
Abstract
Genetic linkage maps based on single nucleotide polymorphisms (SNPs) represent an essential tool for a variety of genomic analyses. Today, next-generation sequencing (NGS) enables rapid genotyping of different mapping populations based on thousands of SNPs and the construction of highly saturated linkage maps. Nevertheless, missing data in the genotyping of the parental lines creates a bottleneck that determines the number of SNPs that can be used for the linkage map. As a proof of concept, a highly saturated genetic linkage map was constructed using the imputed genotypic data of a recombinant inbred line (RIL) population and the limited genotypic information of its parental lines. Two ABH genotype files were created from a pseudo-parental genotypic data set that includes all the SNPs present in the RIL population. In the first ABH file pseudo-parental 1 was considered parental A, while in the second pseudo-parental 1 was considered parental B. These two duplicate ABH genotype files were merged by chromosome and subjected to linkage map analysis. Since the ABH data were duplicated, two mirrored linkage groups were generated per chromosome. The correct linkage map was identified and selected based on the partial genotypic data of the parental lines. This strategy was effective for constructing a highly saturated linkage map of 33,421 SNPs based on the genotyping of 205 RILs and a limited number of 100 SNPs present in the parental lines. This strategy enables the use of all the NGS SNP data obtained from a low-coverage sequencing experiment in the mapping population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The construction of genetic linkage maps is a necessary step for the localization of economically important traits using association with genomic regions through analyses including QTL mapping, genome-wide association studies, genomic selection, positional cloning, de-novo assembly of plant genomes and structural analyses of genomes. The first linkage maps were constructed based on morphological characters (Blixt 1974; Koornneef et al. 1983), isoenzymes (Weeden and Wolko 1990) and later by restriction fragment length polymorphism [RFLP; Botstein et al. (1980), Ellis et al. (1992), Nam et al. (1989)]. The development of molecular markers based on polymerase chain reaction such as random amplified polymorphic DNA [RAPDs; Reiter et al. (1992), Williams et al. (1990)], amplified fragment length polymorphism [AFLP; Vos et al. (1995), Castiglioni et al. (1999)], simple sequence repeats [SSRs; Zietkiewicz et al. (1994), Minamiyama et al. (2006)] led to the construction of linkage maps for many plant species. Nevertheless, the low density of molecular markers in these maps tended to offer too little resolution to dissect complex traits. The development of highly saturated linkage maps with a large number of recombination events is an essential tool for the detection and characterization of loci underlying traits of interest.
Next-generation sequencing (NGS) is a low-cost technology that enables the identification of thousands of single nucleotide polymorphisms (SNPs) (Margulies et al. 2005). The genotyping based on NGS, such as restriction site associated DNA [RAS; Baird et al. (2008)], genotyping-by-sequencing [GBS; Elshire et al. (2011)] and specific-locus amplified fragment sequencing [SLAF-seq; Sun et al. (2013)] enabled the genotyping of large segregating populations with thousands of SNPs in a short period of time. Today, GBS is the most widespread genotyping technique used in plants to characterize diversity panels and mapping populations. In fact, GBS has been used to construct multiple high-density genetic maps for major and minor crops (Bouchet et al. 2017; De Leon et al. 2016; Guajardo et al. 2015; Su et al. 2017; Tello et al. 2019). Despite the cost-effectiveness of GBS, the linkage map construction based on this genotyping platform needs deep sequencing of parental lines and the imputation of large amounts of missing data resulting from incomplete sequencing reactions and sequencing errors.
