
Abstract
Information obtained from Nuclear Magnetic Resonance (NMR) experiments is encoded as a set of constraint lists when calculating three-dimensional structures for a protein. With the amount of constraint data from the world wide Protein Data Bank (wwPDB) that is now available, it is possible to do a global, large-scale analysis using only information from the constraints, without taking the coordinate information into account. This article describes such an analysis of distance constraints from NOE data based on a set of 1834 NMR PDB entries containing 1909 protein chains. In order to best represent the quality and extent of the data that is currently deposited at the wwPDB, only the original data as deposited by the authors was used, and no attempt was made to ‘clean up’ and further interpret this information. Because the constraint lists provide a single set of data, and not an ensemble of structural solutions, they are easier to analyse and provide a reduced form of structural information that is relevant for NMR analysis only. The online resource resulting from this analysis (http://www.ebi.ac.uk/msd/srv/docs/NMR/analysis/results/html/comparison.html) makes it possible to check, for example, how often a particular contact occurs when assigning NOESY spectra, or to find out whether a particular sequence fragment is likely to be difficult to assign. In this respect it formalises information that scientists with experience in spectrum analysis are aware of but cannot necessarily quantify. The analysis described here illustrates the importance of depositing constraints (and all other possible NMR derived information) along with the structure coordinates, as this type of information can greatly assist the NMR community.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The information contained in the world wide Protein Data Bank (wwPDB) (Berman et al. 2007) is growing steadily, with increasing numbers of structures being deposited from both traditional single laboratory sources and recent structural genomics efforts. The two main methods to determine these structures are X-ray crystallography and Nuclear Magnetic Resonance (NMR). NMR structures account for around 15% of all entries in the wwPDB. While inherent size restrictions limit the method to molecules of lower molecular weight, NMR has still made a significant contribution to protein folds and molecular interaction data. However, NMR structures are less straightforward to use than structures determined by X-ray because they are often represented as ‘ensembles’ of structures, where the whole ensemble (and not individual structures by themselves) represents the solution of the structure determination problem based on the experimental data. The reason for this is that information derived from NMR is insufficient with respect to the structure calculation process and thus cannot lead to a single exact solution. For example, measured distance-related NOE data is ensemble and time averaged, so that the final observed NOE data for a set of multiple distinct conformations in fast exchange will be a degenerate mix of the distance information in each of those conformations. All calculated structures that conform to a set of criteria based on the fit to the experimental data (NMR derived constraints) and physical characteristics encoded in the calculation process (overall energy minimum, inter-atomic packing, …) are therefore in principle equally valid (Spronk et al. 2003). However, there is no ‘standard’ set of criteria, and different programs and researchers use different sets when selecting the ‘best’ structures out of the calculated ones. The quality of the NMR structures deposited at the wwPDB therefore varies widely (Nabuurs et al. 2006). This problem is being addressed by novel structure determination methods like Inferential Structure Determination (ISD) (Rieping et al. 2005) that perform more objective structure calculations and select statistically relevant ensembles as a representative solution to the experimental data. These methods are, however, computationally expensive.
The observed NMR data is used in structure calculations to constrain inter-atomic distances, relative bond orientations and/or dihedral angles. Only just over half of the NMR entries in the wwPDB were deposited together with the constraint lists used in the structure calculation process. These constraint lists provide very valuable information, as, for example, they enable recalculation of the structures with better or different protocols, and a globally consistent comparison of the constraints to the original coordinates for validation purposes. However, a major problem in using this information is that constraint files come in many file formats, and that the atom naming and residue numbering in the coordinate and the constraint files often differs. To handle the problem of different file formats, constraint lists are now continuously converted into the NMR-STAR format at the BioMagResBank (BMRB) (Doreleijers et al. 2003; Ulrich et al. 1989). A further step, as part of the DOCR project (Doreleijers et al. 2005), addresses the atom naming and residue numbering problem by directly relating the constraint atoms to the coordinate atoms and molecular system using the FormatConverter and CCPN data model (Fogh et al. 2002; Vranken et al. 2005). This data is then checked for consistency and redundant constraints are removed using the WATTOS software as part of the FRED project (Doreleijers et al. 2005). In the original implementation the DOCR/FRED project resulted in a set of more than 500 internally consistent constraint lists, molecular system information and structure coordinates. This data led to the construction of the RECOORD (Nederveen et al. 2005) and DRESS (Nabuurs et al. 2004) databanks, where structures from the PDB were respectively recalculated and re-refined based on the cleaned up constraint lists.
