
Abstract
Assisted gene flow can restore genetic diversity when genetic drift has driven deleterious alleles to high frequencies in small, isolated populations. Previous crosses among 20 populations of Gymnadenia conopsea documented the strongest heterosis and the weakest inbreeding depression in sparse and small populations, consistent with fixation of mildly deleterious alleles by genetic drift. We genotyped the populations used for crosses, and used 1200–1728 SNPs to test the following predictions: (1) heterosis increases with genetic differentiation (FST) to donor populations and decreases with genetic diversity in the recipient population, (2) inbreeding depression increases with genetic diversity, and (3) genetic diversity increases, and mean FST to other populations decreases, with population size and density. Pairwise FST ranged from very low to moderate (0.005–0.20) and genetic diversity varied moderately among populations (proportion of polymorphic loci = 0.52–0.75). However, neither FST between populations, nor genetic diversity in the recipient population, were related to the strength of heterosis. There was also no association between genetic diversity and the strength of inbreeding depression. Genetic diversity increased and mean FST decreased with population size, consistent with reduced diversity and increased differentiation of small populations by genetic drift. The results indicate that the loci conferring heterosis are not mirrored by overall population differentiation, and limited additional information on potential source populations for genetic rescue is gained by the genetic data. Instead, the use of controlled crosses can directly reveal positive effects of introducing new genetic material, and is a simple method with high potential in conservation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Habitat destruction and fragmentation may cause populations to become small and isolated. Small population size can lead to reduction in fitness because of fixation of deleterious alleles by genetic drift (Keller and Waller 2002), or because of inbreeding depression following mating between relatives (Ellstrand and Elam 1993). Both drift and inbreeding increase homozygosity, and reduced genetic diversity with lower population size is a common situation (Frankham 1996; Lammi et al. 1999; Leimu et al. 2006; Peterson et al. 2008; Ellwanger et al. 2022). Assisted gene flow has the potential to increase fitness by bringing in new alleles that will mask deleterious recessives, i.e., heterosis (Crow 1948; Whitlock et al. 2000). Heterosis can be expressed via increased embryo survival and number of offspring, and via higher offspring performance at later life stages (Fenster and Galloway 2000; Escobar et al. 2008; Groszmann et al. 2014; Oakley et al. 2019). The combined fitness effects are often strong and can persist to the second generation (reviewed in Frankham 2015), suggesting a high potential to increase population growth rate and persistence via genetic rescue (Ingvarsson 2001; Whiteley et al. 2015; Bell et al. 2019). Assisted gene flow is expected to have the highest fitness effect when the recipient population is small, due to the accumulation of a high drift load (Willi et al. 2007; Escobar et al. 2008; Oakley and Winn 2012; Pickup et al. 2013), and also when there is substantial genetic differentiation between populations, due to a high likelihood of differences in the identity and frequency of fixed deleterious recessive alleles (Charlesworth and Willis 2009).
The actual use of assisted gene flow in conservation has been limited (Bell et al. 2019), mainly due to concerns of negative fitness outcomes, i.e., outbreeding depression (Tallmon et al. 2004; Groom et al. 2006; Chung et al. 2023). It has been recommended that assisted gene flow between populations should be restricted to situations where there is evidence of inbreeding depression in a population, and that genetic information should be used to identify appropriate source populations (Edmands 2006; Hedrick and Fredrickson 2010; Hohenlohe et al. 2021). The suggested rationale is often "the closer, the better”, where genetically and adaptively similar individuals from populations in close proximity, located in similar habitats, are thought to be the most suitable sources (McKay et al. 2005; Edmands 2006; Chung et al. 2023; Frankham et al. 2011). However, if drift is the main cause of reduced genetic variation, the most suitable sources could be more distant populations, that exhibit stronger genetic differentiation towards the target population. It is thus unclear how genetic differentiation should be used to predict the effect of crosses between populations. Indeed, several studies have failed to find a link between genetic differentiation among populations and the fitness effect when crossing them (Willi et al. 2007; Pickup et al. 2013; Oakley et al. 2019). To clarify how well both heterosis (or outbreeding depression) and inbreeding depression actually can be predicted from genetic information, we need studies that link genetic data with the fitness effects of crosses between natural populations.