High-density linkage maps have been constructed based on low-coverage sequencing of mapping populations (Bilton et al. 2018; Xie et al. 2010). These strategies depend on the precise imputation of the parental lines of the population (Xie et al. 2010) or the selection of high-quality SNPs for linkage map construction (Bilton et al. 2018). To our knowledge, most of the linkage maps constructed in plants are limited by the number of high-quality SNPs that are present in both parental lines and the mapping populations. Consequently, many SNPs are discarded, because they were missing in the genotyping of the parental lines or the parental lines were considered heterozygous. Even if the inbreeding and phased haplotype present in mapping populations [e.g. recombinant inbred lines (RILs), double haploid lines (DHL)] is being used to produce highly accurate imputed genotypes (Swarts et al. 2014), the haplotypes of parental lines may contain up to 15% of SNPs coded as missing or heterozygous. Typically, the parental lines have higher coverage than the segregating population (e.g. ten samples of each parental line vs. one sample for each progeny). However, the number of missing SNPs tends to still be high, mainly due to the inherently low coverage and multiplex approach of this kind of sequencing method. In maize, the average of SNPs coded as missing in three biparental populations ranged from 4 to 9% after the data imputation (Wang et al. 2020).
Here we present an approach to construct a linkage map using the imputed genotypic data of a plant mapping population when the parental genotypic information is limited or absent. This strategy enabled us to include in the linkage map the SNPs that were missing in the parental lines as well as SNPs for which the parents were heterozygous. To validate this approach, the sorghum [Sorghum bicolor (Moench)] RIL population derived from SC1103 and RTx430 genetically characterized with 33,421 SNPs (Bouchet et al. 2017; Hu et al. 2019) was used to construct highly saturated linkage maps using a subset of just 100 SNPs from the parental lines. This strategy is generally applicable to all types of mapping populations and genetic markers but will be particularly beneficial for linkage map construction in those instances where a low sequence coverage is used for the parental and mapping population.
Material and methods
Genotype (i.e. GBS data) and phenotype (flowering time and plant height) information used in this study is a sorghum community resource (Hu et al. 2019) available at the Dryad Data Repository (https://datadryad.org/stash/dataset/doi:10.5061/dryad.63h8fd4).
RIL population and GBS
The 205 RIL population derived from a cross between SC1103 × RTx430 sorghum belonging to the nested association mapping population (Bouchet et al. 2017) was used as model in this study. The most recent SNP genotyping of this population was based on the BTx623 sorghum genome (version 3.1) which resulted in the identification of 66,376 SNPs with a minor allele frequency greater than 0.05 (MAF > 0.05) (Hu et al. 2019).
Linkage maps constructions
SNP filters
The 66,376 SNPs were filtered according to MAF > 0.40 to retain 42,271 SNPs. These SNPs were filtered for heterozygosity (< 20%) and segregation distortion against a 1:1 expected ratio [χ2 P(value) < 0.05)], which resulted in a total of 33,421 SNPs.
ABH genotype
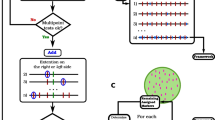
The 33,421 SNPs were transformed to ABH format in Tassel 5.1. (Glaubitz et al. 2014). This format assigns two alleles, A or B, to each SNP locus based on the assignment of parental 1 and 2, respectively. The H indicates heterozygosity. First, two pseudo-parental genotypes of SC1103 and RTx430 were created that were both homozygous for contrasting SNP alleles among the 33,421 SNPs. Second, two duplicate ABH data sets were created assuming: (1) SC1103 and RTx430 were considered parental 1 and 2, respectively; or (2) RTx430 and SC1103 were considered parental 1 and 2, respectively (Fig. 1). These two duplicated ABH data sets were merged (66,842 SNPs) and sorted by chromosome.

Construction of a linkage map with incomplete genotypic data from the parental lines. Step 1 Make two hapmaps that include pseudo-parental lines with either genotype (option 1 and 2). Step 2 Make two ABH Genotype files using each hapmap file and label each SNP based on the hapmap from which it originated (e.g. Ph1_S1_1972 versus Ph2_S1_1972). Step 3 Merge the two ABH Genotype files. Step 4 Construct a linkage map by chromosome using the merged ABH genotypic data. Since the genotypic data are duplicated, two mirrored linkage groups are generated. Step 5 Select the correct linkage map based on the partial genotypic data of the parental lines. In this example, Linkage map 2 was the correct match
Linkage maps
The linkage map was constructed with MSTmap software for Linux (Wu et al. 2008) using Kosambi mapping distance and genotyping error detection. The duplicate ABH data of each chromosome were subjected to linkage analysis by adjusting the cut off p-values (i.e. LOD criterion) to produce two mirrored linkage groups (i.e. same recombination pattern with the replacement of A with B and B with A; Fig. 1) per chromosome. The initial cup off p-value was 1E-11 as implemented in MSTmap software.