These efforts, as well as validation software like AQUA (Doreleijers et al. 1998) or Procheck-NMR (Laskowski et al. 1996), are applied on a per-entry basis, where the original or recalculated coordinates are related to the original or standardised constraint lists. With the amount of constraint data that is now available it has become possible to do a global, large-scale analysis using only the information from the constraints, without taking the coordinate information into account. The constraint lists are the ‘final product’ of the usually human analysis of NMR spectra, and as such they represent the experimental NMR-derived information that is relevant for the structure calculation. The constraint lists also provide a single set of data, whereas NMR structures are usually represented as ensembles that can be calculated in many different ways, which complicates their interpretation. Analysing the constraint data on a large scale can thus provide insights into the NMR data analysis process (e.g. which type of inter-atomic contact is often derived from the spectra), and the relation of the constraints to the coordinate data (e.g. does the structure calculation process add any distance information that is relevant for NMR).
The analysis described here relates only to distance constraints derived from NOE data, with a base set of 1834 NMR PDB entries containing 1909 protein chains. Only constraints between protons in protein chains were retained for analysis, and for validation purposes the base set was further divided into subsets for entries that contain intra-residue constraints and entries where all the original constraint and coordinate information was recognised and linked to each other. A coordinate data set based on the original coordinate files was also generated and used for comparison. This article explores some of the issues surrounding distance constraints and the NMR data they are derived from, and hopes to highlight the importance of depositing the constraint lists used for structure calculations along with the molecular coordinates.
Materials and methods
The data was obtained from two sources: the molecular system, coordinate, secondary structure and author information from the wwPDB, and the original constraint information as NMR-STAR files from the NMR Restraints Grid at the BioMagResBank. Each file was directly parsed and combined into the CCPN data model via the FormatConverter software (Vranken et al. 2005), in a process that extends the procedure used in the original DOCR/FRED project. To handle the larger number of entries an automatic mapping procedure was developed that maps the molecule sequence as derived from the PDB file to the atom information and sequence code numbering used within the constraint lists. For some entries manual mapping between the information from the PDB file and the constraint file was required. The original 409 mappings from the RECOORD project (by Aart Nederveen) and a further mappings 124 by the author and 17 by Jurgen Doreleijers were used to correctly set the PDB-constraint file mapping for the entries in the base set. Using the automatic or manual mapping the atom information from the constraints was then connected to that for the molecule. During this process the dependence on string-based atom names for the assignment (as used in all constraint files) was also removed, since the CCPN data model is object-based. The final CCPN project, in which all the information from the original files is now highly organised, was then written out. This process was completed for 2643 PDB entries.
Three subsets of the base set were generated for comparison and validation purposes (Table 1). Sets HIP and AHIP contain only entries with intra-residue constraints, sets AHP and AHIP only include entries where over 99% of relevant constraint information was assigned to atoms. In addition a set was generated based on the coordinates for the entries included in the HP set. Only the HP, HPC and AHIP sets are further referred to in this article, the HIP and AHP sets are available online for reference purposes.
A workflow based on Python (van Rossum 2003) scripts and dictionaries was then developed to handle the information from the CCPN project files for the 2643 entries (Fig. 1). This workflow was run separately for the base set and each subset. In the first step, the original information was filtered so that only valid constraints between protons in protein chains were retained (Fig. 1). Entries were removed for the following reasons (Table 2): (1) No valid protein chains: the entry contained only chains that are sequence fragments or duplicates of other chains (only the chain with the highest number of valid constraints linked to it is retained in the data set), (2) No valid constraint lists: the originally deposited constraint lists could not be parsed or handled correctly, or (for sets HIP and AHIP), did not contain any intra-residue constraints, (3) Insufficient linking: less than 80% (sets HP, HIP) or 99.9% (sets AHP, AHIP) of the constraint information could be linked to the atom information (constraint information belonging to non-protein chains was ignored for this purpose), (4) Insufficient valid constraints: less then 20% of constraints remained for all lists after removing invalid constraints. Constraints were considered invalid if they did not have any valid items, no upper distance limit, an upper distance limit of larger than 10 Å, or had only items between invalid atoms (non-proton, unlinked or non-protein). Note that some entries had specific distance constraint lists and/or constraints removed for these reasons, but were still included in the analysed set.

Overview of the workflow employed in the analysis. Grey boxes indicate files, white boxes Python scripts
In the second step, the data from all relevant entries was collated (Fig. 1). After reading in the CCPN projects, non-relevant information was removed based on the filtering information, and the data reorganised into a set of Python dictionaries that contain the overall information on the entry, residue and constraint levels. In the final analysis step these dictionaries were read in, areas and data of interest were marked, and organised HTML output was produced for browsing. A Python dictionary with highly reduced information that can be further used for validation or constraint filtering purposes was also generated.