The orchid Gymnadenia conopsea is common in Scandinavia, but currently declining in large parts of its range. Regional extinctions and rapid population size reductions are especially evident in management-dependent meadow populations in southeastern Sweden (Tyler 2007; Jonsell 2010; Johansson and Petersson 2016). The island Öland, Fig. 1, represents a core distribution area with low genetic differentiation and high genetic diversity (Söderquist et al., unpublished results). Crosses between populations on Öland documented variable fitness effects at the seed stage, ranging from strong heterosis (96% increase in seed production in crosses between populations relative to outcrossing within population) to moderate outbreeding depression (22% decrease), and estimates of heterosis and inbreeding depression were negatively correlated across populations (Söderquist et al. 2020), consistent with a history of genetic drift (Oakley and Winn 2012; Lohr and Haag 2015). This indicates that, even in a region with low genetic differentiation and high genetic diversity, a significant drift load exists. On average, heterosis was strongest and inbreeding depression weakest in sparse and small populations, but the relationship with population size was weak (Söderquist et al. 2020). This could be because census and effective population size are only weakly correlated, e.g., due to fluctuating population size, overlapping generations, or among-individual variation in reproductive output (Frankham 1995; Vucetich et al. 1997), and if so, genetic diversity may provide a better estimate of effective population size (Lohr and Haag 2015; Spigler et al. 2017).

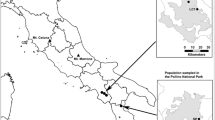
Map of the island Öland with locations of the 30 Gymnadenia conopsea populations included in this study. A solid dot represents a recipient population and a star a donor population used in controlled crosses in Söderquist et al. (2020). An open circle represents an additional population with estimates of genetic statistics and population size. For additional information see Table S1
To clarify the link between population census size and density, genetic diversity and differentiation, and the expression of heterosis and inbreeding depression, we genotyped all populations involved in the previous crossing experiment (Söderquist et al. 2020) using 1,200 SNPs. We evaluated the influence of genetic drift by examining whether (i) genetic diversity increases and mean genetic differentiation of a population decreases with census population size or density. We further tested whether (ii) the strength of heterosis estimated in the crossing experiment increases with genetic differentiation to pollen donor populations and with reduced genetic diversity in the recipient population, and (iii) the strength of inbreeding depression increases with genetic diversity.
Material and methods
Study species
Gymnadenia conopsea (L.) Br. R. is a long-lived terrestrial orchid, distributed across Eurasia (Hultén and Fries 1986). Flowering individuals produce one inflorescence with ca. 10–60 nectar-rewarding flowers. Individual flowers open successively from the bottom of the inflorescence up, and the flowering period lasts up to a month. Each flower can remain open for at least one week if not pollinated (L. Söderquist, personal observation). Pollinia are attached to pollinators in pairs or individually, and each pollinium can pollinate multiple flowers (Sletvold et al. 2012). Gymnadenia conopsea is self-compatible, but mainly outcrossing (Sletvold et al. 2012; Söderquist et al. 2020). Self-pollen deposition is higher at low compared to high population density (Söderquist et al. 2020), consistent with more inbreeding in sparse populations. Main pollinators in the study area are diurnal and nocturnal hawkmoths, moths, and butterflies, and natural fruit set is typically 70–90% (Chapurlat et al. 2015, 2019; Joffard et al. 2022). Fruits mature after 3–6 weeks, and each fruit contains hundreds to thousands of dust-like, wind-dispersed seeds. Germination occurs in spring, as seeds require more than 8 weeks of cold stratification to break seed dormancy (S. Malmgren, personal communication, October 13, 2017). Individuals flower 4–5 years after germination in favorable conditions in cultivation (S. Malmgren, personal communication, October 13, 2017), and are known to live for more than 30 years in natural populations (Tye et al. 2018).
Study site and populations
To examine if estimates of genetic differentiation and diversity were associated with heterosis and inbreeding depression, and related to population size and density, we included a total of 30 populations on the Baltic island Öland, Sweden. We defined a population as a collection of individuals separated from other groups of conspecifics by a minimum of 100 m. Twenty-three of the populations are identical to the 20 recipient populations and the three donor populations included in a previous crossing experiment where we quantified heterosis and inbreeding depression (Söderquist et al. 2020, Fig. 1). In that study, we conducted self-pollination (S), outcross pollination within the population (W, using multiple donors), and outcross pollination between populations (B). For the between-population crosses, we used a pollen mix from the three large donor populations (ca. 10 plants from each population) in all 20 recipient populations. The number of pollen donors was similar for both within- and between-population outcrossings. The experiment included 16 plants per recipient population, with all cross types conducted on each individual. For each cross type, fitness was estimated as number of viable seeds per fruit produced. Inbreeding depression (δID) and heterosis/outbreeding depression (δOD) were estimated as relative performance for each maternal family: δID = (WW-WS)/max(WW, WS) and δOD = (WB-WW)/max(WB, WW). Relative performance gives a value between -1 and 1 (Ågren and Schemske 1993). The mean across the 16 maternal families was used as the estimate of inbreeding depression and outbreeding depression in each population (see Söderquist et al. 2020 for more details). Because the three donor populations were spread across Öland, there was limited variation in mean geographic distance from donors to recipient populations (range from 34 to 67 km, with a mean of 46 km). The seven additional populations were included to clarify the genetic structure on Öland. The 30 populations were separated by 1.2 to 124.1 km (Fig. 1).
