
Abstract
Cellulose microfibrils in conifers, as in other woody materials, are aggregated into loose bundles called macrofibrils. The centre-to-centre spacing of the microfibrils within these macrofibrils can be estimated from the position of a broad diffraction peak in small-angle neutron scattering (SANS) after deuteration. A known spacing of 3.0 nm, increasing with moisture content, is consistent with direct microfibril to microfibril contact. However recent evidence indicates that conifer microfibrils are partially coated with bound xylan chains, and possibly with lignin and galactoglucomannan, implying a wider centre-to-centre spacing as found in angiosperm wood. Delignification of spruce wood allowed a weak SANS peak to be observed without measurable change in spacing. By deuterating spruce wood in mildly alkaline D2O and then re-equilibrating with ambient H2O, deuterium atoms were trapped in a position that gave a 3.8 nm microfibril spacing under dry conditions as in angiosperm wood, instead of the 3.0 nm spacing normally observed in conifers. After conventional vapour deuteration of spruce wood a minor peak at 3.8 nm could be fitted in addition to the 3.0 nm peak. These observations are consistent with some microfibril segments being separated by bound xylan chains as in angiosperms, in addition to the microfibril segments that are in direct contact.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose from higher plants forms partially crystalline microfibrils that, at the time of their formation, appear to contain about 18 chains (Newman et al. 2013), which would give a diameter just under 3 nm (Jarvis 2018). Detailed microfibril structures remain unclear but modelling suggests that both hydrophilic (110, 1–10) and hydrophobic (200) crystallographic faces are exposed (Kubicki et al. 2018). Diameters across the hydrophobic faces can be estimated from Scherrer broadening of the 200 reflection in wide-angle X-ray scattering (WAXS), with corrections for disorder (Jarvis 2018). Where microfibrils cluster into aggregates (macrofibrils), diameters across the hydrophilic faces can be estimated from the centre-to-centre spacing of the aggregated microfibrils measured by small-angle neutron scattering (SANS), with surface deuteration to provide neutron scattering contrast (Jarvis 2018). These scattering methods give broad agreement with other techniques such as atomic force microscopy (Zhang et al. 2016) and electron microscopy (Xu et al. 2007). Partial coalescence of adjacent microfibrils increases the diameter across the hydrophobic faces, estimated by WAXS, to 4–6 nm in hemicellulose-poor fibres like flax (Thomas et al. 2013a) and cotton (Martinez-Sanz et al. 2017) and to about 4 nm in grasses like bamboo (Thomas et al. 2015). In conifers (Fernandes et al. 2011) and bamboo (Thomas et al. 2015) the estimated diameter across the hydrophilic faces, as measured by SANS, remains around 3.0 nm in the dry state. In angiosperms, xylans with a regularly alternating substitution pattern adopt a 21 helical conformation similar to cellulose and bind edge-on to hydrophilic faces of microfibrils (Dupree et al. 2015). The apparent microfibril diameter across the hydrophilic faces, as measured by SANS, is then increased to 3.5–4.2 nm (Thomas et al. 2014; Langan et al. 2014).
Conifer xylans, although structurally different and less abundant than angiosperm xylans, share the alternating substituent pattern that allows binding to hydrophilic cellulose faces (Busse-Wicher et al. 2016; Martinez-Abad et al. 2017). Recently it was shown by multidimensional 13C spin-diffusion that conifer xylans too are indeed bound to cellulose surfaces as in angiosperms, with an associated change in the C6 conformation of the underlying cellulose chain to tg as in crystalline cellulose (Terrett et al. 2019). Terrett et al. (2019) also showed that some galactoglucomannan and lignin are in close (< 1 nm) spatial association with cellulose. Their detailed 13C NMR assignments allow earlier 1H spin-diffusion experiments, which are better suited to estimating longer spatial separations (Newman 1992; Altaner et al. 2006; Fernandes et al. 2011) to be more fully interpreted: specifically, implying that much of the lignin and acetylated galactoglucomannan are located several nm from cellulose and are thus more likely to lie in the sheath-like or beaded (Terashima et al. 2009) matrix between macrofibrils. This model reverses the previous picture, where galactoglucomannans were thought to be associated closely with cellulose whereas a xylan-lignin matrix separated the macrofibrils (Akerholm and Salmen 2001).