The same 1834 entries from the HP set were used to generate the HPC set. The original coordinates were analysed using r −6 distance averaging for equivalent and prochiral atom sets over all structures in the ensemble (Nilges 1995). Only individual distances of less then 7 Å were retained, and final averaged distances of less than 5 Å were considered to be equivalent to an observable constraint contact. Atoms without coordinates were ignored in the analysis.
Highlights from the analysis are described in the results section, and complete details are available from a web site (http://www.ebi.ac.uk/msd-srv/docs/NMR/analysis/results/html/comparison.html). For all statistical operations, the R package (Bates et al. 2007) was accessed via the RPy Python module (Moreira and Warnes 2006). Since a contact is either observed or not observed, it was possible to use a binomial analysis to determine, for example, which secondary structure specific contacts were significantly less or more likely to be observed. Binomial analysis was used throughout with a confidence level of 0.99, meaning that only 1 out of every 100 determined outliers is a false positive. The correlations between the coordinate and the constraint data within a data set and correlations between data sets were plotted via RPy, with linear correlation coefficients determined by both the Spearman and Pearson methods. The Spearman method (Spearman 1904) is a non-parametric measure of correlation, which does not make assumptions about the frequency distribution of the variables, and does not require a linear relationship between the variables. The Pearson method (Pearson 1896) on the other hand assesses if the relationship between the variables is linear.
In the analysis, a residue is marked as ‘assigned’ when at least one proton belonging to it is linked to a constraint. The total number of times a particular inter-atomic contact is observed can be a fraction, as for ambiguous constraints each constraint item contributes a fraction of 1 to the total:
The total number of relevant distances for a particular inter-atomic contact (n dist) is always an integer. The occurrence for a particular inter-atomic contact is calculated as:
where n actual is the number of times the contact occurs (with ambiguity taken into account), and n possible the total number of times this contact could occur between the relevant residues, either for all residues or only between ‘assigned’ residues. The occurrence is given in percent or as a fraction. The ‘ambiguity’ of an inter-atomic contact is defined as:
If the ambiguity is 0, this means that all contributing contacts are unambiguously assigned. The more highly ambiguous the contributing contacts are, the closer this number will be to 1, which also means it is less dependable. Within secondary structure combinations, the same definitions are used, except that n possible and n actual are now determined within that secondary structure combination as n possible,ss and n actual,ss. The uniqueness of a contact for a secondary structure combination, which indicates how unique the contact is within that combination, is defined as:
For prochiral and possible non-equivalent atom sets, the contacts are divided into the individual atom names (e.g. Hβ2 and Hβ3, or Hδ1 and Hδ2 for Phe) if they occur by themselves in a constraint. If both the atom names occur in different items within the same constraint to the same other atom, then they are grouped (e.g. Hβ*, or Hδ* for Phe). If they occur as a group in one constraint item they are used as is. In order to obtain total statistics for these contacts, they were also combined (e.g. (Hβ2 + Hβ3), or (Hδ1 + Hδ2) for Phe). To get the combined occurrence, the n actual for the group is added to the individual contribution with the highest n actual to obtain the n actual used to calculate the occurrence for the combined contact. The combined distance information is obtained from all contributions, and the ambiguity is then calculated from the total n actual of all contributions.
Results
Information from analysis
The analysis of the data for each set is divided into separate categories (visible on the left-hand side menu in Fig. 2): (1) Contact analysis: Arranges inter-atomic distances by residue–residue combination, secondary structure of those residues, and contact type (intra-residue (i–i), sequential (i–i + 1), medium-range defined here as up to 6 residues separation (i–i + n), and long range (i–i + 6<)). (2) Protein secondary structure analysis: Groups inter-atomic distances by secondary structure combinations. (3) Residue atom analysis: Shows assignment percentages for the atoms in each amino acid. (4) Unassigned fragments breakdown: Lists unassigned sequence fragments. (5) Fragment analysis by residue: Analyses tripeptide fragments based on the assignment status of the central residue.

