
Abstract
Necroptosis, a type of programmed cell death that resembles necrosis, is now known to depend on a different molecular mechanism from apoptosis, according to several recent studies. Many efforts have reported the possible influence of necroptosis in human disorders and concluded the crucial role in the pathophysiology of various diseases, including liver diseases, renal injuries, cancers, and others. Fibrosis is the most common end-stage pathological cascade of several chronic inflammatory disorders. In this review, we explain the impact of necroptosis and fibrosis, for which necroptosis has been demonstrated to be a contributing factor. We also go over the inhibitors of necroptosis and how they have been applied to fibrosis models. This review helps to clarify the role of necroptosis in fibrosis and will encourage clinical efforts to target this pathway of programmed cell death.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fibrosis overview
Up to 45% of deaths in the industrialised world are caused by fibrosis, which can damage any organ. Preclinical models and clinical experiments in a variety of organ systems have revealed that fibrosis is a highly dynamic process, contrary to long-held beliefs that it is relentlessly progressing and irreversible. A translational gap still exists between the identification of prospective anti-fibrotic targets and their conversion into efficient therapy despite tremendous advancements in our understanding of the pathobiology of fibrosis [1, 2].
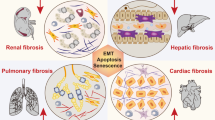
Fibrosis is the most common end-stage pathological cascade of numerous chronic inflammatory disorders. Fibrosis is characterized by an overabundance of fibrous connective tissue (ECM components such as collagen and fibronectin) in the inflamed or destroyed tissue, which might lead to irreversible scar tissue formation, organ failure, and subsequently death [3,4,5]. Infections, autoimmune reactions, allergies, chemical assaults, radiation, and tissue damage contribute to chronic inflammation and fibrosis induction [2]. Fibrosis results after long-term exposure to various stimuli and involves inflammation. Despite their varied etiologies and clinical presentation, most chronic fibrotic disorders promote the release of growth factors and fibrogenic cytokines that severely damage normal anatomical structures [6, 7]. The recruitment of ECM-producing myofibroblasts is a hallmark common to all organ fibrosis [8, 9]. (As shown Fig. 1).

The main mechanism of fibrosis
Liver fibrosis
Liver fibrosis is an immediate and long-term reaction to liver damage [3]. The activation of HSCs, which have a role in myofibroblasts phenotyping, is a critical event in liver fibrosis [10, 11]. Active HSCs are proliferative, expressing alpha-smooth muscle actin (SMA), collagen type I and III secretion, and expression of matrix metalloproteinases (MMPs) [12,13,14,15,16]. The stimulation of HSCs is concluded in two key steps, initiation and perpetuation, followed by fibrosis [17]. Activating HSCs causes ECM buildup and chronic inflammation. Other ECM-producing cells, such as portal fibroblasts, myofibroblasts from bone marrow, and epithelial cells undergoing endothelial mesenchymal transition (EMT), also contribute to liver scarring [18,19,20]. Nevertheless, in the presence of transforming growth factor (TGF), oval cells may undergo EMT, enhancing the expression of HSC markers [21]. Moreover, throughout all stages, inflammatory cell recruitment is critical manifested by the presence of macrophages, which increased cytokine production, such as interleukin-13, causing liver fibrosis [22].
Lung fibrosis
Chronic damage to the alveolar epithelium may be caused by various causes, including smoking and viral infections [23]. Studies have shown that the immune system plays a significant role in the development of lung fibrosis. Interestingly, there is overwhelming evidence that innate and adaptive immunity plays a significant role in the etiology of pulmonary fibrosis. Idiopathic fibroblast proliferation and ECM modification are hallmarks of pulmonary fibrosis, a progressive and generally fatal lung disease [24, 25]. Innate and adaptive immune systems are linked by the pattern recognition receptors (PRRs) expressed on toll-like receptors (TLRs), which may be seen as master regulators of tissue structural and functional integrity [26].
Renal fibrosis
Chronic kidney disease (CKD) is a significant epidemiological clinical problem with a high prevalence and mortality rate. End-stage renal disease can be developed from CKD and result in serious complications [27]. Diabetic nephropathy, hypertension, and chronic interstitial glomerulonephritis are the most common causes of CKD. These diseases can cause structural and functional changes in the kidney. Chronic inflammation can cause renal fibrosis and is a major predisposing factor in CKD [28]. In addition, various cells, like macrophages, participate in renal fibrosis [29,30,31]. Renal fibrosis frequently leads to renal interstitial fibrosis with tubular atrophy and abnormal ECM deposition as the main pathological causes [32]. Renal fibrosis characterized by inflammatory cell infiltration, fibroblast activation, ECM component deposition, and microvascular thinning [33]. Many molecules and cells, such as angiotensin II (Ang II), are linked to the progression of renal fibrosis [34]. Animal models such as surgical, chemical, physical, genetic, and in vitro models are essential for understanding renal fibrosis biopathology and evaluating new therapies [35]. However, there are no available drugs for renal fibrosis. As a result, improving our understanding of renal fibrosis's cellular and molecular mechanisms is critical to eliminating renal fibrosis [36]. Alleviation of fibrosis alone is not sufficient to repair kidney function without restoring lost nephron tissue after damage. It is worth noting that encouraging endogenous tissue regeneration could be a promising treatment option for kidney disease [37].
Cardiac fibrosis
Cardiovascular diseases (CVDs) approximately cause 31% of all deaths worldwide [38], and cardiac fibrosis (CFs) impacts heart failure and end-stage remodeling of ECM. CFs differentiate into myofibroblasts (myoFbs) during cardiac injury [39]. MyoFbs show proliferative and invasive properties in response to disease or other stimuli. MyoFbs also remodel the interstitium by secreting MMPs that degrade ECM, increase collagen turnover, and enhance collagen net formation [40]. CFs, a scarring event in the cardiac muscle, occurs in nearly every type of heart disease, such as myocardial infarction (MI), diabetic cardiomyopathy, and aortic stenosis [41, 42]. ECM components' turnover is important in the fibrosis process, which is pathologically defined by increased deposition of type I and III collagens [43, 44].
Necroptosis overview
There are numerous differences between apoptosis and necroptosis. Apoptotic cells preserve the viability of their cell membranes morphologically. In contrast, cells experiencing necroptosis demonstrate the breakdown of their cell membranes, a significant feature of necrosis. As a result, using traditional histologic methods, necroptotic cells are indistinguishable from necrotic ones despite the same stimuli [45]. Apoptosis and necroptosis have different intracellular signaling mechanisms that lead to their implementation. As caspases play an important role in apoptosis, receptor-interacting protein kinases (RIPKs) play a significant role in necroptosis. There is a prominent cross-talk between apoptosis and necroptosis [46, 47].
Necroptosis is a non-caspase-dependent cell death that has been involved in the pathogenic mechanisms of distinct diseases. It is an exciting area closely related to apoptosis. It is controlled by a number of genes that trigger cell death in a predictable and orderly manner. It shares normal necrosis features, such as loss of metabolic function and subcellular alterations, by activating specialized death signaling pathways [48, 49]. The first signaling molecule discovered in the necrosome was RIPK1 [50]. RIPK1 and RIPK3 interact with the receptor protein, which phosphorylates the mixed lineage kinase domain-like protein (MLKL) [51,52,53].