Xylans bound to the hydrophilic faces of softwood microfibrils would be expected to increase the microfibril diameter beyond the 3 nm expected for bare microfibrils, as they do in angiosperm wood (Thomas et al. 2014; Langan et al. 2014). Bound galactoglucomannan or lignin would also increase the microfibril diameter, although not necessarily in the same orientation as is probed by SANS. However Fernandes et al. (2011) observed only a 3.0 nm characteristic spacing for spruce microfibrils. The present paper addresses this anomaly.
The small-angle scattering approach requires contrasting scattering properties of the microfibrils and the interstitial or interface domains. For SANS, the large neutron scattering length of deuterium allows contrast to be generated from deuteration of the microfibril surface or from interstitial D2O. In SAXS, contrast comes from differences in electron density and is normally insufficient for coherent scattering to be observed for wood, where the microfibrils are closely packed and the non-cellulosic polymers between them are quite similar in electron density to cellulose (Jungnikl et al. 2008). For hydrated primary cell walls, the difference in density between the partially crystalline microfibrils and the interstitial aqueous phase gives the required X-ray scattering contrast (Kennedy et al. 2007), and enough contrast in density for SANS even without deuteration (Thomas et al. 2013b).
When cellulose microfibrils aggregate they are arranged much more loosely and irregularly than a conventional crystalline lattice and the small-angle scattering patterns are correspondingly diffuse. A range of microfibril spacings is present (Xu et al. 2007). The lower limit of the range is fixed by contact between microfibrils and gives rise to the one, broad, coherent equatorial scattering peak observed (Kennedy et al. 2007). Here we call this minimum microfibril spacing, equal to the mean diameter of the two microfibrils in contact, the characteristic spacing, retaining the crystallographic symbol d derived from the position q of the coherent scattering peak by d = 2π/q.
Unexpectedly, Jungnikl et al. (2008) found what appeared to be a coherent SAXS peak at d = 3.9 nm for spruce wood from which lignin and other polymers had been removed by H2O2. The reasons were not clear, but it is possible that the H2O2 delignification treatment opened up the microfibril structure somewhat. It was therefore of interest whether coherent small-angle neutron scattering might be observable after delignification but without deuteration.
A second way of introducing SANS contrast was also explored. Surface deuteration of cellulose for SANS is normally done by liquid- or vapour-phase exchange with D2O and is reversible (Thomas et al. 2013b). However, deuteration in mildly alkaline D2O or at elevated temperature gives a small amount of permanent deuteration stable to re-exchange with H2O (Altaner et al. 2014). The O-D stretching region of the Fourier-transform infrared (FTIR) spectrum indicated that the O-D groups thus introduced were on cellulose chains in the conformation of crystalline cellulose (Altaner et al. 2014). A sub-surface location was suggested. The deuteration conditions were mild enough that no change in composition was detected by FTIR (Altaner et al. 2014).
Here we report SANS experiments on spruce that had been subjected to chlorite delignification or mild alkaline deuteration.
Results and discussion
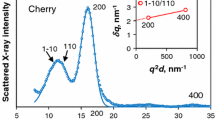
Figure 1 shows that the delignified spruce wood gave a very weak equatorial SANS peak centred on d = 3 nm, similar to the original deuterated and dried spruce wood (Fernandes et al. 2011). Dry spruce wood showed no coherent neutron scattering without deuteration (Fernandes et al. 2011). Therefore, delignification introduced a small amount of neutron scattering contrast, perhaps by causing a small degree of additional dissociation of microfibrils and introducing additional voids within the macrofibril structure. The characteristic spacing did not appear to differ from the unmodified cell walls, but the intensity of the SANS peak was too low for any minor change in characteristic spacing to be detected.

Equatorial SANS profile for Sitka spruce wood after stable deuteration under mildly alkaline conditions, and after chlorite delignification with no deuteration. With an exponential model for non-coherent scattering, the centre of the diffraction peak corresponded to microfibril spacings of 3.8 nm (q = 1.6 nm−1) for the alkaline-deuterated wood and 3.0 nm (q = 2.0 nm−1) for the delignified wood
The weak SANS peak at d = 3 nm after delignification is consistent with the idea that much of the lignin is located outside the macrofibrils (Altaner et al. 2006; Hill et al. 2009; Langan et al. 2014; Terashima et al. 2009), but it does not exclude the possibility that lignin is bound to hydrophobic faces of the microfibrils.