Example web page for Ala → Ala i → i + 2 contact information from http://www.ebi.ac.uk/msd-srv/docs/NMR/analysis/results/html/comparison.html. The different data sets, comparisons between them, and analysis categories can be accessed via the left hand side menu. In this contact information page, overall statistics for the individual Ala residues involved in this contact are shown in the ‘Residue 1’ and ‘Residue 2’ tables at the top, statistics for the combined Ala–Ala residues in the ‘Residue combination’ table. The information on an atom level is listed in the ‘Breakdown per contact’ table. Outliers per secondary structure combination based on a binomial analysis are highlighted in red (higher than expected) and blue (lower than expected). This colour coding is used throughout the web pages for other types of analyses
Validation of data sets
The relevance of the base set (HP) was validated by comparison with the most restricted AHIP set. This is necessary because the HP set contains entries without intra-residue constraints and entries where less constraint information is linked to atoms. The correlation between the occurrences of contacts between both sets is very high (0.97) (Fig. 3), and in a breakdown per contact type no correlation is less than 0.91 (results on web pages). A further detailed analysis shows that out of 43,984 compared contacts only 5 differ significantly at a confidence level of 0.999. Of these, 3 are His → His contacts where the differences are most likely introduced by changes in the number of His residues that are assigned in the data sets (from 71.66% in HP to 68.47% in AHIP). The other 2 are long-range contacts involving side chain protons between Leu → Phe and Phe → Val, which are uncommon, and these differences can be attributed to accidental variations in the number of entries that have these contacts and are included in the data set. Overall, this data shows that including the additional 688 entries does not introduce major changes in the occurrence levels. The HP set was therefore chosen as the reference set, as estimates of, for example, significant differences between contacts in secondary structure elements tend to become more reliable as more data is included. Information on correlations between all data sets is available from the web pages (Fig. 2, the ‘Comparison’ link in the left-hand side menu).

Correlation between the f occurrence for the HP and AHIP data sets (correlation Spearman 0.971, Pearson 0.996)
A further way to validate the constraint information is that it should reproduce the typical contacts that are observed in secondary structure elements. The secondary structure definitions as determined by DSSP (Kabsch and Sander 1983) were taken from the PDB file header for each entry. Because of the size of the data that is generated by the analysis, only the Ala residue is used here as an example. The complete information can be accessed via the web pages (Fig. 2). The numbers that are relevant in order to determine whether a contact is significant for a particular secondary structure element are the occurrence within that particular secondary structure element (which has to be significantly higher than expected from the overall occurrence), the ambiguity of the contact (if it is high it comes, by definition, from highly ambiguous constraints and is therefore unreliable), and the uniqueness of the contact (which indicates how often the contact is seen within that secondary structure element compared to the total number of times it is observed).
For intra-residue contacts there are not many significant differences in the observed occurrence within different secondary structure elements, although overall fewer contacts are observed when secondary structure is absent (Table 3). This is likely due to higher signal overlap for atoms in ‘random coil’ fragments, which complicates assignment. The information from the coordinate-derived HPC set is provided throughout for comparison: there are, as expected when using a distance cutoff of 5 Å, no differences for this HPC set between the occurrence for different secondary structure elements, and only minor differences in the average and mean distance are observed (data available on web pages).
For sequential Ala–Ala contacts, the H–H contact is observed significantly more in α-helices than in β-sheets (Table 3), and is also highly unique (0.65). This situation is, as expected, reversed for the Hα–H contact, which is observed significantly less in α-helices and more in β-sheets. Overall, however, this contact is more uniquely observed in α-helices (0.49 compared to 0.08 for β-sheets), which is due to the prevalence of Ala–Ala fragments in α-helices (56.2% compared to 5.35%). Interestingly, however, an H–Hβ* contact is also observed significantly more in α-helices than in β-sheets or when no secondary structure is present, and has a uniqueness of 0.71, so within a sequential Ala–Ala fragment this type of contact is highly predictive of α-helical structure. The sequential Hα–Hβ* contact is, on the other hand, highly predictive of β-sheet with a uniqueness of 0.20, although it is on average mostly observed when no secondary structure is present (0.38). This illustrates that the uniqueness of a contact is strongly related to the prevalence of a particular sequence combination in a particular secondary structure element, and is not necessarily indicative of the kind of secondary structure element a particular contact usually occurs in. The information from the constraints and the coordinates show clear differences, as illustrated by comparison with some of the contacts described earlier. The H–Hβ* contact occurs quite often in β-sheets based on the coordinate data, with a median distance that is slightly higher than in α-helices (4.63 Å compared to 4.31 Å). The Hα–Hβ* contact, which would generally be difficult to identify due to overlap in the aliphatic region of a NOESY spectrum, is always present based on the coordinate data, but is only seen in 0.30 cases based on the constraints. These differences illustrate that the NOE constraint data not only incorporates distance information, but also encodes NMR-specific information such as the difficulty with which a particular contact can be assigned.