Necroptosis mainly regulates several signals, such as the caspase-8, nuclear factor-κB (NF-κB), and the mitogen-activated protein (MAP) kinase cascade. Many efforts have investigated the possible influence of necroptosis on human disorders. Interestingly, necroptosis plays a key role in the pathophysiology of various diseases, including hepatic diseases, renal injuries, human cancers, and others [54].
Necroptosis activation and signaling
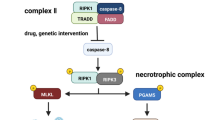
Necroptosis signals have been thoroughly explored with the discovery of necrostatins as specialized inhibitors targeting RIPK1 [55,56,57]. Among the different models, the precious model is tumor necrosis factor (TNF)-α-induced necroptosis. After TNF-α binds to TNF receptor (TNFR)1, the adaptor molecules Fas-associated death domain (FADD) and TNF-receptor-associated death domain recruit RIPK1, which then binds RIPK3 to create the 'necrosome' complex [51, 52, 58, 59]. The oligomerization of RIPK3 and RIPK1 via the RIPK homotypic interaction motif (RHIM) domain causes autophosphorylation of RIPK3, which culminates in RIPK3 stimulation. Further early players, such as Toll/IL-1 receptor domain-containing adaptor-inducing IFN- (TRIF) and DAI, employ the RHIM domain to stimulate RIPK3, suggesting that the RHIM domain is involved in necroptosis. RIPK3 activation phosphorylates MLKL, which is responsible for necroptosis execution [53, 60]. When MLKL is phosphorylated, a molecular switch is set off that allows MLKL to travel to the plasma membrane and disrupt it [61, 62]. Moreover, IFNs cause necroptosis by overexpression of protein kinase R (PKR). This PKR interacts with RIPK1 and promotes the creation of the PKR necrosome, which consists of PKR, RIPK1, and RIPK3 [63]. Notably, IFNs are key in maintaining the RIPK1–RIPK3 complex activation [64].
Necroptosis is divided into three types according to the causes that trigger it: TNF-α stimulates extrinsic necroptosis, reactive oxygen species (ROS) induce intrinsic necroptosis, and ischemia stimulates intrinsic necroptosis. TNF-α -mediated necroptosis is a kind of necroptosis in which TNF-α binds to a complementary receptor, creating a short-lived membrane signalling complex (complex I) [65, 66]. Consequently, TRAF2/3/5 and cIAPs are then recruited to Complex I [67]. During activation, cIAP1/2 and TRAF2/5 cause RIPK1 to be ubiquitinated, creating stable complex I and starting an alternate route that leads to cell survival via the NF-кB and MAPK signals [68]. (As shown in Fig. 2).

Molecular mechanisms of necroptosis
Necroptosis and inflammation
In contrast to apoptosis, cytokine production is a primary process leading to substantial inflammation following necroptosis. Necroinflammation, or the loss of plasma membrane integrity, which leads to the release of damage-associated molecular patterns (DAMPs) and the aggravation of tissue damage, is a unique characteristic of necroptosis [61]. After MLKL insertion, RIPK3 activates the inflammatory response mostly through the production of DAMP from cells. According to recent research, RIPK3 also triggers caspase-1 and caspase-11 by directly activating inflammasome in response to cellular stress or microbial infection [69, 70]. RIPK3 has been demonstrated in different investigations to increase cytokines release and inflammasome activation [71,72,73]. MLKL has been shown to be required for RIPK3-dependent inflammation [74]. However, it is uncertain how RIPK3 promotes NLPR3-mediated inflammasome production and whether or not MLKL is involved. According to certain theories, RIPK3 is a scaffold for complexes comprising RIPK1, FADD, and caspase-8 [70, 75, 76]. RIPK1 stimulates cytokine production independently of RIPK3 despite the presence of RIPK3. RIPK1 works as a scaffold in some models, particularly during TNF-mediated NF-κB and JNK activation, which leads to cytokine release [77, 78]. Following TLR4 activation, it was recently shown that RIPK1 is required to raise the circulating IL-1β, which is required to activate NF-κB, FOS, and ERK [79].
Necroptosis and oxidative stress
Oxidative stress can lead to necroptosis, which has been proven by several investigations [80,81,82,83]. Hydrogen peroxide induces necroptosis in RPE cells [84], and oxidative injury caused by paraquat produces necroptosis in cardiac muscles [85], indicating that oxidative stress plays a role in necroptosis. In mice, acetaminophen toxicity resulted in the formation of ROS and necroptosis [86]. Hyperoxia-induced oxidative injury resulted in necroptotic cell death in the pulmonary tissues of rats [87]. Researchers discovered that indicators of necroptosis are elevated in Sod1−/− mice, which have a high level of oxidative stress [88, 89]. The age-related rise in oxidative stress is associated with an increase in necroptosis, based on an existing study indicating oxidative stress may trigger necroptosis mediated by a decrease in redox-sensitive pathway Nrf2 [90].
Some evidence demonstrated that ROS generation by mitochondrial respiratory complex I was critical for the necrotic response of L929 cells to TNF in the early 1990s. This was the first-time mitochondrial energy metabolism related to necrosis execution [91]. Cell death-related ultrastructural alterations in the mitochondria and endoplasmic reticulum (ER) are also mediated by mitochondrial ROS [91, 92]. RIPK3 action may connect TNFR1 signaling, mitochondrial bioenergetics, and ROS overproduction, even though ROS production is not required for all cases of TNF-induced necrosis [92]. TNF-induced necrotic cell death is additionally accelerated by the creation of non-mitochondrial ROS by the plasma membrane NADPH oxidase NOX1 (which is attracted by RIPK1) [93]. NOX1 is activated by TNF, and NOX1-induced ROS may induce or maintain the mitochondrial respiratory chain's further generation of ROS [94]. RNS, like ROS, are powerful oxidants that can begin or amplify lipid, protein, and peroxidation oxidation [95, 96]. Nitric oxide acts as a second messenger in a variety of signaling pathways at low intracellular concentrations. Nitric oxide is particularly detrimental when overproduced, and it produces RNS with different chemical and biological features [97]. Nitric oxide has recently been demonstrated to cause RIPK1- and RIPK3-mediated necroptosis [98].
Role of necroptosis in the progression of organ fibrosis
Liver fibrosis
Interestingly, necroptosis may play a critical role in different liver diseases. As explained previously, necroptosis is commonly seen in hepatocytes, and it is always accompanied by inflammation [99,100,101]. MLKL deficiency reduces glucose intolerance and hepatic insulin resistance [102]. By inhibiting hepatocyte autophagy, MLKL knockout protects against NASH induced by a high-fat, fructose, and cholesterol diet [103]. Targeting MLKL may be a successful strategy for treating liver fibrosis because MLKL-mediated signalling plays a significant role in liver damage and fibrosis [104]. Therefore, we addressed the concerns about necroptosis and its important role in liver inflammation and fibrosis.