Selection of correct linkage maps
To identify and select the correct linkage group representing each chromosome, we used incomplete genotypic data of each parental line. A subset of 100 selected SNPs spaced at regular intervals across each chromosome (10 SNPs per chromosome) was transformed to ABH format in Tassel 5.1 using SC1103 and RTx430 as parental 1 and 2, respectively. The ten linkage groups with similar ABH matches with this subset were selected and grouped as the SC1103-RTx430 linkage map.
QTL mapping
To evaluate the effect of using a linkage map constructed without knowing the full parental genotype, we performed and compared the results of three QTL mapping analyses. Based on the random selection of one of the two mirrored linkage group per chromosome, we could create two linkage maps (referred to as pseudo-linkage map 1 and 2, respectively).
The flowering time and plant height of the RIL population (Hu et al. 2019) were utilized to identify QTLs using inclusive composite interval mapping (ICIM) and interval mapping (IM) as implemented in QTL IciMapping software (Meng et al. 2015). The three linkage maps (SC1103-RTx430 linkage map and pseudo-linkage maps 1 and 2) were scanned using a walking speed of 1 cM and threshold for determining a significant QTL were calculated with 1,000 permutation for experiment-wise error of α = 0.05.
Results
Linkage maps
The duplicated ABH dataset produced mirrored linkage groups per chromosome (Table 1). Three chromosomes (1, 2 and 3) required strengthening of cut off p-values to produce two mirrored linkage groups. The SNPs on chromosomes 1, 4, 6 and 10 were grouped into more than two mirrored linkage groups. However, for chromosomes 1, 6, and 10, most of the SNPs (99.8%) were sorted into just two mirrored linkage groups. For chromosome 4, the SNPs were distributed into two mirrored linkage groups, one of 1,366 SNPs and another of 309 SNPs. The map lengths of mirrored linkage groups were similar, with the largest difference being 6 cM observed for chromosome 3.
The subset of 100 SNPs from the parental lines SC1103 and RTx430 was sufficient to identify and select the correct linkage group for each chromosome. This ABH subset was identical to those obtained from the pseudo-parental genotype. Moreover, we found that the ABH matrix generated from parental lines including the 33,421 SNPs was identical to those obtained from the pseudo-parental genotype. As expected, pseudo-linkage maps 1 and 2 contained four and six correct linkage groups, respectively (Table 1). The SC1103-RTx430 linkage map was collinear with the BTx623 reference genome, with centromeric regions having most of the SNPs with segregation distortion (Supplementary Fig. S1).
QTL mapping
The genome scan based on ICIM for flowering time detected three regions on chromosome 3 and 6 using either of the three linkage maps (Fig. 2 and Table 2). Likewise, a genome scan based on IM detected the two largest QTL on chromosomes 3 and 6. The LOD value, percent of variance explained (PVE) and additive effects were slightly different between the linkage maps.