The dry, alkaline-deuterated spruce wood also showed a SANS peak, but not in the same position (Fig. 1). Instead, the corresponding characteristic spacing was 3.8 nm. Whatever the location of the introduced deuterium atoms, this implies microfibrils packed at a 3.8 nm centre-to-centre spacing similar to that observed for hardwoods (Thomas et al. 2014) where it was attributed to microfibrils 3.0 nm in diameter separated by a xylan chain interposed edge-on between their hydrophilic faces (Oehme et al. 2015; Dupree et al. 2015). The intensity of the d = 3.8 nm SANS peak, although small, was of the same order as the d = 3.0 nm peak observed for dry, vapour-deuterated spruce wood by Fernandes et al. (2011).
The observation of a SANS peak at d = 3.8 nm after alkaline deuteration implies that SANS intensity at similar q should also be observable in spruce wood deuterated conventionally in pure D2O, in addition to the intensity corresponding to the main 3.0 nm characteristic spacing. That is, for a 3.8 nm characteristic spacing to be observed, deuteration around the interfaces is required to provide SANS contrast but its exact location does not matter. Fernandes et al. (2011) commented that the SANS peaks reported for spruce wood saturated with D2O or H2O were visibly asymmetric, but that fitting two overlapping diffraction peaks would have required too many adjustable parameters. We revisited the SANS data sets reported by Fernandes et al. (2011), using 2D fitting to obtain a better separation of the coherent scattering from the non-coherent baseline which has a wider azimuthal distribution. It was assumed that the 3.8 nm characteristic spacing was relatively insensitive to hydration as in hardwoods (Thomas et al. 2014). At intermediate D2O contents it was possible to fit overlapped coherent scattering peaks at q values corresponding to the minor 3.8 nm characteristic spacing and to a major characteristic spacing of 3.1 nm at 5% D2O content or 3.3 nm at 10% D2O content (Figure SI 1). The major characteristic spacings were 0.1 nm less than when a single coherent scattering peak was fitted at these D2O levels by Fernandes et al. (2011). 2D fitting was not possible in the dry state due to the low intensity of coherent neutron scattering, nor could two peaks be separated at saturation with D2O or H2O because the major characteristic spacing, expanded by hydration, was then too close to the 3.8 nm characteristic spacing. The fitting process was challenging and due to the number of fitted variables the separation of the two fitted peaks should not in itself be taken as unambiguous evidence for the 3.8 nm characteristic spacing, but it is consistent with the observation of this characteristic spacing after alkaline deuteration.
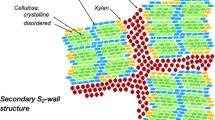
Cellulose aggregates in Sitka spruce wood thus appear to have microfibrils spaced at two centre-to-centre distances as measured by SANS: a major spacing at 3.0 nm in the dry state, corresponding to two microfibrils in direct contact (Fernandes et al. 2011), and a minor spacing about 0.8 nm wider than this. The minor spacing would be consistent with the insertion of one or more glucuronoxylan chains between adjacent microfibrils as in angiosperms and as described for conifer wood by Terrett et al. (2019). Softwood xylans with alternating substituted and unsubstituted xylosyl residues are considered to bind edge-on in a 21 helical conformation like an additional cellulose chain (Terrett et al. 2019) (Fig. 2). With no C-6 on the substituted xylosyl residues, there is a gap that could be filled by water (Langan et al. 2014; Jarvis 2018). If several xylan chains stack on one hydrophilic face of a microfibril in this way (Busse-Wicher et al. 2016), H2O or D2O molecules might enter or leave with some difficulty under ambient conditions (Jarvis 2018), but more readily at mildly alkaline pH, giving stable deuteration when the conditions are returned to ambient. Deuteration in this way would affect only the underlying cellulose hydroxyls, since the substituted xylan residue has no hydroxyls suitably placed for intermolecular hydrogen bonding (Fig. 2). The tg C-6 conformation of the cellulose chain may then be favoured as observed by FTIR, presumably leading to increased longitudinal stiffness compared to a bare microfibril surface (Altaner et al. 2014).

For a SANS characteristic spacing to be observed, the spacing must repeat, approximately at least, across an array of microfibrils. This implies separate arrays with microfibrils in direct contact (d = 3.0 nm dry) and with interposed xylan chains (d = 3.8 nm). A further implication is that microfibrils are separated by the width of only one xylan chain, as suggested by Terrett et al. (2019) in the sequence Mx..Mx..Mx where M is the cellulose microfibril and x is the xylan with its unsubstituted face towards M. Physical properties of the microfibrils would then differ between the alternating bare and coated faces.