Hepatic RIPK3 has been investigated as a factor in NAFLD severity in people and mice by Afonso et al. [105]. In this study, TNF-α promotes RIPK3-dependent oxidative stress through hepatocyte necroptosis. Hepatic RIPK3, MLKL, and TNF-α expression levels were increased in HFCD and MCD diet-fed animals. Strikingly, RIPK3 deficiency reduced MCD diet-induced liver fibrosis [105]. In line with this study, Majdi [106] and colleagues showed that the inhibition of RIPK1 improved NASH characteristics in HFD-fed mice and reversed steatosis through an MLKL-dependent mechanism that mainly affects mitochondrial respiration. They revealed that RIPA-56 inhibits RIPK1 to decrease liver damage, inflammation, fibrosis, and steatosis in animal models in either a curative or preventative manner. RIPA-56 or necrosulfonamide, a selective inhibitor of human MLKL, and the KO of MLKL in fat-loaded AML-12 murine hepatocytes were used. In steatotic hepatocytes, MLKL-KO activated mitochondrial respiration and increased β-oxidation. Additionally, in experimental NASH, RIPK3-KO animals showed higher activity of the liver mitochondrial respiratory chain complexes, which is consistent with lowered MLKL activation [106]. In the same context, Mohammed et al. [107] used a mouse model that produces spontaneous NAFLD/NASH and develops into fibrosis and HCC with age. MLKL and p-MLKL were markedly upregulated in the livers of Cu/Zn superoxide dismutase deficient (Sod1/ or Sod1KO) compared to WT mice. Similarly, RIPK3 and p-RIPK3 also increased. Also, NLRP3 inflammasome and transcript levels of proinflammatory cytokines and chemokines (TNF-α, IL-6, IL-1β, and Ccl2) linked with human NASH were upregulated in this study. In contrast, necrostatin-1 (Nec-1) treatment reversed these effects on Sod1KO mice [107]. In (2021), Afonso and co-authors explored the function of RIPK3 in controlling liver metabolism, damage, and inflammation. To hinder NASH, RIPK3 and its complex signalling have emerged as a new viable treatment. NASH patients' RIPK3 levels were elevated in both cohorts, and this was shown to be associated with both inflammation and fibrosis of the liver. As a result, RIPK3 impairment reduced CDAA-induced inflammation and fibrosis and inhibited tumorigenesis in mice at both 32 and 66 weeks of age. There were nodules and enhanced hepatocellular proliferation in WT mice fed a CDAA diet for six months, which were decreased in RIPK3−/− animals [108].
Besides, the kinase activity of the RIPK1 protein, particularly in hematopoietic-derived macrophages, has been shown to participate in the pathogenesis of NASH, according to Xu et al. [109]. The NASH phenotype of hepatic steatosis, liver damage, fibrosis, and decreased hepatic cell death and inflammation in Rip1K45A/K45A mice was dramatically relieved compared to WT mice. RIPK1 kinase activation induced inflammasome activation, necrosis, and cell death in both bone marrow-derived macrophages and mice primary Kupffer cells following induction of kinase activation by lipotoxicity and in vitro saturated fatty acids (palmitic acid) [109].
Importantly, it has been observed that necroptosis is mediated by RIPK3 as well as neutrophil-driven inflammation of the alcoholic liver in patients with end-stage cirrhosis. Patients with alcoholic cirrhosis may have higher RIPK3 and MPO levels as a sign of a bad prognosis. The liver levels of MPO and RIPK3 were shown to be associated with a higher Ishak score [110]. A new study by Zhang et al. [111] identifies OGT as an important inhibitor of necroptosis in hepatocytes, and OGT-LKO mice may serve as an efficient spontaneous genetic model of liver fibrosis in humans. Hepatocyte necroptosis and the onset of hepatic fibrosis are prevented by O-GlcNAc alteration. Patients with alcoholic liver cirrhosis and mice with liver damage caused by ethanol both have lower levels of O-GlcNAc. Overt necroptosis and increased protein expression levels of RIPK3 and MLKL were seen in OGT-deficient liver cells [111].
It was discovered for the first time in human and rodent acute-on-chronic liver failure (ACLF) by Kondo et al. [112] that RIPK1-mediated cell death is critical, and it was also shown that inhibiting RIPK1 might be a new therapeutic method for preventing the progression of vulnerable individuals from ACLF. LPS was provided to bile-duct ligated rats, and galactosamine (CCL4/GalN) was delivered to carbon tetrachloride (CT)-induced fibrosis mice. Nec-1 therapy lowered the severity of ACLF by reducing liver, kidney, and brain damage, paralleled by reduced hepatic and renal cell deaths. A mouse model of ACLF induced by CCL4/GalN showed similar hepatoprotective effects with RIPA56 [112]. Non-ACLF patients at risk of developing ACLF may benefit from targeting RIPK1 as a therapeutic target, as shown by these findings. According to a recent study by Bai et al. [113], M2-like macrophages protect the liver by reducing necroptosis-S100A9-necroinflammation in ACLF [113].
Interestingly, melatonin (MLT) was found to reduce hepatic hydroxyproline content, TGF-β1, smooth muscle actin expression levels, and hepatocellular damage in chronic CCl4 administration. By impeding necroptosis-associated inflammatory signalling, MLT reduced these increases and prevented liver fibrosis by downregulating RIPK1 and MLKL. Serum high-mobility group box 1 (HMGB1) and IL-1β were both reduced by MLT, as was the interaction between HMGB1 receptors for advanced glycation end products and these two other CCl4-induced changes (RAGE) [114].
Another study conducted liver samples of human primary biliary cholangitis patients, coinciding with thioflavin T labelling, and indicating stimulation of necroptosis through elevated RIPK3 and p-MLKL levels. BDL resulted in apparent markers of necroptosis. In this study, RIPK3 impairment inhibited BDL-induced necroinflammation and oxidative stress reduced in RIPK3−/− animals at 3 days following BDL. RIPK3 deficiency also related with increased hepatic expression of HO-1 indicating bile acid toxicity and buildup of iron in BDL mice. In PBC patients, necroptosis is initiated and facilitates hepatic necrosis in acute cholestasis produced by BDL. However, it may be necessary to use other strategies to prevent chronic liver disease development in necroptosis-targeted acute cholestasis [115].
Finally, Mohammed et al. [116] demonstrated that liver aging is accompanied with an increase in necroptosis, and necroptosis leads to chronic liver inflammation and fibrosis. Changes in necroptotic indicators in the liver were shown to correlate with changes in the expression of TNF-α, IL6, and IL1β, as well as the expression of markers of inflammation (TGF-β1, IFN-γ). Necroptosis markers and proinflammatory cytokines were enhanced in aged mice hepatocytes and liver macrophages compared to young mice. In contrast, Nec-1 decreased necroptosis, M1 macrophage markers, cell senescence, and the cytokines released in the livers of aged mice [116].
Lung fibrosis
Recently, CoV-2 has been found to stimulate caspase-8, which causes apoptosis and inflammation of lung epithelial cells in response to SARS-CoV-2 infection. The virus-induced necroptosis mechanism releases the processed inflammatory cytokines. It has been shown that the SARS-CoV-2 virus causes apoptosis, necroptosis, and inflammatory activation in HFH4-hACE2 transgenic mice. In addition to necroptosis and apoptosis, a study of postmortem lung sections from COVID-19 patients found extensive inflammatory cell infiltration, necrotic cell debris, and pulmonary interstitial fibrosis. A treatment strategy for COVID-19 might benefit from these findings [117]. Latterly, Tao et al. [118] reported that macrophage-mediated necroptosis aids the course of silicosis by increasing pulmonary inflammation as well as fibrogenesis in their mouse model of the disease. Mice infected with silicosis show a substantial increase in necroptotic macrophage signaling in their lungs. In this silicosis model, the authors observed an increase in M1 macrophage infiltration and proinflammatory cytokines (TNF-α, IL-6). TGF-β1, fibrosis biomarkers α-SMA, and collagen I were likewise deregulated; however, Nec-1 was able to restore these effects [118].