Genome scan for flowering time of 205 recombinant inbred lines derived from the cross between SC1103 and RTx430 (Hu et al. 2019). A Inclusive composite interval mapping and B interval mapping using: 1 pseudo-linkage map 1, 2 pseudo-linkage map 2, and 3 linkage map of SC1103. Horizontal dashed lines mark the significance threshold (P < 0.05) based on 1000 permutations
The genome scan based on ICIM for plant height varied between pseudo-linkage maps (Table 3). The genome scan with pseudo-linkage map 1 detected six genomic regions located on chromosomes 4, 7, 9 and 10, whereas five genomic regions were detected on chromosomes 4, 5, 9 and 10 using pseudo-linkage map 2. The difference among each analysis was the detection of three minor QTLs on chromosome 5 (pseudo-linkage map 2), 7 and 10 (pseudo-linkage map 1). The SC1103-RTx430 map validated four QTLs located on chromosomes 4, 9 (two QTLs) and 10. A genome scan based on IM detected three of these QTLs (chromosomes 9 and 10) using either of the three linkage maps (Fig. 3 and Table 3). The LOD value, PVE and additive effects for plant height were slightly different among linkage maps using ICIM, but similar when IM was used. The additive effects calculated based on pseudo-linkage maps 1 and 2 were positive or negative depending on whether SC1103 was considered parental 1 or 2.

Genome scan for plant height of 205 recombinant inbred lines derived from the cross between SC1103 and RTx430 (Hu et al. 2019). A Inclusive composite interval mapping and B interval mapping using: 1 pseudo-linkage map 1, 2 pseudo-linkage map 2, and 3 linkage map of SC1103. Horizontal dashed lines mark the significance threshold (P < 0.05) based on 1000 permutations
Discussion
Linkage map construction is an essential step for the genomic dissection of agronomically important traits. Here we presented a simple approach to construct a linkage map using limited genotypic information from the parental lines. This strategy allowed us to use most of the genotypic information obtained from the low-coverage sequencing of a RIL population. Moreover, in the absence of a known parental genotype, this approach enabled a complete genome scan to detect QTLs.
Inferring parental genotypes has been a successful strategy to construct linkage maps and to impute genotypes of progeny from biparental populations (Gonen et al. 2020; Xie et al. 2010). The parental genotype is essential information in livestock breeding, human inheritance studies and genomic selection programs where the target populations may involve multiple parents (Grossi et al. 2018; Tubbs et al. 2021). However, in plants the homozygosity of parental lines and the use of biparental populations simplifies the construction of linkage maps. Typically, low-coverage sequencing of RIL populations results in missing data at a large portion of SNP sites. In addition, the linkage map is limited to the SNPs present in the parental genotypes. Here, we demonstrated that the SNPs present in the RIL population with acceptable genotypic data (before or after the imputation process) can be used for linkage map construction regardless of whether or not they are present in the parental lines. Therefore, it is not necessary to devote resources to obtaining high-coverage sequence data of the parental lines. Instead, the SNPs present in the RILs and the recombination pattern can be used to construct mirrored linkage groups for each chromosome, which can later be identified and selected based on a limited number of SNPs present in parental lines.
Today, the most effective algorithm to impute missing genotypes from GBS in breeding populations is the full-sib family haplotype (FSFHap) (Swarts et al. 2014), while Beagle v.4–5 (Browning and Browning 2016) is preferable for diverse genetic panels. The procedure by Xie et al. (2010) first imputes the genotype of the parental lines using the SNPs of the entire mapping population in order to subsequently construct the linkage map. This requires identification of all the recombination break points across the genome. The accuracy of imputation of FSFHap for bi-parental mapping populations is higher than the method from Xie et al. (2010), because it first infers the two parental haplotypes for a genomic region (e.g. 50 SNPs), and subsequently imputes the genotypes of the lines comprising the mapping population for these particular segments. However, the imputation of parental genotypes could include a high number of heterozygous and missing genotypes that limit the number of SNPs for linkage map construction. In this regard, we imputed the genotypic data of the 205 RILs for chromosome 1 using FSFHap including a single sequence read of each parental line (i.e. RTx430 and SC1103). The imputation for the RILs was precise, but the low sequence coverage used for the parental lines created an excess of heterozygosity in parental line SC1103 (data not shown). Since the main goal of genotyping biparental mapping populations is to construct linkage maps for QTL analysis, our approach is direct and makes the maximum use of the precise genotype imputation of the mapping populations.
