori A, Ishii KJ, Saito J, Singer DS, Klinman DM, et al. Activation of target-tissue immune-recognition molecules by double-stranded polynucleotides. Proc Natl Acad Sci. 1999;96:2285–90.
Park JH, Chang SH, Kim MC, Shin SH, Youn HJ, Kim JK, et al. Up-regulation of the expression of major histocompatibility complex class I antigens by plasmid DNA transfection in non-hematopoietic cells. FEBS Lett. 1998;436:55–60.
Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, et al. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol. 2005;174:6129–36.
Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med. 2005;202:1333–9.
Yasuda K, Rutz M, Schlatter B, Metzger J, Luppa PB, Schmitz F, et al. CpG motif-independent activation of TLR9 upon endosomal translocation of “natural” phosphodiester DNA. Eur J Immunol. 2006;36(2):431–6.
Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 2016;44:739–54.
Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–65.
Kaufman T, Magosevich D, Moreno MC, Guzman MA, D’Atri LP, Carestia A, et al. Nucleosomes and neutrophil extracellular traps in septic and burn patients. Clin Immunol. 2017;183:254–62.
De Meyer SF, Suidan GL, Fuchs TA, Monestier M, Wagner DD. Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol. 2012;32:1884–91.
Huang H, Chen H-W, Evankovich J, Yan W, Rosborough BR, Nace GW, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol. 2013;191:2665–79.
Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187:160–9.
Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012;23:1375–88.
Shin SH, Joo HW, Kim MK, Kim JC, Sung YK. Extracellular histones inhibit hair shaft elongation in cultured human hair follicles and promote regression of hair follicles in mice. Exp Dermatol. 2012;21:956–8.
Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243:206–14.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109.
Zhu H, Sun A. Programmed necrosis in heart disease: molecular mechanisms and clinical implications. J Mol Cell Cardiol. 2018;116:125–34.
Ibáñez B, Heusch G, Ovize M, Van De Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–71.
Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-κB signaling in heart. J Mol Cell Cardiol. 2006;41:580–91.
Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-κB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93.
Dick MS, Sborgi L, Rühl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun. 2016;7:11929.
Stutz A, Horvath GL, Monks BG, Latz E. ASC speck formation as a readout for inflammasome activation. Methods Mol Biol. 2013;1040:91–101.
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–206.
Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–87.
Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–84.
Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J Immunol. 2016;197:1353–67.
Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–67.
Dinarello CA. A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–17.
Schroder K, Tschopp J. The Inflammasomes. Cell. 2010;140:821–32.
Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604.
Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:1–9.
Nagareddy P, Smyth SS. Inflammation and thrombosis in cardiovascular disease. Curr Opin Hematol. 2013;20:457–63.
Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15(4):203–14.
Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2013;99:164–74.
Ge L, Zhou X, Ji W-J, Lu R-Y, Zhang Y, Zhang Y-D, et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308:500–9.
Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. 2014;123(1):141–8.
Yang XM, Cui L, White J, Kuck J, Ruchko MV, Wilson GL, et al. Mitochondrially targeted endonuclease III has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Basic Res Cardiol. 2015;110:3.
Vinten-Johansen J, Zatta AJ, Jiang R, Shi W. Lethal myocardial reperfusion injury. Manag Myocard Reperfus Inj 2013;51–85.
Eltzschig HK, Collard CD. Vascular ischaemia and reperfusion injury. Br Med Bull. 2004;70:71–86.
Piper HM, García-Dorado D, Ovize M. A fresh look at reperfusion injury. Cardiovasc Res. 1998;38:291–300.
Alcantara FF, Iglehart DJ, Ochs RL. Heparin in plasma samples causes nonspecific binding to histones on Western blots. J Immunol Methods. 1999;226:11–8.
Thourani VH, Brar SS, Kennedy TP, Thornton LR, Watts JA, Ronson RS, et al. Nonanticoagulant heparin inhibits NF-kappaB activation and attenuates myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2000;278:2084–93.
Kouretas PC, Myers AK, Kim YD, Cahill PA, Myers JL, Wang N, et al. Heparin and nonanticoagulant heparin preserve regional myocardial contractility after ischemia-reperfusion injury: role of nitric oxide. J Thorac Cardiovasc Surg. 1998;115:440–9.
Kouretas PC, Kim YD, Cahill PA, Myers AK, To LN, Wang YN, et al. Nonanticoagulant heparin prevents coronary endothelial dysfunction after brief ischemia-reperfusion injury in the dog. Circulation. 1999;99:1062–8.
Pevni D, Frolkis I, Shapira I, Schwartz D, Yuhas Y, Schwartz IF, et al. Heparin added to cardioplegic solution inhibits tumor necrosis factor-α production and attenuates myocardial ischemic-reperfusion injury. Chest. 2005;128:1805–11.
Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Kadota K, Sato K. Heparins attenuated histone-mediated cytotoxicity in vitro and improved the survival in a rat model of histone-induced organ dysfunction. Intensive Care Med Exp. 2015;3:36.
Nagano F, Mizuno T, Mizumoto S, Yoshioka K, Takahashi K, Tsuboi N, et al. Chondroitin sulfate protects vascular endothelial cells from toxicities of extracellular histones. Eur J Pharmacol. 2018;826:48–55.
Collino M, Pini A, Mastroianni R, Benetti E, Lanzi C, Bani D, et al. The non-anticoagulant heparin-like K5 polysaccharide derivative K5-N,OSepi attenuates myocardial ischaemia/reperfusion injury. J Cell Mol Med. 2012;16:2196–207.
Funding
The authors acknowledge the support of the British Heart Foundation (PG/19/51/34493, PG/18/44/33790, PG/16/85/324710), BHF Clinical Research Training Fellowship (FS/18/80/33937 to Mohammed Shah), and Biomedical Research Council (BRC429/CV/SG/101320 to SD).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shah, M., Yellon, D.M. & Davidson, S.M. The Role of Extracellular DNA and Histones in Ischaemia-Reperfusion Injury of the Myocardium. Cardiovasc Drugs Ther 34, 123–131 (2020). https://doi.org/10.1007/s10557-020-06946-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-020-06946-6