In BLM-induced lung fibrosis and inflammation, Lee et al. (2018) found that necroptosis is linked to lung fibrosis via the release of damage-associated molecular patterns (DAMP). In idiopathic pulmonary fibrosis (IPF) lungs, RIPK3 expression was elevated, apoptosis, and necroptosis were mostly seen in alveolar epithelial cells (AECs). Knockout studies in AECs of Nec-1 and RIPK3 demonstrated that necroptosis is involved in BLM and hydrogen peroxide-induced cell death in necroptosis. AECs treated with BLM showed increased RIPK3, IL-1β, and HMGB1 expression levels. In RIPK3 mutant mice, Nec-1, as well as decreased HMGB1 and IL-1β levels, attenuated BLM-induced lung inflammation and fibrosis [119].
Renal fibrosis
Several studies discussed the role of RIPK-mediated necroptosis in kidney disorders, including AKI and fibrosis [120]. In (2015), Zhu et al. found that necroptotic cell death, a higher level of RIPK1, and RIPK3 appeared eight weeks after subtotal nephrectomy surgery. Nec-1 treatment significantly improved renal functions. Nec-1 inhibited necroptosis as indicated by downregulating RIPK1, RIPK3, and MLKL [121]. Also, necroptosis has an important role in gentamicin-induced kidney injury progression; therefore, it is considered a potential target to alleviate AKI by inhibiting necroptosis. Upregulation of MLKL, RIPK3, p-MLKL was observed in gentamicin-treated mice and cultured renal tubule cells. In contrast, Nec-1 ameliorated gentamicin-induced necrosis and upregulated MLKL and RIPK3 in mice and in vitro cultured cells [122]. In cisplatin-induced nephropathy, disruption of TGF-β II receptor suppressed Smad2/3 activation and mitigated kidney injury. Smad2 evoked AKI by increasing apoptosis and inflammation, according to these findings. Smad2 knockout in TECs prevented renal function loss and reduced p53-mediated cell death, RIPK-evoked necroptosis, and NF-κB-induced inflammation. Lentivirus-mediated Smad2 knockdown mitigated kidney injury and inflammatory response while enhancing renal function [123].
In UUO-induced renal fibrosis, Dai et al. and co-workers showed the antifibrotic activity of fluorofenidone (AKF-PD) and Nec-1, which is mediated by the necroptosis suppression in TNF-α and Z-VAD stimulated HK-2 cells. They found that these agents ameliorate renal tubular damage and expression of IL-1β, TNF-α, and chemokines, as well as the deposition of collagen. Treatment with AKF-PD or Nec-1 protects renal tubular epithelial cells from necrosis and reduces the serum level of LDH-mediated by suppressing MLKL and RIPK3 phosphorylation [124]. In line with this study, Xiao et al. also reported that the levels of RIPK1/RIPK3/MLKL protein increased in the obstructed kidneys seven days after UUO. Interestingly, Nec-1 decreased TNF-α, IL-1β, and monocyte chemotactic protein-1 expression levels as well as TGF-β and α-SMA, indicating renal fibrosis suppression [125].
Zhu et al. [126] suggested that Ang-II exposure mediated necroptosis in renal tubular epithelial cells. They assess the necroptosis in the renal tubular cell in vivo by adding Nec-1 and in vitro with Ang-II and RIPK1/3 or MLKL inhibitors in HK-2 cells. Fas and FasL proteins have a key role in Ang-II-induced necroptosis, and FasL disruption reduced the percentage of necroptotic cells, implying that Fas and FasL are likely important signal regulators in Ang-II-induced necroptosis [126]. In addition, Zhu et al. speculated that necroptotic cell death might play a role in the loss of renal tubular cells in SNx rats. Effectively inhibiting necroptosis and apoptosis in CKD's early and intermediate phases improves renal function and tubular damage. Nec-1 and zVAD treatment significantly reduced necroptosis and apoptosis in SNx rats. Also, Nec-1 inhibited necroptosis and reduced the proportion of the TUNEL-positive cells [127].
Cardiac fibrosis
Several investigations reported the potential impact of necroptosis heart diseases. Yue et al. [128] found that alliin could protect cardiomyocytes against necroptosis by maintaining cardiac function, decreasing myocardial lesions, and attenuating MI. In vitro and in vivo studies demonstrated that alliin prevented necroptosis while promoting autophagy. Alliin downregulated RIPK1, RIPK3, and TRAF2, while increasing Beclin-1 and LC-3 levels in a dose-dependent manner. In addition, alliin raises the level of PPAR-γ [128]. Zhang et al. have demonstrated that NaHS has anti-necrosis activity by reducing the expression of RIPK1 and RIPK3. NaHS also reduced the number of cardiac fibroblasts and the levels of α-SMA, proliferating cell nuclear antigen (PCNA), collagen I, and collagen III. In hypoxia-induced cardiac fibroblasts, NaHS boosted Sirtuin 3 (SIRT3) expression. Furthermore, the necroptosis inhibitory and antioxidant effects were decreased following SIRT3 siRNA transfection [129]. Also, Xu et al. reported the cardiac protective anti-inflammatory effects of irbesartan. The protective effects of irbesartan were related to inhibition of necroptosis as indicated by the downregulation of RIPK1, RIPK3, and MLKL levels [130]. Furthermore, Sharifi et al. and colleagues demonstrated that nesfatin-1 dose-dependently could exert a cardioprotective effect against MI/R by reducing oxidative stress and collagen deposition. Nesfatin-1 can suppress necroptosis by downregulating RIPK1, RIPK3, and MLKL [131].
Many studies discuss the cardioprotective effects of anti-necroptosis agents necrostatin-7 (Nec-7) and Nec-1 through inhibition of RIPK pathway. In 2022, Qiao et al. demonstrated that a modest dose of Nec-1 and GSK872 (GSK) inhibited RIPK1/RIPK3 in HGF-induced cardiac fibrosis. HGF activates the RIPK pathway, causing autophagic-related proteins such LC3-II, P62, and active-cathepsin D to be upregulated. The levels of RIPK3/p-RIPK3 and RIPK1/p-RIPK1 were both reduced when RIPK1/RIPK3 was inhibited. P62 forms a compound with both kinases and stimulates RIPK1 and RIPK3 binding. In a diabetic rat model, Nec-1 effectively reduced CF, decreased autophagic proteins, and enhanced heart function [132]. Furthermore, Wang et al. and co-workers found that Nec-1 ameliorates myocardial cell death by inhibiting fibrosis in rats with myocardial ischemia/late reperfusion. Nec-1 decreased creatine kinase and downregulated autophagy within 24 h after reperfusion. These findings imply that anti-necroptosis therapy may improve the therapeutic outcomes of ischemic heart patients [133]. In the same context, another study found that administration of Nec-7 before 1 h of left coronary arterial occlusion in the rat model reduced the scar length in the left ventricle on the 21st day after surgery. Nec-7 decreased N-terminal pro-brain natriuretic peptide, which improved left ventricular function [134]. In (2021) Fu et al. proved that necroptosis is a novel mechanism in AF pathogenesis in mice. Administration of CaCl2-Ach enhanced AF susceptibility and fibrosis in HFD model mediated by upregulating of RIPK1, RIPK3, MLKL, and CaMKs II. However, Nec-1 administration partially attenuated CaCl2-Ach or HFD-induced fibrosis, linking necroptosis to AF pathogenesis [135] (Table 1).