The SANS data reported here are consistent with a model of the softwood cell wall (Terrett et al. 2019) in which xylan chain segments in a cellulose-like conformation are bound to hydrophilic faces of the cellulose microfibrils and increase their spacing to 3.8 nm, as in angiosperms (Thomas et al. 2014). The difference from angiosperm cell walls is that the lower xylan content in conifers permits direct contact between other hydrophilic microfibril surfaces, giving a dominant 3.0 nm spacing as in grasses (Thomas et al. 2015). These findings have implications for the mechanisms by which softwoods deform under mechanical stress and show recalcitrance to deconstruction.
Experimental
Longitudinal-radial microtome sections of mature Sitka spruce (Picea sitchensis) wood, with a nominal thickness of 20 μm, were prepared as described by Altaner et al. (2014) and deuterated in 0.1 M KOH/D2O (Altaner et al. 2014) or delignified at 70 °C with four successive additions of acetic acid and sodium chlorite as described by Wise (1946) followed by vacuum drying. Approximately 20 sections were mounted together to give sample dimensions of 0.5 mm × 30 mm × 8 mm for SANS. SANS experiments were carried out on Beamline D33 at ILL, Grenoble and data processed in the ILL GRASP software package as described by Thomas et al. (2014). Peak fitting was carried out as described in the Supplementary Methods.
References
Akerholm M, Salmen L (2001) Interactions between wood polymers studied by dynamic FT-IR spectroscopy. Polymer 42:963–969. https://doi.org/10.1016/s0032-3861(00)00434-1
Altaner C, Apperley DC, Jarvis MC (2006) Spatial relationships between polymers in Sitka spruce: proton spin-diffusion studies. Holzforschung 60:665–673. https://doi.org/10.1515/hf2006.112
Altaner CM, Thomas LH, Fernandes AN, Jarvis MC (2014) How cellulose stretches: synergism between covalent and hydrogen bonding. Biomacromol 15:791–798. https://doi.org/10.1021/bm401616n
Busse-Wicher M, Li A, Silveira RL, Pereira CS, Tryfona T, Gomes TCF, Skaf MS, Dupree P (2016) Evolution of Xylan substitution patterns in gymnosperms and angiosperms: implications for xylan interaction with cellulose. Plant Physiol 171:2418–2431. https://doi.org/10.1104/pp.16.00539
Dupree R, Simmons TJ, Mortimer JC, Patel D, Iuga D, Brown SP, Dupree P (2015) Probing the molecular architecture of Arabidopsis thaliana secondary cell walls using two- and three-dimensional C-13 solid state nuclear magnetic resonance spectroscopy. Biochemistry 54:2335–2345. https://doi.org/10.1021/bi501552k
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley DC, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci USA 108:E1195–E1203. https://doi.org/10.1073/pnas.1108942108
Hill SJ, Franich RA, Callaghan PT, Newman RH (2009) Nature’s nanocomposites: a new look at molecular architecture in wood cell walls. NZ J For Sci 39:251–257
Jarvis MC (2018) Structure of native cellulose microfibrils, the starting point for nanocellulose manufacture. Philos Trans R Soc A Math Phys Eng Sci 376:20170045. https://doi.org/10.1098/rsta.2017.0045
Jungnikl K, Paris O, Fratzl P, Burgert I (2008) The implication of chemical extraction treatments on the cell wall nanostructure of softwood. Cellulose 15:407–418. https://doi.org/10.1007/s10570-007-9181-5
Kennedy CJ, Sturcova A, Jarvis MC, Wess TJ (2007) Hydration effects on spacing of primary-wall cellulose microfibrils: a small angle X-ray scattering study. Cellulose 14:401–408. https://doi.org/10.1007/s10570-007-9129-9
Kubicki JD, Yang H, Sawada D, O’Neill H, Oehme D, Cosgrove D (2018) The shape of native plant cellulose microfibrils. Sci Rep 8:1–8. https://doi.org/10.1038/s41598-018-32211-w