To quantify genetic diversity and differentiation among populations, we sampled eight individuals per population, except for the smallest population Resmo, where we only found seven. This resulted in a total of 239 individuals from the 30 populations. From each plant, we collected leaf tissue on silica gel for DNA extraction with the Qiagen Plant DNeasy kit (Qiagen, Germantown, MD). All samples were collected in the summer of 2017. We quantified census population size as the number of flowering individuals. We counted all flowering individuals in populations with less than approximately 1,000 individuals. In larger populations, we extrapolated a count of the first thousand to the total area, keeping account of local density variation. For 22 of the populations, population size estimates were available for three or four years (2017–2020), and we used the mean of the available years (Table S1). Density estimates were available for 18 of the populations, and the mean across years was used (for more details, see Söderquist et al. (2020) and Table S1). Population size and density were significantly positively correlated (r = 0.50, p = 0.04, n = 18).
Sequencing and genotyping
We genotyped all DNA samples (n = 239) using a custom designed Illumina iSelect chip with 2,803 SNP assays (hereafter referred to as the Orchid3K chip). To construct the Orchid3K chip, we initially used double-digest RAD sequencing (ddRAD-seq) to generate genome-wide polymorphism data (Söderquist et al. unpublished results). Genotyping was performed using the Illumina Infinium assay and the results were analyzed using the software GenomeStudio 2.0.3 (Illumina Inc.) at the SNP&SEQ Technology Platform of SciLifeLab, Uppsala, Sweden. The SNP conversion rate was 77.5%, resulting in 2,108 SNPs with an average call rate of 93.6%. Reproducibility was 100% (0 conflicts in 54,953 duplicate tests). Only biallelic SNPs were retained. We found six duplicated pairs of genotypes (two in Mensalvaret, and one each in Enerumsalvaret, Högby, Kristinelund, and Vedby), indicating clonality, and removed one individual from each pair (n = 6). We removed 380 invariant loci across the data set. The resulting dataset contained 1728 loci from 233 individuals in 30 populations, and was used in estimation of FST, principal component analysis (PCA), and conStruct. For population-level analyses of genetic diversity estimates we further removed 250 loci and three individuals with > 5% missing values and 278 loci with minor allele frequency < 2.5%, resulting in a filtered dataset including 1200 loci in 230 individuals from 30 populations.
Genetic structure
To account for potential confounding factors that could influence the crossing effect, we investigated the genetic structure of the 30 populations using the R-package conStruct v1.0.4 (Bradburd et al. 2018) in R version 4.1.2 (R Core Team 2021), based on both a discrete (non-spatial) and a continuous (spatial) model for one to four clusters (K). Cross-validation of conStruct runs suggested highest predictive accuracy for the spatial model with K equal to one (Fig. S1), i.e. all populations belonging to the same genetic cluster, and we do not discuss these results further.
Genetic differentiation and diversity
All summary statistics were calculated in R version 4.1.2 (R Core Team 2021), using the filtered dataset. We first calculated genetic differentiation between populations using FST (Weir and Cockerham 1984), and the proportion of polymorphic loci (P). We further calculated the allelic richness (AR) using the allelic.richness function in hierfstat (Goudet 2005). All invariant loci in each population were subsequently removed before calculating expected heterozygosity (HE), observed heterozygosity (HO), and the inbreeding coefficient (FIS). We estimated HE, HO, and FIS, using the basic.stats function from the hierfstat package. For the calculation of HE and HO, we excluded any loci that significantly deviated from Hardy–Weinberg equilibrium (HWE) and randomly sampled the same number of loci in each population, with the number set by the smallest number of HWE loci observed in a population (n = 1,149). When calculating FIS, we randomly sampled the same number of loci in each population (n = 1,200). Values for AR, HE, HO, and FIS were averaged across all loci. As P, AR, HE and HO were all highly positively correlated (all rs ≥ 0.7, p < 0.001, n = 30), we focused on P as our measure of genetic diversity in statistical analyses. As nine individuals, from three different populations, were removed in the filtering step, we examined the effect of variation in sample size on P by comparing the original analyses (n = 6–8 individuals per population) with those based on a dataset with six randomly chosen individuals per populations. Estimates of P were highly correlated (rs = 0.91, p < 0.001), and we report results based on the dataset with original sample sizes. Genetic differentiation with the donor populations was calculated as the mean FST between recipient populations and the three donor populations. As we did not know whether the donor populations contributed equally to seed siring, we also explored models using maximum FST between recipient and donor populations. Results were nearly identical, and we report those based on mean FST. We tested for isolation-by-distance (IBD) using a Mantel test with 1,000,000 replicates from the R package vegan (Oksanen et al. 2019). We chose not to estimate effective population size (Ne). This is because estimation methods are based on assumptions (e.g., discrete generations, no age structure, constant Ne) that will yield biased estimates, in particular for long-lived iteroparous species such as Gymnadenia conopsea (lifespan > 15 years common, can live more than 40 years; Tholstrup et al. 2024), where number of cohorts included in the sample (n = 8 per population) is expected to deviate strongly from generation length (Waples et al. 2014).