Conclusions and future recommendations
Cell death can originate from pathological situations and is a natural process for replacing old cells. It is understood that cell death is a crucial component of both acute and chronic illnesses. Necroptosis is a mode of cell death that differs from apoptosis morphologically and biochemically. Necroptosis is mediated by intracellular signaling molecules, including RIPK1, RIPK3, MLKL, caspase-8, and others. Necroptosis is involved in many different diseases, according to preclinical research. Necroptosis has been shown to be a contributing factor in fibrosis. Consequently, liver, kidney, lung, and heart fibrosis have reportedly benefited from necroptosis inhibition. The inhibition of necroptosis motivates therapeutic efforts to pharmacologically target this mechanism of programmed cell death.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Henderson NC, Rieder F, Wynn TA (2020) Fibrosis: from mechanisms to medicines. Nature 587(7835):555–566. https://doi.org/10.1038/s41586-020-2938-9
Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. J Pathol 214(2):199–210. https://doi.org/10.1002/path.2277
Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Investig 115(2):209–218. https://doi.org/10.1172/jci24282
Wynn TA (2011) Integrating mechanisms of pulmonary fibrosis. J Exp Med 208(7):1339–1350. https://doi.org/10.1084/jem.20110551
Quan TE, Cowper SE, Bucala R (2006) The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep 8(2):145–150. https://doi.org/10.1007/s11926-006-0055-x
Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Investig 117(3):524–529. https://doi.org/10.1172/jci31487
Friedman SL (2004) Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol 1(2):98–105. https://doi.org/10.1038/ncpgasthep0055
Gabbiani G (2003) The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 200(4):500–503. https://doi.org/10.1002/path.1427
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6(5):392–401. https://doi.org/10.1038/nrc1877
Hahn E, Wick G, Pencev D, Timpl R (1980) Distribution of basement membrane proteins in normal and fibrotic human liver: collagen type IV, laminin, and fibronectin. Gut 21(1):63–71. https://doi.org/10.1136/gut.21.1.63
Brown B, Lindberg K, Reing J, Stolz DB, Badylak SF (2006) The basement membrane component of biologic scaffolds derived from extracellular matrix. Tissue Eng 12(3):519–526. https://doi.org/10.1089/ten.2006.12.519
Maher JJ, McGuire RF (1990) Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Investig 86(5):1641–1648. https://doi.org/10.1172/jci114886
Schmitt-Gräff A, Krüger S, Bochard F, Gabbiani G, Denk H (1991) Modulation of alpha smooth muscle actin and desmin expression in perisinusoidal cells of normal and diseased human livers. Am J Pathol 138(5):1233–1242
Neubauer K, Knittel T, Aurisch S, Fellmer P, Ramadori G (1996) Glial fibrillary acidic protein–a cell type specific marker for Ito cells in vivo and in vitro. J Hepatol 24(6):719–730. https://doi.org/10.1016/s0168-8278(96)80269-8
Niki T, De Bleser PJ, Xu G, Van Den Berg K, Wisse E, Geerts A (1996) Comparison of glial fibrillary acidic protein and desmin staining in normal and CCl4-induced fibrotic rat livers. Hepatology (Baltimore, MD) 23(6):1538–1545. https://doi.org/10.1002/hep.510230634
Benyon RC, Arthur MJ (2001) Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis 21(3):373–384. https://doi.org/10.1055/s-2001-17552
Friedman SL (2008) Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88(1):125–172. https://doi.org/10.1152/physrev.00013.2007
Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF et al (2006) Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol 45(3):429–438. https://doi.org/10.1016/j.jhep.2006.04.014
Lemoinne S, Cadoret A, El Mourabit H, Thabut D, Housset C (2013) Origins and functions of liver myofibroblasts. Biochem Biophys Acta 1832(7):948–954. https://doi.org/10.1016/j.bbadis.2013.02.019
Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H et al (2007) Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 282(32):23337–23347. https://doi.org/10.1074/jbc.M700194200
Wang P, Liu T, Cong M, Wu X, Bai Y, Yin C et al (2009) Expression of extracellular matrix genes in cultured hepatic oval cells: an origin of hepatic stellate cells through transforming growth factor beta? Liver Int 29(4):575–584. https://doi.org/10.1111/j.1478-3231.2009.01992.x
Chiaramonte MG, Cheever AW, Malley JD, Donaldson DD, Wynn TA (2001) Studies of murine schistosomiasis reveal interleukin-13 blockade as a treatment for established and progressive liver fibrosis. Hepatology (Baltimore, MD) 34(2):273–282. https://doi.org/10.1053/jhep.2001.26376
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183(6):788–824. https://doi.org/10.1164/rccm.2009-040GL
Gross TJ, Hunninghake GW (2001) Idiopathic pulmonary fibrosis. N Engl J Med 345(7):517–525. https://doi.org/10.1056/NEJMra003200
Strieter RM (2005) Pathogenesis and natural history of usual interstitial pneumonia: the whole story or the last chapter of a long novel. Chest 128(5 Suppl 1):526s-s532. https://doi.org/10.1378/chest.128.5_suppl_1.526S
Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R (2001) Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2(10):947–950. https://doi.org/10.1038/ni712
Michael B, Burke JF Jr (1994) Chronic renal disease: new therapies to delay kidney replacement. Geriatrics 49(8):33–38
Shanley PF (1996) The pathology of chronic renal ischemia. Semin Nephrol 16(1):21–32
Guiteras R, Flaquer M, Cruzado JM (2016) Macrophage in chronic kidney disease. Clin Kidney J 9(6):765–771. https://doi.org/10.1093/ckj/sfw096
Martínez-Arias L, Panizo S, Alonso-Montes C, Martín-Vírgala J, Martín-Carro B, Fernández-Villabrille S et al (2021) Effects of calcitriol and paricalcitol on renal fibrosis in CKD. Nephrol Dial Transpl 36(5):793–803. https://doi.org/10.1093/ndt/gfaa373
Cao Q, Harris DC, Wang YJP (2015) Macrophages in kidney injury, inflammation, and fibrosis. Physiology 30(3):183–194
Sun YB, Qu X, Caruana G, Li J (2016) The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differ Res Biol Div 92(3):102–107. https://doi.org/10.1016/j.diff.2016.05.008
Allinovi M, De Chiara L, Angelotti ML, Becherucci F, Romagnani P (2018) Anti-fibrotic treatments: a review of clinical evidence. Matrix Biol 68–69:333–354. https://doi.org/10.1016/j.matbio.2018.02.017
Klahr S, Morrissey JJ (2000) The role of vasoactive compounds, growth factors and cytokines in the progression of renal disease. Kidney Int Suppl 75:S7-14
Yang HC, Zuo Y, Fogo AB (2010) Models of chronic kidney disease. Drug Discov Today Dis Model 7(1–2):13–19. https://doi.org/10.1016/j.ddmod.2010.08.002
Nogueira A, Pires MJ, Oliveira PA (2017) Pathophysiological mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo 31(1):1–22. https://doi.org/10.21873/invivo.11019