The most critical step is the creation of mirrored linkage groups per chromosome. The quality of the SNP data (e.g. segregation distortion, excess of missing data, etc.) and parameters adjusted for the analysis (e.g. LOD values) affect the clustering of markers into linkage groups (Zuo et al. 2019). Since the physical location of the SNPs is known, we clearly observed when two mirrored linkage groups joined incorrectly. For chromosomes 1, 2 and 3 both mirrors joined at the end of each group without mixed recombination pattern when using the initial cut-off p-value (1E-11). The use of a more stringent cut-off p-value separated both mirrors. The objective of mirrored linkage groups is to cluster SNPs into linkage groups that identify the correct ABH format. A chromosome can be fragmented into multiple mirrored linkage groups, which can later be identified and selected based on parental genotypes. After the corrected ABH data matrix is assembled, the analysis should be directed to clustering SNPs into one linkage group per chromosome or to assigning the order of SNPs based on physical location. The analysis presented here was limited to demonstrate that all SNPs were correctly assigned to ABH format and were not further examined for other genotyping errors (e.g. unlikely recombination events and marker order within the centromeric regions). Hence, the SC1103-RTx430 map is longer than previously reported in the NAM population (Bouchet et al. 2017).
In most plant breeding programs the parental lines of the RIL populations are available, but frequently one or both parents are absent in breeding programs for perennial crops. Likewise, it might be possible that parental lines are highly heterozygous. An effective strategy to overcome the absence or heterozygosity of parental genotypes is to use both mirrored linkage maps for QTL analysis. Our analysis detected the major QTLs previously identified for flowering time and plant height (Hu et al. 2019) using either mirrored linkage map. The false plant height QTLs detected with ICIM using pseudo-linkage map 1 and 2 are due to the fact that the algorithm used most-significant regressions across the linkage map for background control (Li et al. 2007). To overcome this problem, it is necessary to select the QTLs identified in both pseudo-linkage maps or use IM for the detection of major QTLs. The subsequent fine mapping of these QTLs could be completed by crossing two lines from the mapping population with contrasting genotypes in the genomic region of interest. The resulting segregating population can then also be used to determine the additive effect of the parental alleles.
Although the cost of most of the commercially available NGS platforms has decreased over the last 10 years (Wang et al. 2020), only the genomes of the most important varieties within each crop has been subjected to whole-genome resequencing. This means that individual breeding programs still rely heavily on the use of high-density molecular markers rather than whole-genome sequences. The advancement of plant breeding programs relies on the recombination of different parental genotypes followed by the selection of superior progenies. In this regard, the number of parental genotypes to be recombined each year could be over 100 (large-scale) depending on the resources of the breeding program. The integration of NGS has transformed traditional plant breeding programs by enabling marker-assisted selection (MAS) and genomic selection (Meuwissen et al. 2001). The rapid development of molecular markers in early generations is needed for the subsequent implementation of MAS. Optimizing the low-sequence coverage by using the maximum number of the SNPs identified in the segregating population(s) and parental lines, using the methodology presented here, increases the likelihood of identifying relevant molecular markers that can be used in high-throughput screening of progenies, in both small-scale and large-scale breeding programs.
Conclusion
Parental genotypes are not necessary for the construction of linkage maps from biparental populations. The two alleles existing in the segregating populations enable construction of two mirrored linkage groups per chromosome based on pseudo-parental genotypes. Using a limited number of SNP presents in parental lines it is possible to identify and select the correct linkage group per chromosome to construct a linkage map for QTL analysis. In the absence or heterozygosity of parental genotypes, the mirrored linkage groups could also be used for subsequent QTL analysis by the fine mapping of the region(s) of interest through the intercross of selected lines from the mapping populations. Our approach offers the benefit of being able to use all SNP information obtained from low-coverage sequencing (after imputation) in a segregating population for the genomic dissection of any trait of interest.