The relationships of population size and density, with genetic diversity and differentiation
To test if genetic diversity increased and mean genetic differentiation (FST) decreased with population size and density, we used multiple linear regressions. We calculated mean genetic differentiation as the mean population pairwise FST between the recipient population and all other populations. To improve normality, population size and density were both ln transformed. We illustrated the relationships using added variable plots (Cook and Weisberg 1989). It should be noted that genetic diversity and differentiation (mean pairwise FST) were strongly negatively correlated (r = − 0.98, p < 0.001, n = 18).
Effects of genetic differentiation and diversity on heterosis and inbreeding depression
We used univariate linear regressions to test if heterosis in the recipient population increased with mean genetic differentiation to the donor populations and decreased with genetic diversity in the recipient population, and if inbreeding depression increased with genetic diversity. The smallest population, Resmo, had an extreme estimate of inbreeding depression (-0.53), and analyses were also run excluding this population (Figs. S2 and 4). It should again be noted that genetic diversity and mean differentiation (FST) to donor populations in the crossing experiment were strongly negatively correlated (r = -0.95, p < 0.001, n = 18).
Results
Genetic differentiation and diversity
Pairwise population genetic differentiation (FST) ranged from very low to moderate (0.005–0.20, Fig. S3), and mean pairwise FST across all population pairs was 0.09. The weakest differentiation was found between Hörninge and Störlinge (6.5 km apart) and the strongest between Klinta and Mensalvaret (97.3 km apart, Figs. 1 and S3). Two populations, Hörninge and Störlinge, showed low differentiation to most other populations, with a mean FST of 0.047 and 0.048, respectively (Figs. 1 and S3). Three populations showed a high mean value towards all other populations (Mensalvaret: FST = 0.14, Klinta: FST = 0.13, Västra alvaret: FST = 0.13). The proportion of polymorphic loci (P) within each population ranged from 0.52 to 0.75 with a mean of 0.66 (Table S1). Allelic richness (AR) ranged from 1.35 to 1.69 and averaged 1.57 across all populations (Table S1). Observed heterozygosity (HO) ranged from 0.19 to 0.22 (mean 0.21), and expected heterozygosity (HE) from 0.18 to 0.22 with a mean of 0.20 (Table S1). The inbreeding coefficient (FIS) ranged from -0.15 to 0.04 with an average of -0.002 (Table S1). Population pairwise genetic differentiation (FST) increased with geographic distance, indicating significant isolation-by-distance (p < 0.001, Mantel’s r = 0.21, n = 30; Fig. S4).
The relationships of population size and density, with genetic diversity and differentiation
Genetic diversity (P) significantly increased with increasing population size (β = 0.02, p = 0.01), but was not related to density of flowering plants (β = -0.04, p = 0.13, Fig. 2a, b). The full model, including both population size and density, explained 38% of the variation in genetic diversity (R2 = 0.38, F2,15 = 4.67, p = 0.03, n = 18), while a model including only population size explained 28% of the variation in genetic diversity (R2 = 0.28, F1,16 = 6.28, p = 0.02, n = 18). Mean genetic differentiation (FST) decreased with population size (β = -0.01, p = 0.003), but was not significantly related to density of flowering plants (β = 0.02, p = 0.06). Together, both factors explained 45% of the variation in mean genetic differentiation (R2 = 0.45, F2,15 = 6.13, p = 0.01, n = 18, Fig. 2c, d), while population size alone explained 30% of the variation in mean genetic differentiation (R2 = 0.30, F1,16 = 6.89, p = 0.018, n = 18). Due to the strong negative correlation between P and mean FST, the associations between population size and genetic diversity (Fig. 2a) and between population size and genetic differentiation (Fig. 2c) show similar but inverted relationships.

Added variable plots of proportion of polymorphic loci (a, b), and mean pairwise population genetic differentiation (FST) (c, d) in relation to population size and population density (both ln transformed). Trend line indicates a significant relationship
Effects of genetic differentiation and diversity on heterosis and inbreeding depression
Heterosis was not significantly related to mean genetic differentiation to the three donor populations (β = 2.67, R2 = 0.07, F1,18 = 1.25, p = 0.28, n = 20, Fig. 3a), or to genetic diversity in the recipient population (β = -1.50, R2 = 0.13, F1,18 = 2.65, p = 0.12, n = 20, Fig. 3b). Inbreeding depression was not related to genetic diversity, neither when including the smallest population, Resmo, (β = 0.89, R2 = 0.06, F1,18 = 1.09, p = 0.31, n = 20, Fig. S2), nor when excluding it (β = -0.20, R2 = 0.01, F1,17 = 0.11, p = 0.74, n = 19, Fig. 4).