For i–i + 2 and i–i + 3 contacts, as expected, the typical H–H, Hα–H, Hα–Hβ* and Hβ*–H contacts are highly prevalent for α-helices (Table 3), with generally high uniqueness and low ambiguity. An H–Hβ* contact is also more often observed than average (respectively in 0.09 and 0.12 cases), while Hβ*–Hβ* contacts are very rarely seen for i–i + 2 contacts in an α-helix. Note that based on the coordinate data the i–i + 2 Hβ*–Hβ* contact is highly relevant for β-sheets (0.70), but it is in practice rarely observed (0.05), probably because it falls in a densely populated region of a typical NOESY spectrum. For 310 helices the percentages are often similar to the α-helical ones, but there are often not enough data to determine whether a difference is significant (data on web pages). Also of interest is the i–i + 3 contact between Hβ*–H, which occurs in 0.04 cases for β-sheets and 0.06 cases where secondary structure is absent. The binomial analysis indicates that the 0.06 fraction is observed significantly less, while it does not mark the 0.04 fraction. This is because this type of analysis is dependent on sample size (only 1 sample for the 0.04 fraction, 12 for the 0.06 fraction).
For the i–i + 4 contacts, Hα–H and Hβ*–H connections are again highly prevalent for α-helices, while Hα–Hβ* and Hβ*–Hβ* contacts are present in the coordinate set but are seldom observed in practice (Table 3). All contacts with a separation of 5 residues or higher are very rarely observed in α-helices, but become highly prevalent for β-sheets (data not shown). The only exception to this are long range Hα–Hβ* and Hβ*–Hβ* contacts, which are seen in significantly higher percentages between α-helices as compared to the average.
Contact data highlights
Traditionally, and as described above, identifying secondary structure contacts is based on the commonly observed contacts between protons from the backbone and the β position (Wüthrich 1986). However, with experience in assigning NOE peaks comes the knowledge that other contacts are also often observed (e.g. i → i + 2 contacts between side chain protons in a β-sheet). In this analysis, such contacts are readily observed (Table 4). For example, the sequential Trp Hε3–Gly H contact is quite common based on the coordinate data, but is in practice particularly observed in a β-sheet. The sequential Trp Hε3–Phe Hα contact is seemingly more often observed in an α-helix, but the differences are not significant. This is a case that could be clarified if more relevant data were available. The Thr Hγ2*–Tyr Hε* i–i + 2 contact is clearly observed in mostly β-sheet. Interesting in this case is that according to the constraint data it can occur in an α-helix, while this is not the case based on the coordinate data, even though the ambiguity of the contact is 0.00. This is possible because this data point is based on one contact with an upper distance limit of 6 Å, whereas the cutoff used for coordinates is 5 Å. Generally speaking this type of situation can occur for highly ambiguous constraints, where other constraint items satisfy the upper distance limit. An Hα–Hδ1* i–i + 3 contact between Ala and Ile is observed almost exclusively in an α-helix (and never in a β-sheet), and is very unique (0.78). To be able to discern whether this contact is very common from the Hα of any amino acid to the Hδ1* of an Ile, the data was joined as Xxx residues to produce generic information to and from each amino acid (data on web pages). The specific information for the i–i + 3 Hα–Hδ1* contact from all residues to Ile is shown in Table 4 and shows that this contact is common in α-helices. Finally, some other interesting i–i + 4 side chain contacts that often occur in helices are listed in Table 4.
It is not possible to describe all information in detail in this article, and the web pages serve as the reference resource for any investigations. However, to provide a better overview of the overall trends in secondary structure elements, all backbone contacts were grouped by atom type (H, Hα, Hβ) and secondary structure combinations (Table 5, full data from web site). For intra-residue contacts, more contacts involving Hβ protons are defined in β sheets. Sequential contacts in α helices originating from the H and Hβ protons are observed significantly more often, whereas ones originating from the Hα proton are less common. This situation is reversed in β sheets (except for Hβ–H contacts). There are, only for sequential contacts, some discrepancies between the data from the HP and HPC sets, with, for example, the rate at which sequential H–Hα contacts are observed being reversed in the HPC set as compared to the HP set. The reasons for this are not immediately clear, but are likely related to overlap. Most i–i + 2 contacts are more common in α helices, except for ones to the Hα proton and between Hβ protons. The latter contact is more often observed in β sheets. As expected most i–i + 3 contacts are commonly observed in α helices, except for Hβ–Hα and H–Hα. The latter is again more frequently seen for β sheets, but in this case this is expected to be between different strands in the hairpin area. The trends are not as clear for i–i + 4 contacts, where Hα–H, Hα–Hβ and Hβ–H contacts are more frequent in α helices, and all other ones more frequent in β sheets. This is again likely related to hairpin contacts. All contacts from i → i + 5 and more are very infrequent for α helices but relatively very frequent for β sheets.