Becherucci F, Mazzinghi B, Allinovi M, Angelotti ML, Romagnani P (2018) Regenerating the kidney using human pluripotent stem cells and renal progenitors. Expert Opin Biol Ther 18(7):795–806. https://doi.org/10.1080/14712598.2018.1492546
Murtha LA, Schuliga MJ, Mabotuwana NS, Hardy SA, Waters DW, Burgess JK et al (2017) The processes and mechanisms of cardiac and pulmonary fibrosis. Front Physiol 8:777. https://doi.org/10.3389/fphys.2017.00777
Porter KE, Turner NA (2009) Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther 123(2):255–278. https://doi.org/10.1016/j.pharmthera.2009.05.002
Jellis C, Martin J, Narula J, Marwick TH (2010) Assessment of nonischemic myocardial fibrosis. J Am Coll Cardiol 56(2):89–97. https://doi.org/10.1016/j.jacc.2010.02.047
Segura AM, Frazier OH, Buja LM (2014) Fibrosis and heart failure. Heart Fail Rev 19(2):173–185. https://doi.org/10.1007/s10741-012-9365-4
Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC (2013) Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 10(1):15–26. https://doi.org/10.1038/nrcardio.2012.158
Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S (2014) Myocardial extracellular matrix: an ever-changing and diverse entity. Circ Res 114(5):872–888. https://doi.org/10.1161/circresaha.114.302533
Schellings MW, Pinto YM, Heymans S (2004) Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res 64(1):24–31. https://doi.org/10.1016/j.cardiores.2004.06.006
Gupta K, Phan N, Wang Q, Liu B (2018) Necroptosis in cardiovascular disease: a new therapeutic target. J Mol Cell Cardiol 118:26–35. https://doi.org/10.1016/j.yjmcc.2018.03.003
Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13(19):2514–2526. https://doi.org/10.1101/gad.13.19.2514
Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N et al (2007) Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 19(10):2056–2067. https://doi.org/10.1016/j.cellsig.2007.05.016
Galluzzi L, Vitale I, Abrams J, Alnemri E, Baehrecke E, Blagosklonny M et al (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death. Cell Death Diffe 19(1):107–20
Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK (2014) The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity 41(4):567–578. https://doi.org/10.1016/j.immuni.2014.09.016
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S et al (2000) Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1(6):489–495. https://doi.org/10.1038/82732
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137(6):1100–1111. https://doi.org/10.1016/j.cell.2009.05.021
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M et al (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137(6):1112–1123. https://doi.org/10.1016/j.cell.2009.05.037
Sun L, Wang H, Wang Z, He S, Chen S, Liao D et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148(1–2):213–227. https://doi.org/10.1016/j.cell.2011.11.031
Jouan-Lanhouet S, Riquet F, Duprez L, Vanden Berghe T, Takahashi N, Vandenabeele P (2014) Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 35:2–13. https://doi.org/10.1016/j.semcdb.2014.08.010
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N et al (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1(2):112–119. https://doi.org/10.1038/nchembio711
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X et al (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4(5):313–321. https://doi.org/10.1038/nchembio.83
Linkermann A, Green DR (2014) Necroptosis. N Engl J Med 370(5):455–465. https://doi.org/10.1056/NEJMra1310050
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC et al (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science (New York, NY) 325(5938):332–336. https://doi.org/10.1126/science.1172308
Vandenabeele P, Declercq W, Van Herreweghe F, Vanden BT (2010) The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Science signaling. 3(115):4. https://doi.org/10.1126/scisignal.3115re4
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J et al (2012) Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA 109(14):5322–5327. https://doi.org/10.1073/pnas.1200012109
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517(7534):311–320. https://doi.org/10.1038/nature14191
Silke J, Rickard JA, Gerlic M (2015) The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol 16(7):689–697. https://doi.org/10.1038/ni.3206
Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M et al (2013) Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci USA 110(33):E3109–E3118. https://doi.org/10.1073/pnas.1301218110
McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB et al (2014) Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci USA 111(31):E3206–E3213. https://doi.org/10.1073/pnas.1407068111
Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114(2):181–190. https://doi.org/10.1016/s0092-8674(03)00521-x
Han J, Zhong CQ, Zhang DW (2011) Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol 12(12):1143–1149. https://doi.org/10.1038/ni.2159
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11(10):700–714. https://doi.org/10.1038/nrm2970
Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D et al (2014) Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science (New York, NY) 343(6177):1357–1360. https://doi.org/10.1126/science.1249361
Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE (2013) Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1β production (Baltimore, Md : 1950). J Immunol 191(10):5239–46. https://doi.org/10.4049/jimmunol.1301581
Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L et al (2012) Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36(2):215–227. https://doi.org/10.1016/j.immuni.2012.01.012
Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D (2013) Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38(1):27–40. https://doi.org/10.1016/j.immuni.2012.09.015
Yabal M, Müller N, Adler H, Knies N, Groß CJ, Damgaard RB et al (2014) XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep 7(6):1796–1808. https://doi.org/10.1016/j.celrep.2014.05.008
Moriwaki K, Bertin J, Gough PJ, Chan FK (2015) A RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing (Baltimore, Md : 1950). J Immunol 194(4):1938–44. https://doi.org/10.4049/jimmunol.1402167
Wong WW, Vince JE, Lalaoui N, Lawlor KE, Chau D, Bankovacki A et al (2014) cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood 123(16):2562–2572. https://doi.org/10.1182/blood-2013-06-510743
Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA et al (2015) RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 6:6282. https://doi.org/10.1038/ncomms7282
Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G et al (2017) Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA 114(6):E961–E969. https://doi.org/10.1073/pnas.1613305114
Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV (1996) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4(4):387–396. https://doi.org/10.1016/s1074-7613(00)80252-6