The relationship between heterosis and a mean genetic differentiation (FST) to the three donor populations, and b the proportion of polymorphic loci in the recipient population. A positive value indicates heterosis, and a negative value indicates outbreeding depression. Horizontal error bars indicate range of FST, and vertical error bars indicate standard deviation of heterosis

The relationship between inbreeding depression and proportion polymorphic loci, excluding the smallest population, Resmo, showing extreme values of ID, see Fig. S2 for results including it. Error bars indicate standard deviation of inbreeding depression
Discussion
The results are consistent with genetic drift causing small Gymnadenia conopsea populations to have low genetic diversity and strong genetic differentiation compared with other populations. However, the variation in fitness effects observed in the previous crossing experiment (Söderquist et al. 2020) could not be explained by genetic data from the populations. This indicates that the loci conferring heterosis are not necessarily reflected by overall population differentiation, and suggests that management efforts may gain little useful information from the generation of this type of genetic data.
Contrary to expectations, we did not detect any significant relationship between the documented heterosis effect in previous crossings and the genetic differentiation to donor populations. Instead, our results reinforce several previous studies that found genetic distance to be a poor predictor of the outcome of between-population crosses (Willi et al. 2007; Pickup et al. 2013; Oakley et al. 2019). While a stronger pairwise genetic differentiation should imply a higher probability of differing identity of deleterious alleles fixed by random genetic drift in two populations (Charlesworth and Willis 2009), the results of this and previous studies show that FST does not directly reflect differentiation at the loci contributing to fitness effects following crosses. A recent study of the annual Clarkia pulchella constitutes an exception, where both single stage and lifetime heterosis increased with genetic distance between populations, across a range of FST values similar to the present study (Bontrager and Angert 2019). In the Clarkia system, genetic and environmental differences were correlated, but it is unlikely that large environmental differences between populations would be beneficial. It seems more likely that other system-specific differences can explain the inconsistent results, i.e., differences in the genetic basis for the observed heterosis. Taken together, current studies suggest a weak link between genetic differentiation and the effects of assisted gene flow, indicating a limited potential for FST estimates to guide management in the design of genetic rescues.
We expected to see stronger heterosis and weaker inbreeding depression in populations with less genetic diversity, but the strength of both heterosis and inbreeding depression was unrelated to genetic diversity within G. conopsea populations. This suggests that overall diversity is not strongly correlated with the occurrence of fixed deleterious alleles, at least not for those responsible for effects at the examined life stage. In Söderquist et al. (2020), we only quantified heterosis and inbreeding depression at a single life stage (seed production), and we cannot dismiss the possibility of an association between FST or genetic diversity and the crossing effects on lifetime fitness in G. conopsea.
The study was designed without prior knowledge of genetic structure, and a stronger experimental design could have been achieved by maximizing the range of pairwise genetic differentiation, and by including populations with particularly low genetic diversity. However, this would have required including populations outside of the island of Öland, thereby also increasing environmental differences. Populations on Öland belong to a southeastern Scandinavian genetic group, that in general has high genetic diversity and moderate genetic differentiation (Söderquist et al., unpublished results). To substantially increase the range of pairwise population differentiation, we would need to cross Öland populations with populations from the northwestern Scandinavian group, where genetic diversity in general is lower, and where differentiation can be very strong (max FST = 0.5; Söderquist et al., unpublished results). It would be interesting to test whether associations between genetic metrics and phenotypic effects of crosses would emerge from such crosses, and also whether effects were reciprocal. We would expect the strongest heterosis in small, northern populations, although potential negative effects of habitat and climatic differences would be counteractive. For potential reinforcements and restoration efforts on Öland, local, large populations should be optimal pollen or seed sources.
We used a mix of three donor populations in all between-population crosses, which means that we covered a range in genetic differentiation towards the recipient population. Using max donor FST instead of mean FST did not have any effect on the results, but it is still possible that variation in how much different donor populations contributed to seed siring blurs relationships.
The predicted associations between genetic diversity, heterosis, and inbreeding depression are based on populations at demographic equilibrium, and will not necessarily hold if bottlenecks, fragmentation, or fluctuating population sizes have occurred (Spigler et al. 2017). All of these processes may have influenced the G. conopsea populations included in this study. Land use change has been dramatic during the 1900’s, increasing fragmentation and reducing the number and size of populations across a large part of the historical range (Rydberg et al. 2001; Fröberg 2006; Johansson and Petersson 2016; Länsstyrelsen i Jönköpings län 2017). In addition, variation in the fraction of both dormant and flowering individuals is common among years in G. conopsea (Tye et al. 2018, Söderquist et al., unpublished data), which could contribute to fluctuating population sizes. Theoretical models have shown that for a given level of genetic diversity, populations experiencing stronger fluctuations in size should express stronger heterosis and lower inbreeding depression, compared to more stable populations (Spigler et al. 2017). We found that the strength of heterosis and inbreeding depression varied substantially among population with similar genetic diversity, but cannot presently link this to any degree of fluctuations. As G. conopsea is a long-lived perennial we would need long-term demographic data to quantify fluctuations and elucidate the interactive effects of demography and genetic diversity on the distribution of genetic load across populations.