An analysis of the percentage of atoms that were assigned within each residue type (Residue atom analysis on the website) shows that generally atoms are significantly higher assigned in α-helices and β-sheets, and lower when no secondary structure is present. This finding is not surprising as the secondary structure elements are defined by constraints, and the atoms have to be assigned to obtain those. Another general trend is that prochiral methylenes are within α-helices significantly more degenerated, i.e. the HB2 and HB3 atoms exists as a QB pseudoatom or an HB* type atom set. This type of analysis can be significantly improved by cleaning up the stereospecific assignment status based on the coordinates.
The unique sequence fragments for which no assignments were found are also listed on the website (Unassigned fragments breakdown). An example from this data is that often no constraints are found for His tags. The LEHHHHH fragment, for example, was not assigned in 13 entries. A general overview of the percentage of residues that are assigned confirms that His residues, for example, are assigned in only 71.66% of cases, while Trp is assigned in 97.87% of cases (Fragment analysis by residue).
To examine this in more detail, all tripeptide fragments where a particular amino acid is the central residue are listed (for the N- and C-terminus these are dipeptide fragments). Listed for each fragment are (1) the total number of times the tripeptide fragment occurs in the data set, (2) the percentage of times it is unassigned compared to the total number of times the amino acid occurs, (3) the number of times the amino acid is not assigned, (4) the number of times the tripeptide fragment occurs overall, and (5) the assignment percentages per secondary structure element. The entries in which the fragments are unassigned are also listed. Continuing with His as an example, it is not assigned when part of the EHH fragment in 69% of cases, and when part of a C-terminal HH fragment it is not assigned in 96% of all times the fragment occurs. To get a better overall view of the sequence fragments that are difficult to assign, they were grouped by joining, respectively, the i − 1 and i + 1 residues. The results for His and some selected other fragments are shown in Table 6. A Ser residue preceded or followed by a Gly, for example, is often unassigned. This type of information could be useful for predicting which areas of a protein sequence are difficult to assign from an NMR perspective.
Discussion
In this analysis only the original data as deposited by the authors was used, and no attempt was made to ‘clean up’ and further interpret this information, except for linking the constraint with the coordinate data and removing identical sequences from the data set pool (where only the entry with the highest number of constraints linked to atoms was kept). This approach is intentional, as it best represents the quality and extent of the data that is currently deposited at the PDB. Only the distance constraint information was included in the analysis, and the information from dihedral, H-bond and RDC constraints was ignored. Even though these constraints contain important structural information, they were, as experimental data, recorded independently from the NOE data. They are used in the structure determination process, however, and it was not investigated whether their presence influences the quality of the final distance constraint lists. There are several other issues that can still be addressed, and although these are likely to improve some of the aspects of this type of study, it is also important to start with the original information so that a comparison point is available.
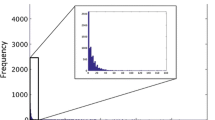
The first issue is that stereospecific assignments can be swapped or deassigned based on the original coordinates, similar to the approach in the RECOORD project. This could in principle reveal preferences related to stereospecifically assigned atoms in secondary structure elements. The second issue concerns the distances that were provided with the constraints. These are often ‘binned’ in weak/medium/strong classes with fixed distance cutoffs, so that the resulting distance distributions often show spikes at these distances. In Fig. 4, for example, it is clear that spikes occur at 3.0, 3.5 and 5.0 Å. Recalibrating the distances based on the deposited coordinates should improve the quality of the resulting information, and reveal relationships between distance and occurrence. The third issue concerns the sequences that are included: the current data sets include protein sequences with a high homology. This is clearly not an ideal situation, but there is currently not enough data available for a cleaner analysis. It is therefore important to check whether a particular contact appears in a large amount of entries, or the observed occurrence might be due to systematic error from homologous proteins produced by the same laboratory. The fourth problem is the identification of the secondary structure fragments. This is now based on the PDB DSSP analysis from the original coordinates for only the first or representative model so that secondary structure elements are not always identified properly. If chemical shifts were available an identification based on CSI (Wishart et al. 1992) would become possible so secondary structure stretches can be included that are more flexible and less defined on the coordinate level. More refined secondary structure identifications from the coordinates could also reveal patterns related to, for example, turns. Overall, the best way to improve this analysis remains to increase the sample size by encouraging deposition of constraint lists and all related NMR information (peak lists, chemical shifts, spectra), and ensuring that the data is consistent when deposited by the authors. Efforts like the CCPN project (Fogh et al. 2002, 2005), which allow data harvesting from NMR data collection to structure calculation, should provide this kind of data without requiring any additional effort by the scientists who produce the data.