Langan P, Petridis L, O’Neill HM, Pingali SV, Foston M, Nishiyama Y, Schulz R, Lindner B, Hanson BL, Harton S, Heller WT, Urban V, Evans BR, Gnanakaran S, Ragauskas AJ, Smith JC, Davison BH (2014) Common processes drive the thermochemical pretreatment of lignocellulosic biomass. Green Chem 16:63–68. https://doi.org/10.1039/c3gc41962b
Martinez-Abad A, Berglund J, Toriz G, Gatenholm P, Henriksson G, Lindstrom M, Wohlert J, Vilaplana F (2017) Regular motifs in xylan modulate molecular flexibility and interactions with cellulose surfaces. Plant Physiol 175:1579–1592. https://doi.org/10.1104/pp.17.01184
Martinez-Sanz M, Pettolino F, Flanagan B, Gidley MJ, Gilbert EP (2017) Structure of cellulose microfibrils in mature cotton fibres. Carbohyd Polym 175:450–463. https://doi.org/10.1016/j.carbpol.2017.07.090
Newman RH (1992) Nuclear magnetic resonance study of spatial relationships between chemical components in wood cell walls. Holzforschung 46:205–210. https://doi.org/10.1515/hfsg.1992.46.3.205
Newman RH, Hill SJ, Harris PJ (2013) Wide-Angle X-Ray scattering and solid-state nuclear magnetic resonance data combined to test models for cellulose microfibrils in mung bean cell walls. Plant Physiol 163:1558–1567. https://doi.org/10.1104/pp.113.228262
Oehme DP, Doblin MS, Wagner J, Bacic A, Downton MT, Gidley MJ (2015) Gaining insight into cell wall cellulose macrofibril organisation by simulating microfibril adsorption. Cellulose 22:3501–3520. https://doi.org/10.1007/s10570-015-0778-9
Terashima N, Kitano K, Kojima M, Yoshida M, Yamamoto H, Westermark U (2009) Nanostructural assembly of cellulose, hemicellulose, and lignin in the middle layer of secondary wall of ginkgo tracheid. J Wood Sci 55:409–416. https://doi.org/10.1007/s10086-009-1049-x
Terrett OM, Lyczakowski JJ, Yu L, Iuga D, Franks WT, Brown SP, Dupree R, Dupree P (2019) Molecular architecture of softwood revealed by solid-state NMR. Nat Commun 10:1–11. https://doi.org/10.1038/s41467-019-12979-9
Thomas LH, Altaner CM, Jarvis MC (2013a) Identifying multiple forms of lateral disorder in cellulose fibres. J Appl Crystallogr 46:972–979. https://doi.org/10.1107/s002188981301056x
Thomas LH, Forsyth VT, Sturcova A, Kennedy CJ, May RP, Altaner CM, Apperley DC, Wess TJ, Jarvis MC (2013b) Structure of cellulose microfibrils in primary cell walls from collenchyma. Plant Physiol 161:465–476. https://doi.org/10.1104/pp.112.206359
Thomas LH, Forsyth VT, Martel A, Grillo I, Altaner CM, Jarvis MC (2014) Structure and spacing of cellulose microfibrils in woody cell walls of dicots. Cellulose 21:3887–3895. https://doi.org/10.1007/s10570-014-0431-z
Thomas LH, Forsyth VT, Martel A, Grillo I, Altaner CM, Jarvis MC (2015) Diffraction evidence for the structure of cellulose microfibrils in bamboo, a model for grass and cereal celluloses. BMC Plant Biol. https://doi.org/10.1186/s12870-015-0538-x
Wise LE, Murphy M, d’Addieco AA (1946) Chlorite holocellulose, its fractionation and bearing on summative wood analysis and on studies on the hemicelluloses. Pap Trade J 122:35–43
Xu P, Donaldson LA, Gergely ZR, Staehelin LA (2007) Dual-axis electron tomography: a new approach for investigating the spatial organization of wood cellulose microfibrils. Wood Sci Technol 41:101–116. https://doi.org/10.1007/s00226-006-0088-3
Zhang T, Zheng YZ, Cosgrove DJ (2016) Spatial organization of cellulose microfibrils and matrix polysaccharides in primary plant cell walls as imaged by multichannel atomic force microscopy. Plant J 85:179–192. https://doi.org/10.1111/tpj.13102
Acknowledgments
We thank ILL (Grant No. 9-13-300), Grenoble for a grant of beamtime and C.M. Altaner (University of Christchurch, NZ) and V.T. Forsyth (ILL) for useful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thomas, L.H., Martel, A., Grillo, I. et al. Hemicellulose binding and the spacing of cellulose microfibrils in spruce wood. Cellulose 27, 4249–4254 (2020). https://doi.org/10.1007/s10570-020-03091-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-020-03091-z