Genetic diversity increased with population size, in line with theoretical predictions and previous empirical studies (Frankham 1996; Lammi et al. 1999; Leimu et al. 2006; Peterson et al. 2008; Ellwanger et al. 2022). Population size explained 28% of the variation in genetic diversity, which indicates a fair correlation between census and effective population size. However, particularly small populations (< 160 flowering individuals) cover a broad range in genetic diversity (Table S1; Fig. 2a), perhaps because some have been small for a long time, while others have experienced recent size reductions. Population flowering density was, however, not associated with genetic diversity. In a previous study, we found higher self-pollen deposition in sparse compared to dense patches within a population (Söderquist et al. 2020), suggesting that sparse populations experience more inbreeding than dense populations. It could be that the variation documented among patches within a population does not scale up to variation among populations, but the lack of a density effect could also be due to a weak link between self-pollen deposition and fertilization rate (Goodwillie and Weber 2018), or to inbred individuals being removed by selection before reaching the flowering stage (Sletvold et al. 2012; 2024). In sum, the data suggest that population size alone can serve as a crude proxy for genetic differentiation and diversity at this spatial scale, but the variance was high among small populations.
Gymnadenia conopsea is not a very rare species in immediate need of assisted gene flow, especially not on Öland. However, species in the early phases of decline can provide general insights regarding the relationships between population size and genetic diversity and differentiation. We suggest that the use of controlled crosses can serve as a feasible, low-cost method for directly evaluating effects of assisted gene flow, without a need for genetic analyses. This is important for managers working with conservation of plant species, as genetic data are expensive, and analyses and interpretations usually require external expert knowledge. To determine the full potential of genetic rescue, fitness effects should ideally be followed beyond the seed stage. However, similar studies have shown that benefits are greater as more of the life cycle is included, and also as environments become more stressful (Frankham et al. 1995; Frankham 2015). Conducting crosses will clearly not be practical in all plant systems of conservation interest, but should be applicable in a wide range of herbaceous species. We therefore encourage managers to consider the use of controlled crosses to determine fitness effects when selecting suitable source populations for genetic rescue, and to use population size as a proxy for genetic diversity.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Ågren J, Schemske DW (1993) Outcrossing rate and inbreeding depression in two annual monoecious herbs Begonia hirsuta and B. Semiovata. Evolution 47:125–135
Bell DA, Robinson ZL, Funk WC, Fitzpatrick SW, Allendorf FW, Tallmon DA, Whiteley AR (2019) The exciting potential and remaining uncertainties of genetic rescue. Trends Ecol Evol 34:1070–1079. https://doi.org/10.1016/j.tree.2019.06.006
Bontrager M, Angert AL (2019) Gene flow improves fitness at a range edge under climate change. Evol Lett 3:55–68. https://doi.org/10.1002/evl3.91
Bradburd GS, Coop GM, Ralph PL (2018) Inferring continuous and discrete population genetic structure across space. Genetics 210:33–52. https://doi.org/10.1534/genetics.118.301333
Chapurlat E, Ågren J, Sletvold N (2015) Spatial variation in pollinator-mediated selection on phenology, floral display and spur length in the orchid Gymnadenia conopsea. New Phytol 208:1264–1275. https://doi.org/10.1111/nph.13555
Chapurlat E, Ågren J, Anderson J, Friberg M, Sletvold N (2019) Conflicting selection on floral scent emission in the orchid Gymnadenia conopsea. New Phytol 222:2009–2022. https://doi.org/10.1111/nph.15747