Availability of data and materials
Genotype (i.e. GBS data) and phenotype (flowering time and plant height) information used in this study is a sorghum community resource (Hu et al. 2019) available at the Dryad Data Repository (https://datadryad.org/stash/dataset/doi:10.5061/dryad.63h8fd4).
References
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA et al (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE. https://doi.org/10.1371/journal.pone.0003376
Bilton TP, Schofield MR, Black MA, Chagne D, Wilcox PL, Dodds KG (2018) Accounting for errors in low coverage high-throughput sequencing data when constructing genetic maps using biparental outcrossed populations. Genetics 209:65–76. https://doi.org/10.1534/genetics.117.300627
Blixt S (1974) The pea. In: King RC (ed) Handbook of genetics. Plenum Press, New York, pp 181–221
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Bouchet S, Olatoye MO, Marla SR, Perumal R, Tesso T, Yu J et al (2017) Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population. Genetics 206:573–585. https://doi.org/10.1534/genetics.116.198499
Browning BL, Browning SR (2016) Genotype imputation with millions of reference samples. Am J Hum Genet 98:116–126. https://doi.org/10.1016/j.ajhg.2015.11.020
Castiglioni P, Ajmone-Marsan P, van Wijk R, Motto M (1999) AFLP markers in a molecular linkage map of maize: codominant scoring and linkage group distribution. Theor Appl Genet 99:425–431. https://doi.org/10.1007/s001220051253
De Leon TB, Linscombe S, Subudhi PK (2016) Molecular dissection of seedling salinity tolerance in rice (Oryza sativa L.) Using a high-density GBS-based SNP linkage map. Rice 9:52. https://doi.org/10.1186/s12284-016-0125-2
Ellis THN, Turner L, Hellens RP, Lee D, Harker CL, Enard C et al (1992) Linkage maps in pea. Genetics 130:649–663
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES et al (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q et al (2014) TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE. https://doi.org/10.1371/journal.pone.0090346
Gonen S, Wimmer V, Gaynor R, Byrne E, Gorjanc G (2020) Phasing and imputation of single nucleotide polymorphism data of missing parents of biparental plant populations. Crop Sci. https://doi.org/10.1002/csc2.20409
Grossi DA, Brito LF, Jafarikia M, Schenkel FS, Feng Z (2018) Genotype imputation from various low-density SNP panels and its impact on accuracy of genomic breeding values in pigs. Animal 12:2235–2245. https://doi.org/10.1017/S175173111800085X
Guajardo V, Solis S, Sagredo B, Gainza F, Munoz C, Gasic K et al (2015) Construction of high density sweet cherry (Prunus avium L.) linkage maps using microsatellite markers and SNPs detected by genotyping-by-sequencing (GBS). PLoS ONE. https://doi.org/10.1371/journal.pone.0127750
Hu ZB, Olatoye MO, Marla S, Morris GP (2019) An integrated genotyping-by-sequencing polymorphism map for over 10,000 sorghum genotypes. Plant Genome. https://doi.org/10.3835/plantgenome2018.06.0044
Koornneef M, van Eden J, Hanhart CJ, Stam P, Braaksman FJ, Feenstra WJ (1983) Linkage map of Arabidopsis thaliana. J Hered 74:265–272. https://doi.org/10.1093/oxfordjournals.jhered.a109781
Li HH, Ye GY, Wang JK (2007) A modified algorithm for the improvement of composite interval mapping. Genetics 175:361–374. https://doi.org/10.1534/genetics.106.066811
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380. https://doi.org/10.1038/nature03959
Meng L, Li HH, Zhang LY, Wang JK (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3:269–283. https://doi.org/10.1016/j.cj.2015.01.001
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Minamiyama Y, Tsuro M, Hirai M (2006) An SSR-based linkage map of Capsicum annuum. Mol Breeding 18:157–169. https://doi.org/10.1007/s11032-006-9024-3