Distance distribution from the constraint information for sequential Ala–Ala contacts between backbone H protons
In this analysis a particular inter-atomic contact between two residues from one PDB entry is either observed or not observed. The reason why a contact is observed (or not) implicitly includes distance information, peak overlap, water exchange line broadening, and all other factors that can lead to not observing or assigning a contact during analysis of a spectrum. This is different from the traditional meaning of an ‘assigned atom’ on the chemical shift level, where it means that the chemical shift value for the resonance that arises from the atom is known. However, this does not necessarily mean that these assigned atoms produce any valid inter-atomic distance information. Thus, an ‘assigned atom’ (or residue) on the constraint level means that a chemical shift assignment also produced useful and valid information related to the inter-atomic distances within the molecule.
The original study that defined the inter-atomic contacts relevant for assigning secondary structure elements with NMR used a set of 19 high-resolution protein crystal structures comprising about 3,200 residues (Wüthrich et al. 1984). In this study the extent of identification was defined as the percentage of times a distance was smaller than a particular cutoff value within a secondary structure element, while the uniqueness of identification is the percentage of times the distance is observed within a particular secondary structure element out of the total number of times it is observed. The extent is equivalent to the f occurence used in this study for the constraint sets, with the exception that no distance cutoff is used (although some distance information is, as mentioned, implicitly included because the reason a constraint is observed or not is very dependent on distance), and that values are labelled as significant based on a binomial analysis. The uniqueness has the same definition, except that again it is based on the amount of constraints that are observed.
Also of interest is the relationship between the information that comes directly from the deposited constraints and the information that comes from the deposited coordinates. Here, the constraint information is compared to the distances from the originally deposited coordinates. Although a set of recalculated coordinates (as in RECOORD) or X-ray structures could have equally well been used, the originally deposited coordinates were chosen because they should best match the content of the constraint lists. All comparisons between constraint and coordinate information are intended for informative purposes only: the constraints represent the experimental NMR side of the information contained in the coordinates, and are in effect only a subset of the information contained therein. However, a dependable determination of whether a particular NOE contact is observed or not is not possible based on an NMR structural ensemble, but is trivial based on the constraints because they inherently contain NMR-specific information like signal overlap, dynamics, etc.
From Fig. 5 it is clear that the contact occurrence is almost always higher for the HPC set compared to the HP set. This is related to the use of a direct distance cutoff of 5.0 Å in the HPC set: contacts with long distances could give rise to peaks that are too weak to be seen in a real spectrum but are still included. Also, many contacts have an f occurrence of 1.0 in the HPC set because of conformational constraints from covalent bonds. Not all of these contacts are seen in real spectra because of, for example, peak overlap or line broadening. The correlations between the occurrences overall are not very high (Spearman 0.770, Pearson 0.694), with especially the intra-residue contacts giving bad correlations (Spearman 0.451, Pearson 0.356, see web site), and i–i + 2 (Spearman 0.720, Pearson 0.783) and i + 3 (Spearman 0.711, Pearson 0.659) contacts giving the best results. An indication that the main reason for the bad correlation between the occurrences is distance related comes from the large improvement that is observed in the overall correlations if only coordinate distances of less than 3.6 Å are considered (Spearman 0.906, Pearson 0.916). However, results from using both the HP and HPC information to filter ambiguous constraints lists show that both sets essentially give the same results (personal data), even though the constraints are available in a much more ‘compressed’ form than the coordinates, and no force field information was used.

Correlation between the f occurrence from the HPC (Coordinates) and HP (Constraint) sets
In the KNOWNOE (Gronwald et al. 2002) X-ray structure based approach to obtain probabilities for assignments, the distance distributions for inter-atomic contacts are used to generate volume-based probabilities in addition to the atom identity based probabilities. This approach improves the probabilities that are generated, but it does require that the original peak list with volumes is available. This is not possible within the current analysis, although this will be pursued if a meaningful way to recalibrate the distance constraint bounds is available. This would also allow a better comparison between the NMR constraint data and any coordinate data (from NMR or X-ray structures).
Conclusion
A resource is now available where it is possible to check how likely a particular contact is when assigning NOESY spectra, or if a particular sequence fragment is likely to be difficult to assign. In this respect it formalises information that scientists with experience in spectrum analysis are aware of but cannot quantify. The amount of information provided here is extensive, however, and is even more useful when used as ‘knowledge based’ probabilities in automatic assignment strategies, to filter and/or validate ambiguous constraint possibilities, and as a tool to rank assignment possibilities in spectrum analysis programs. These are being implemented as part of the CCPN framework.