Charlesworth D, Willis JH (2009) The genetics of inbreeding depression. Nat Rev Genet 10:783–796. https://doi.org/10.1038/nrg2664
Chung MY, Merilä J, Li J, Mao K, López-Pujol J, Tsumura Y, Chung MG (2023) Neutral and adaptive genetic diversity in plants: An overview. Front Ecol Evol 11:1116814. https://doi.org/10.3389/fevo.2023.1116814
Cook RD, Weisberg S (1989) Regression diagnostics with dynamic graphics. Technometrics 31:277–291. https://doi.org/10.1080/00401706.1989.10488547
Crow JF (1948) Alternative hypotheses of hybrid vigor. Genetics 33:477–487. https://doi.org/10.1093/genetics/33.5.477
Edmands S (2006) Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation. Mol Ecol 16:463–475. https://doi.org/10.1111/j.1365-294X.2006.03148.x
Ellstrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Ellwanger C, Steger L, Pollack C, Wells R, Fant JB (2022) Anthropogenic fragmentation increases risk of genetic decline in the threatened orchid Platanthera leucophaea. Ecol Evol 12:e8578. https://doi.org/10.1002/ece3.8578
Escobar JS, Nicot A, David P (2008) The different sources of variation in inbreeding depression, heterosis and outbreeding depression in a metapopulation of Physa acuta. Genetics 180:1593–1608
Fenster CB, Galloway LF (2000) Inbreeding and outbreeding depression in natural populations of Chamaecrista fasciculata (Fabaceae). Conserv Biol 14:1406–1412
Frankham R (1995) Effective population size/adult population size ratios in wildlife: a review. Genet Res 66:95–107. https://doi.org/10.1017/S0016672300034455
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508. https://doi.org/10.1046/j.1523-1739.1996.10061500.x
Frankham R (2015) Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow. Mol Ecol 24:2610–2618. https://doi.org/10.1111/mec.13139
Frankham R, Ballou JD, Eldridge MDB, Lacy RC, Ralls K, Dudash MR, Fenster CB (2011) Predicting the probability of outbreeding depression. Conserv Biol 25:465–475. https://doi.org/10.1111/j.1523-1739.2011.01662.x
Fröberg L (2006) Blekinges flora. Svenska Botaniska Föreningen, Uppsala
Goodwillie C, Weber JJ (2018) The best of both worlds? A review of delayed selfing in flowering plants. Am J Bot 105:641–655. https://doi.org/10.1002/ajb2.1045
Goudet J (2005) hierfstat, a package for r to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x
Groom MJ, Meffe GK, Carroll CR (2006) Principles of conservation biology, 3rd edn. Sinauer Associates, Sunderland
Groszmann M, Gonzalez-Bayon R, Greaves IK, Wang L, Huen AK, Peacock WJ, Dennis ES (2014) Intraspecific Arabidopsis hybrids show different patterns of heterosis despite the close relatedness of the parental genomes. Plant Physiol 166:265–280
Hedrick PW, Fredrickson R (2010) Genetic rescue guidelines with examples from Mexican wolves and Florida panthers. Conserv Genet 11:615–626
Hohenlohe PA, Funk WC, Rajora OP (2021) Population genomics for wildlife conservation and management. Mol Ecol 30:62–82. https://doi.org/10.1111/mec.15720
Hultén E, Fries M (1986) Atlas of the North European vascular plants: north of the tropic of cancer. Koeltz Scientific Books, Köningstein
Ingvarsson PK (2001) Restoration of genetic variation lost—the genetic rescue hypothesis. Trends Ecol Evol 16:62–63. https://doi.org/10.1016/S0169-5347(00)02065-6
Joffard N, Olofsson C, Friberg M, Sletvold N (2022) Extensive pollinator sharing does not promote character displacement in two orchid congeners. Evolution 76:749–764
Johansson BG, Petersson J (2016) Gotlands flora. Gotlands Botaniska Förening, Visby
Jonsell L (2010) Upplands flora. Svenska Botaniska Föreningen förlaget, Uppsala
Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17:230–241
Lammi A, Siikamäki P, Mustajärvi K (1999) Genetic diversity, population size, and fitness in central and peripheral populations of a rare plant Lychnis viscaria. Conserv Biol 13:1069–1078. https://doi.org/10.1046/j.1523-1739.1999.98278.x
Länsstyrelsen i Jönköpings län (2017) Floraövervakning i odlingslandskapet, Floraväktarna 1994–2017.