Nam HG, Giraudat J, Denboer B, Moonan F, Loos WDB, Hauge BM et al (1989) Restriction fragment length polymorphism linkage map of Arabidopsis thaliana. Plant Cell 1:699–705. https://doi.org/10.1105/tpc.1.7.699
Reiter RS, Williams JGK, Feldmann KA, Rafalski JA, Tingey SV, Scolnik PA (1992) Global and local genome mapping in Arabidopsis thaliana by using recombinant inbred lines and random amplified polymorphic DNAs. Proc Natl Acad Sci USA 89:1477–1481. https://doi.org/10.1073/pnas.89.4.1477
Su CF, Wang W, Gong SL, Zuo JH, Li SJ, Xu SZ (2017) High density linkage map construction and mapping of yield trait QTLs in maize (Zea mays) using the genotyping-by-sequencing (GBS) technology. Front Plant Sci 8:706. https://doi.org/10.3389/fpls.2017.00706
Sun XW, Liu DY, Zhang XF, Li WB, Liu H, Hong WG et al (2013) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 8:e58700. https://doi.org/10.1371/journal.pone.0058700
Swarts K, Li HH, Navarro JAR, An D, Romay MC, Hearne S et al (2014) Novel methods to optimize genotypic imputation for low-coverage, next-generation sequence data in crop plants. Plant Genome. https://doi.org/10.3835/plantgenome2014.05.0023
Tello J, Roux C, Chouiki H, Laucou V, Sarah G, Weber A et al (2019) A novel high-density grapevine (Vitis vinifera L.) integrated linkage map using GBS in a half-diallel population. Theor Appl Genet 132:2237–2252. https://doi.org/10.1007/s00122-019-03351-y
Tubbs JD, Hwang LD, Luong J, Evans DM, Sham PC (2021) Modeling parent-specific genetic nurture in families with missing parental genotypes: application to birthweight and BMI. Behav Genet 51:289–300. https://doi.org/10.1007/s10519-020-10040-w
Vos P, Hogers R, Bleeker M, Reijans M, Vandelee T, Hornes M et al (1995) AFLP: a new technique for DNA-fingerprinting. Nucl Acids Res 23:4407–4414. https://doi.org/10.1093/nar/23.21.4407
Wang N, Yuan YB, Wang H, Yu DS, Liu YB, Zhang A et al (2020) Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Sci Rep 10:1–12
Weeden NF, Wolko B (1990) Linkage map for the garden pea (Pisum sativum). In: Obrien SJ (ed) Genetic maps: Locus maps of complex genomes. Spring Harbor Laboratory, Cold Spring Harbor, New York, pp 106–112
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic-markers. Nucl Acids Res 18:6531–6535. https://doi.org/10.1093/nar/18.22.6531
Wu Y, Bhat PR, Close TJ, Lonardi S (2008) Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet 4:e1000212. https://doi.org/10.1371/journal.pgen.1000212
Xie WB, Feng Q, Yu HH, Huang XH, Zhao QA, Xing YZ et al (2010) Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc Natl Acad Sci USA 107:10578–10583. https://doi.org/10.1073/pnas.1005931107
Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain-reaction amplification. Genomics 20:176–183. https://doi.org/10.1006/geno.1994.1151
Zuo JF, Niu Y, Cheng P, Feng JY, Han SF, Zhang YH et al (2019) Effect of marker segregation distortion on high density linkage map construction and QTL mapping in Soybean (Glycine max L.). Heredity 123:579–592. https://doi.org/10.1038/s41437-019-0238-7
Funding
This research was funded by the USDA-ARS Current Research Information System (project 6090-21000-053-00-D; HC) and the U.S. Department of Energy Office of Science, Biological and Environmental Research (BER) Grant No. DE-SC0014171 and DE-SC0019097.
Author information
Authors and Affiliations
Contributions
HC and WV designed the method, performed experiments and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The author declared that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cuevas, H.E., Vermerris, W. Linkage map construction using limited parental genotypic information. Euphytica 218, 58 (2022). https://doi.org/10.1007/s10681-022-03005-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-022-03005-z