Finally, the NMR constraint lists encompass the experimental NMR data encoded in the NMR structural ensembles, and comprise a single set of data that is much easier to analyse than an ensemble of solutions. As such, they provide a reduced form of structural information that is relevant for NMR analysis only. For this reason, and to allow a basic level of scientific reproducibility and validation, it is important that constraints, and all other possible NMR derived information, are deposited along with the structure coordinates. It is very likely that a lot more information than described in this article can be gained from it, which in turn can assist the NMR community and can help to understand the relationships between NMR and structure.
References
Bates D, Chambers J, Dalgaard P et al (2007) The R project for statistical computing. http://www.r-project.org/. Cited 10 Apr 2006. University site
Berman H, Henrick K, Nakamura H et al (2007) The worldwide Protein Data Bank (wwPDB): ensuring a single uniform archive of PDB data. Nucleic Acids Res 35:D301–D303
Doreleijers JF, Rullmann JA, Kaptein R (1998) Quality assessment of NMR structures: a statistical survey. J Mol Biol 281:149–164
Doreleijers JF, Mading S, Maziuk D et al (2003) BioMagResBank database with sets of experimental NMR constraints corresponding to the structures of over 1400 biomolecules deposited in the Protein Data Bank. J Biomol NMR 26:139–146
Doreleijers JF, Nederveen AJ, Vranken W et al (2005) BioMagResBank databases DOCR and FRED containing converted and filtered sets of experimental NMR restraints and coordinates from over 500 protein PDB structures. J Biomol NMR 32:1–12
Fogh R, Ionides J, Ulrich E et al (2002) The CCPN project: an interim report on a data model for the NMR community. Nat Struct Biol 9:416–418
Fogh RH, Boucher W, Vranken WF et al (2005) A framework for scientific data modeling and automated software development. Bioinformatics 21:1678–1684
Gronwald W, Moussa S, Elsner R et al (2002) Automated assignment of NOESY NMR spectra using a knowledge based method (KNOWNOE). J Biomol NMR 23:271–287
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637
Laskowski RA, Rullmann JA, MacArthur MW et al (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486
Moreira W, Warnes GR (2006) RPy (R from Python). http://rpy.sourceforge.net/. SourceForge site
Nabuurs SB, Nederveen AJ, Vranken W et al (2004) DRESS: a database of REfined solution NMR structures. Proteins 55:483–486
Nabuurs SB, Spronk CA, Vuister GW et al (2006) Traditional biomolecular structure determination by NMR spectroscopy allows for major errors. PLoS Comp Biol 2:e9
Nederveen AJ, Doreleijers JF, Vranken W et al (2005) RECOORD: a recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59:662–672
Nilges M (1995) Calculation of protein structures with ambiguous distance restraints: automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J Mol Biol 245:645–660
Pearson K (1896) Mathematical contributions to the theory of evolution: III. Regression heredity and panmixia. Phil Trans Royal Soc London Ser A 187:253–318
Rieping W, Habeck M, Nilges M (2005) Inferential structure determination. Science 309:303–306
Spearman C (1904) The proof and measurement of association between two rings. Am J Psych 15:72–101
Spronk CA, Nabuurs SB, Bonvin AM et al (2003) The precision of NMR structure ensembles revisited. J Biomol NMR 25:225–234
Ulrich EL, Markley JL, Kyogoku Y (1989) Creation of a nuclear magnetic resonance data repository and literature database. Protein Seq Data Anal 2:23–37
van Rossum G (2003) The Python language reference manual. Network Theory Ltd Bristol UK
Vranken WF, Boucher W, Stevens TJ et al (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696
Wishart DS, Sykes BD, Richards FM (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31:1647–1651
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley-Interscience, New York
Wüthrich K, Billeter M, Braun W (1984) Polypeptide secondary structure determination by nuclear magnetic resonance observation of short proton–proton distances. J Mol Biol 180:715–740
Acknowledgements
The author thanks Aart Nederveen and Jurgen Doreleijers for the RECOORD and DOCR/FRED collaborations that were instrumental in creating the setup to harmonise constraints and coordinates, and Kim Henrick and Wolfgang Rieping for reading the manuscript and suggesting improvements. Most importantly, this work would not be possible without the members of the NMR community who made the effort to deposit their coordinates and constraints at the PDB. This project was funded by the EU FP6 Extend-NMR grant (18988) and by the Wellcome Trust (WT GR075968MA).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Vranken, W. A global analysis of NMR distance constraints from the PDB. J Biomol NMR 39, 303–314 (2007). https://doi.org/10.1007/s10858-007-9199-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-007-9199-x