Leimu R, Mutikainen P, Koricheva J, Fischer M (2006) How general are positive relationships between plant population size, fitness and genetic variation? J Ecol 94:942–952. https://doi.org/10.1111/j.1365-2745.2006.01150.x
Lohr JN, Haag CR (2015) Genetic load, inbreeding depression, and hybrid vigor covary with population size: an empirical evaluation of theoretical predictions: genetic load and population size in Daphnia. Evolution 69:3109–3122. https://doi.org/10.1111/evo.12802
McKay JK, Christian CE, Harrison S, Rice KJ (2005) “How local is local?”-a review of practical and conceptual issues in the genetics of restoration. Restor Ecol 13:432–440. https://doi.org/10.1111/j.1526-100X.2005.00058.x
Oakley CG, Winn AA (2012) Effects of population size and isolation on heterosis, mean fitness, and inbreeding depression in a perennial plant. New Phytol 196:261–270. https://doi.org/10.1111/j.1469-8137.2012.04240.x
Oakley CG, Lundemo S, Ågren J, Schemske DW (2019) Heterosis is common and inbreeding depression absent in natural populations of Arabidopsis thaliana. J Evol Biol 32:592–603. https://doi.org/10.1111/jeb.13441
Oksanen J, Blanchet F, Friendly M, et al (2019) Vegan: community ecology package. https://cran.r-hub.io/web/packages/vegan/vegan.pdf
Peterson A, Bartish IV, Peterson J (2008) Effects of population size on genetic diversity, fitness and pollinator community composition in fragmented populations of Anthericum liliago L. Plant Ecol 198:101–110. https://doi.org/10.1007/s11258-007-9388-4
Pickup M, Field DL, Rowell DM, Young AG (2013) Source population characteristics affect heterosis following genetic rescue of fragmented plant populations. Proc R Soc B Biol Sci 280:20122058
R Core Team (2021) R: a language and environment for statistical computing
Rydberg H, Wanntorp H-E, Eriksson T (2001) Sörmlands flora. Botaniska Sällskapet, Stockholm
Sletvold N, Grindeland JM, Zu P, Ågren J (2012) Strong inbreeding depression and local outbreeding depression in the rewarding orchid Gymnadenia conopsea. Conserv Genet 13:1305–1315. https://doi.org/10.1007/s10592-012-0373-7
Sletvold N, Joffard N, Söderquist L (2024) Fine-scale genetic structure in the orchid Gymnadenia conopsea is not associated with local density of flowering plants. Am J Bot 111:e16273. https://doi.org/10.1002/ajb2.16273
Söderquist L, Broberg A, Rosenberg V, Sletvold N (2020) Predicting heterosis and inbreeding depression from population size and density to inform management efforts. J Appl Ecol 57:1459–1468. https://doi.org/10.1111/1365-2664.13643
Spigler RB, Theodorou K, Chang S-M (2017) Inbreeding depression and drift load in small populations at demographic disequilibrium: inbreeding depression in small populations. Evolution 71:81–94. https://doi.org/10.1111/evo.13103
Tallmon D, Luikart G, Waples R (2004) The alluring simplicity and complex reality of genetic rescue. Trends Ecol Evol 19:489–496
Tholstrup DW, Sletvold N, Øien D-I, Moen A, Colchero F, Dahlgren JP (2024) Actuarial senescence progresses similarly across sites and species in four boreal orchids. J Ecol 112:443–686. https://doi.org/10.1111/1365-2745.14262
Tye M, Dahlgren JP, Øien D-I, Moen A, Sletvold N (2018) Demographic responses to climate variation depend on spatial- and life history-differentiation at multiple scales. Biol Conserv 228:62–69. https://doi.org/10.1016/j.biocon.2018.10.005
Tyler T (2007) Floran i Skåne: arterna och deras utbredning. Lunds Botaniska Förening, Lund
Vucetich JA, Waite TA, Nunney L (1997) Fluctuating population size and the ratio of effective to census population size. Evolution 51:2017–2021. https://doi.org/10.2307/2411022
Waples RS, Antao JT, Luikart G (2014) Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 197:769–780. https://doi.org/10.1534/genetics.114.164822
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon DA (2015) Genetic rescue to the rescue. Trends Ecol Evol 30:1–49
Whitlock MC, Ingvarsson PK, Hatfield T (2000) Local drift load and the heterosis of interconnected populations. Heredity 84:452–457. https://doi.org/10.1046/j.1365-2540.2000.00693.x
Willi Y, Kleunen MV, Dietrich S, Fischer M (2007) Genetic rescue persists beyond first-generation outbreeding in small populations of a rare plant. Proc R Soc B 274:2357–2364. https://doi.org/10.1098/rspb.2007.0768
Acknowledgements
We thank Julia Braun, Anna Broberg, Paschalis Chatzopolous, Alessio Costa, Emma Hellkvist, Kasper Kupka, Ieva Petrauskaitė, Viktor Rosenberg, Maria Uscka-Perzanowska, Linus Vikström, Mari-Helen Westlund and Fiona White for help with collecting samples, estimating population size and density, and general assistance in the field. Jon Ågren provided helpful comments on the manuscript. Genotyping was performed by the SNP&SEQ Technology Platform in Uppsala (www.genotyping.se). The facility is part of the National Genomics Infrastructure supported by the Swedish Research Council for Infrastructures and Science for Life Laboratory, Sweden.
Funding
Open access funding provided by Uppsala University. The study was funded by Grants 2014-00601 and 2018-01397 from The Swedish Research Council Formas to NS.
Author information
Authors and Affiliations
Contributions
NS, SK and LS conceived the study questions, and LS collected the field data. LS analyzed the data and led the writing of the manuscript. All authors contributed critically to revisions.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Söderquist, L., Karrenberg, S. & Sletvold, N. Genetic differentiation and diversity do not explain variation in heterosis or inbreeding depression: empirical evidence from a long-lived iteroparous plant. Conserv Genet (2024). https://doi.org/10.1007/s10592-024-01641-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10592-024-01641-7