Liu ZG, Hsu H, Goeddel DV, Karin M (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87(3):565–576. https://doi.org/10.1016/s0092-8674(00)81375-6
Bury JJ, Highley JR, Cooper-Knock J, Goodall EF, Higginbottom A, McDermott CJ et al (2016) Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology 36(2):125–134. https://doi.org/10.1111/neup.12240
Bokov A, Chaudhuri A, Richardson A (2004) The role of oxidative damage and stress in aging. Mech Ageing Dev 125(10–11):811–826. https://doi.org/10.1016/j.mad.2004.07.009
Hassanein EHM, Sayed GA, Alzoghaibi AM, Alammar AS, Abdel-Wahab BA, Abd El-Ghafar OAM et al (2022) Azithromycin mitigates cisplatin-induced lung oxidative stress, inflammation and necroptosis by upregulating SIRT1, PPARγ, and Nrf2/HO-1 signaling. Pharmaceuticals (Basel) 16(1):7. https://doi.org/10.3390/ph16010052
Tashkandi HM, Althagafy HS, Jaber FA, Alamri T, Al-Abbas NS, Shaer NA et al (2023) Vinpocetine mitigates methotrexate-induced duodenal intoxication by modulating NF-κB, JAK1/STAT-3, and RIPK1/RIPK3/MLKL signals. Immunopharm Immunotoxicol 4:1–9. https://doi.org/10.1080/08923973.2023.2239491
Hassanein EHM, Bakr AG, El-Shoura EAM, Ahmed LK, Ali FEM (2023) Acetovanillone augmented the cardioprotective effect of carvedilol against cadmium-induced heart injury via suppression of oxidative stress and inflammation signaling pathways. Sci Rep 13(1):5278. https://doi.org/10.1038/s41598-023-31231-5
Hanus J, Anderson C, Wang S (2015) RPE necroptosis in response to oxidative stress and in AMD. Ageing Res Rev 24(Pt B):286–298. https://doi.org/10.1016/j.arr.2015.09.002
Zhang L, Feng Q, Wang T (2018) Necrostatin-1 protects against paraquat-induced cardiac contractile dysfunction via RIP1-RIP3-MLKL-dependent necroptosis pathway. Cardiovasc Toxicol 18(4):346–355. https://doi.org/10.1007/s12012-017-9441-z
Takemoto K, Hatano E, Iwaisako K, Takeiri M, Noma N, Ohmae S et al (2014) Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio 4:777–787. https://doi.org/10.1016/j.fob.2014.08.007
Han CH, Guan ZB, Zhang PX, Fang HL, Li L, Zhang HM et al (2018) Oxidative stress induced necroptosis activation is involved in the pathogenesis of hyperoxic acute lung injury. Biochem Biophys Res Commun 495(3):2178–2183. https://doi.org/10.1016/j.bbrc.2017.12.100
Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R et al (2006) Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radical Biol Med 40(11):1993–2004. https://doi.org/10.1016/j.freeradbiomed.2006.01.036
Zhang S, Wang Y, Li D, Wu J, Si W, Wu Y (2016) Necrostatin-1 attenuates inflammatory response and improves cognitive function in chronic ischemic stroke mice. Medicines (Basel, Switzerland). 3(3):787. https://doi.org/10.3390/medicines3030016
Zhang H, Davies KJA, Forman HJ (2015) Oxidative stress response and Nrf2 signaling in aging. Free Radical Biol Med 88(Pt B):314–336. https://doi.org/10.1016/j.freeradbiomed.2015.05.036
Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W (1992) Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions: evidence for the involvement of mitochondrial radical generation. J Biol Chem 267(8):5317–23
Festjens N, Kalai M, Smet J, Meeus A, Van Coster R, Saelens X et al (2006) Butylated hydroxyanisole is more than a reactive oxygen species scavenger. Cell Death Differ 13(1):166–169. https://doi.org/10.1038/sj.cdd.4401746
Kim YS, Morgan MJ, Choksi S, Liu ZG (2007) TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell 26(5):675–687. https://doi.org/10.1016/j.molcel.2007.04.021
Morgan MJ, Kim YS, Liu ZG (2008) TNFalpha and reactive oxygen species in necrotic cell death. Cell Res 18(3):343–349. https://doi.org/10.1038/cr.2008.31
Goss SP, Singh RJ, Hogg N, Kalyanaraman B (1999) Reactions of *NO, *NO2 and peroxynitrite in membranes: physiological implications. Free Radical Res 31(6):597–606. https://doi.org/10.1080/10715769900301171
Tien M, Berlett BS, Levine RL, Chock PB, Stadtman ER (1999) Peroxynitrite-mediated modification of proteins at physiological carbon dioxide concentration: pH dependence of carbonyl formation, tyrosine nitration, and methionine oxidation. Proc Natl Acad Sci USA 96(14):7809–7814. https://doi.org/10.1073/pnas.96.14.7809
Brüne B (2005) The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid Redox Signal 7(3–4):497–507. https://doi.org/10.1089/ars.2005.7.497
Davis CW, Hawkins BJ, Ramasamy S, Irrinki KM, Cameron BA, Islam K et al (2010) Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radical Biol Med 48(2):306–317. https://doi.org/10.1016/j.freeradbiomed.2009.11.001
Li X, Dong G, Xiong H, Diao H (2021) A narrative review of the role of necroptosis in liver disease: a double-edged sword. Ann Transl Med 9(5):422. https://doi.org/10.21037/atm-20-5162
Shi S, Verstegen MMA, Mezzanotte L, de Jonge J, Löwik C, van der Laan LJW (2019) Necroptotic Cell death in liver transplantation and underlying diseases: mechanisms and clinical perspective. Liver Transpl 25(7):1091–1104. https://doi.org/10.1002/lt.25488
Idrovo JP, Boe DM, Kaahui S, Yang WL, Kovacs EJ (2020) Hepatic inflammation after burn injury is associated with necroptotic cell death signaling. J Trauma Acute Care Surg 89(4):768–774. https://doi.org/10.1097/ta.0000000000002865
Xu H, Du X, Liu G, Huang S, Du W, Zou S et al (2019) The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab 23:14–23. https://doi.org/10.1016/j.molmet.2019.02.003
Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S et al (2020) MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J Hepatol 73(3):616–627. https://doi.org/10.1016/j.jhep.2020.03.023
Guo R, Jia X, Ding Z, Wang G, Jiang M, Li B et al (2022) Loss of MLKL ameliorates liver fibrosis by inhibiting hepatocyte necroptosis and hepatic stellate cell activation. Theranostics 12(11):5220–5236. https://doi.org/10.7150/thno.71400
Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H et al (2015) Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (London, England : 1979) 129(8):721–39. https://doi.org/10.1042/cs20140732
Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F et al (2020) Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J Hepatol 72(4):627–635. https://doi.org/10.1016/j.jhep.2019.11.008
Mohammed S, Nicklas EH, Thadathil N, Selvarani R, Royce GH, Kinter M et al (2021) Role of necroptosis in chronic hepatic inflammation and fibrosis in a mouse model of increased oxidative stress. Free Radical Biol Med 164:315–328. https://doi.org/10.1016/j.freeradbiomed.2020.12.449
Afonso MB, Rodrigues PM, Mateus-Pinheiro M, Simão AL, Gaspar MM, Majdi A et al (2021) RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut 70(12):2359–2372. https://doi.org/10.1136/gutjnl-2020-321767
Xu C, Wu X, Zhang X, Xie Q, Fan C, Zhang H (2018) Embryonic lethality and host immunity of RelA-deficient mice are mediated by both apoptosis and necroptosis (Baltimore, Md : 1950). J Immunol 200(1):271–85. https://doi.org/10.4049/jimmunol.1700859
Zhang Z, Xie G, Liang L, Liu H, Pan J, Cheng H et al (2018) RIPK3-mediated necroptosis and neutrophil infiltration are associated with poor prognosis in patients with alcoholic cirrhosis. J Immunol Res 2018:1509851. https://doi.org/10.1155/2018/1509851
Zhang B, Li MD, Yin R, Liu Y, Yang Y, Mitchell-Richards KA et al (2019) O-GlcNAc transferase suppresses necroptosis and liver fibrosis. JCI Insight 4(21):7. https://doi.org/10.1172/jci.insight.127709
Kondo T, Macdonald S, Engelmann C, Habtesion A, Macnaughtan J, Mehta G et al (2021) The role of RIPK1 mediated cell death in acute on chronic liver failure. Cell Death Dis 13(1):5. https://doi.org/10.1038/s41419-021-04442-9
Bai L, Kong M, Duan Z, Liu S, Zheng S, Chen Y (2021) M2-like macrophages exert hepatoprotection in acute-on-chronic liver failure through inhibiting necroptosis-S100A9-necroinflammation axis. Cell Death Dis 12(1):93. https://doi.org/10.1038/s41419-020-03378-w
Choi HS, Kang JW, Lee SM (2015) Melatonin attenuates carbon tetrachloride-induced liver fibrosis via inhibition of necroptosis. Transl Res 166(3):292–303. https://doi.org/10.1016/j.trsl.2015.04.002
Afonso MB, Rodrigues PM, Simão AL, Ofengeim D, Carvalho T, Amaral JD et al (2016) Activation of necroptosis in human and experimental cholestasis. Cell Death Dis 7(9):e2390. https://doi.org/10.1038/cddis.2016.280
Mohammed S, Thadathil N, Selvarani R, Nicklas EH, Wang D, Miller BF et al (2021) Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 20(12):e13512. https://doi.org/10.1111/acel.13512
Li S, Zhang Y, Guan Z, Li H, Ye M, Chen X et al (2020) SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct Target Ther 5(1):235. https://doi.org/10.1038/s41392-020-00334-0
Tao H, Zhao H, Ge D, Liao J, Shao L, Mo A et al (2022) Necroptosis in pulmonary macrophages promotes silica-induced inflammation and interstitial fibrosis in mice. Toxicol Lett 355:150–159. https://doi.org/10.1016/j.toxlet.2021.11.015
Lee JM, Yoshida M, Kim MS, Lee JH, Baek AR, Jang AS et al (2018) Involvement of alveolar epithelial cell necroptosis in idiopathic pulmonary fibrosis pathogenesis. Am J Respir Cell Mol Biol 59(2):215–224. https://doi.org/10.1165/rcmb.2017-0034OC
Uni R, Choi MEJN (2021) Novel roles of necroptosis mediator receptor-interacting protein kinase 3 in kidney injury. Nephron 146:1–5
Zhu Y, Cui H, Gan H, Xia Y, Wang L, Wang Y et al (2015) Necroptosis mediated by receptor interaction protein kinase 1 and 3 aggravates chronic kidney injury of subtotal nephrectomised rats. Biochem Biophys Res Commun 461(4):575–581. https://doi.org/10.1016/j.bbrc.2015.03.164
Huang H, Jin WW, Huang M, Ji H, Capen DE, Xia Y et al (2020) Gentamicin-induced acute kidney injury in an animal model involves programmed necrosis of the collecting duct. J Am Soc Nephrol 31(9):2097–2115. https://doi.org/10.1681/asn.2019020204
Yang Q, Ren G-L, Wei B, Jin J, Huang XR, Shao W et al (2019) Conditional knockout of TGF-βRII /Smad2 signals protects against acute renal injury by alleviating cell necroptosis, apoptosis and inflammation. Theranostics 9(26):8277–8293. https://doi.org/10.7150/thno.35686
Dai Q, Zhang Y, Liao X, Jiang Y, Lv X, Yuan X et al (2020) Fluorofenidone Alleviates Renal Fibrosis by Inhibiting Necroptosis Through RIPK3/MLKL Pathway. Front Pharmacol 11:534775. https://doi.org/10.3389/fphar.2020.534775
Xiao X, Du C, Yan Z, Shi Y, Duan H, Ren Y (2017) Inhibition of necroptosis attenuates kidney inflammation and interstitial fibrosis induced by unilateral ureteral obstruction. Am J Nephrol 46(2):131–138. https://doi.org/10.1159/000478746
Zhu Y, Cui H, Lv J, Li G, Li X, Ye F et al (2020) Angiotensin II triggers RIPK3-MLKL-mediated necroptosis by activating the Fas/FasL signaling pathway in renal tubular cells. PLoS ONE 15(3):e0228385. https://doi.org/10.1371/journal.pone.0228385
Zhu Y, Cui H, Xia Y, Gan HJPO (2016) RIPK3-mediated necroptosis and apoptosis contributes to renal tubular cell progressive loss and chronic kidney disease progression in rats. PLoS ONE 11(6):0156729
Yue LJ, Zhu XY, Li RS, Chang HJ, Gong B, Tian CC et al (2019) S-allyl-cysteine sulfoxide (alliin) alleviates myocardial infarction by modulating cardiomyocyte necroptosis and autophagy. Int J Mol Med 44(5):1943–51
Zhang Y, Gong W, Xu M, Zhang S, Shen J, Zhu M et al (2021) Necroptosis inhibition by hydrogen sulfide alleviated hypoxia-induced cardiac fibroblasts proliferation via sirtuin 3. Int J Mol Sci 22(21):11893
Xu Q, Tan X, Xian W, Geng J, Li H, Tang B et al (2021) Changes of necroptosis in irbesartan medicated cardioprotection in diabetic rats. Diabetes Metabolic Syndrome Obes 14:3851
Sharifi M, Nazarinia D, Ramezani F, Azizi Y, Naderi N, Aboutaleb NJMBR (2021) Necroptosis and RhoA/ROCK pathways: molecular targets of Nesfatin-1 in cardioprotection against myocardial ischemia/reperfusion injury in a rat model. Mol Biol Rep 48(3):2507–18
Qiao S, Hong L, Zhu Y, Zha J, Wang A, Qiu J et al (2022) RIPK1-RIPK3 mediates myocardial fibrosis in type 2 diabetes mellitus by impairing autophagic flux of cardiac fibroblasts. Cell Death Diseas 13(2):1–11
Wang L, Lv X, Tian J, Wang X, Wu Y, Liu HRJCT (2021) Cardioprotective effect of Nec-1 in rats subjected to MI/R: downregulation of autophagy-like cell death. Cardiovasc Therapeutics 45:1–8
Dmitriev YV, Karpov A, Dracheva A, Minasian S, Chefu S, Vasina L et al (2015) Cardioprotfetive effects of necrostatin-7 in the rat model of permanent coronary occlusion. Ross Fiziol Zh Im I M Sechenova 101(4):408–14
Fu Y, Jiang T, Sun H, Li T, Gao F, Fan B et al (2021) Necroptosis is required for atrial fibrillation and involved in aerobic exercise-conferred cardioprotection. J Cell Mol Med 25(17):8363–8375
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
All authors take public responsibility for the content of the work submitted for review. The authors confirm contribution to the paper as follows: study conception and design: EHMH, HSA; data collection: IMI, MSAE, MKA, EKA; draft manuscript preparation: IMI, MSAE, MKA preparation of final manuscript: EHMH, IMI, MSAE, MKAE, EKA, HSA. All authors reviewed and approved the final version of the manuscript. Authors declare that this manuscript is original, has not been published before and is not currently being considered for publication elsewhere.
Corresponding author
Ethics declarations
Conflict of interests
There are no conflicts of interest declared by the authors.
Ethical approval
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hassanein, E.H.M., Ibrahim, I.M., Abd El-Maksoud, M.S. et al. Targeting necroptosis in fibrosis. Mol Biol Rep 50, 10471–10484 (2023). https://doi.org/10.1007/s11033-023-08857-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08857-9