
Abstract
Background
Insufficient patient enrollment per month (=accrual) is the leading cause of cancer trial termination.
Objective
To identify and quantify factors associated with patient accrual in trials leading to the US Food and Drug Administration (FDA) approval of new cancer drugs.
Data
All anti-cancer drugs with FDA approval were identified in the Drugs@FDA database (2000–2022). Data on drug indication’s background-, treatment-, disease-, and trial-related factors were collected from FDA labels, clinicaltrials.gov, and the Global Burden of Disease study. The association between patient accrual and collected variables was assessed in Poisson regression models reporting adjusted rate ratios (aRR).
Results
We identified 170 drugs with approval in 455 cancer indications on the basis of 292 randomized and 163 single-arm trials. Among randomized trials, median enrollment per month was 38 patients (interquartile range [IQR]: 26–54) for non-orphan, 21 (IQR: 15–38, aRR 0.88, p = 0.361) for common orphan, 20 (IQR: 10–35, aRR 0.73, p <0.001) for rare orphan, and 8 (IQR 6–12, aRR 0.30, p < 0.001) for ultra-rare orphan indications. Patient enrollment was positively associated with disease burden [aRR: 1.0003 per disability-adjusted life year (DALY), p < 0.001), trial sites (aRR: 1.001 per site, p < 0.001), participating countries (aRR: 1.02 per country, p < 0.001), and phase 3 vs. 1/2 trials (aRR: 1.64, p = 0.037). Enrollment was negatively associated with advanced-line vs. first-line treatments (aRR: 0.81, p = 0.010) and monotherapy vs. combination treatments (aRR: 0.80, p = 0.007). Patient enrollment per month was similar between indications with and without a biomarker (median: 27 vs. 32, aRR 0.80, p = 0.117). Patient enrollment per month was substantially lower in government-sponsored than industry-sponsored trials (median: 14 vs. 32, aRR 0.80, p = 0.209). Enrollment was not associated with randomization ratios, crossover, and study blinding.
Conclusions
Disease incidence and disease burden alongside the number of study sites and participating countries are the main drivers of patient enrollment in clinical trials. For rare disease trials, greater financial incentives could help expedite patient enrollment. Novel trial design features, including skewed randomization, crossover, or open-label masking, did not entice patient enrollment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This study identifies factors associated with the speed of patient enrollment using data from 170 drugs with FDA approval in 455 cancer indications (2000–2022). |
Disease incidence, disease burden, the FDA orphan designation, the number of clinical trial sites, and the number of participating countries were the main drivers of fast patient accrual. |
Contemporary trial designs believed to entice patient enrollment, e.g., skewed randomization, crossover, or open-label masking, were not associated with faster patient enrollment. |
Greater financial incentives leading to more study sites and closer international collaboration could expedite patient enrollment for (ultra-)rare orphan drugs. |
1 Introduction
Clinical trials are the fundament of evidence-based medicine. Trials inform physicians which treatments improve patient outcomes. Although 70% of US citizens are inclined to participate in trials [1], merely 3–5% of cancer patients are enrolled [2]. As a result, 20–40% of trials close owing to insufficient patient enrollment [3, 4]. Insufficient patient enrollment is the leading cause of early termination in cancer trials [5, 6]. Therefore, there is great clinical and economic interest from academia, governments, and industry to increase trial participation and expedite patient enrollment.
Systematic reviews sought to identify barriers to clinical trial participation [7,8,9,10,11,12,13,14,15,16]. These studies found that eligibility criteria, industry sponsorship, number and location of study sites, underlying cancer diseases, disease burden, disease incidence, trial phase, and the number of enrolled patients may influence patient participation and clinical trial completion. On the basis of these findings, scholars conceptualized that patient enrollment is moderated by a unique mix of background, treatment-related, disease-related, trial design, and patient-related factors [7,8,9]. However, prior studies examining clinical trial enrollment and patient accrual are often limited in their scope of analyzed clinical trials, analyzed time horizon, examined variables, frequently only examine enrollment barriers at single study sites, and tend to focus on government-sponsored rather than industry-sponsored trials [17,18,19,20,21,22].
Furthermore, pharmaceutical companies frequently argue that certain clinical trial design features may entice patients to participate in clinical trials. It is believed that the option to crossover from the control to the treatment arm for nonresponders, active comparators, skewed randomization that increases the probability of being allocated to the treatment arm, and open-label blinding encourage patients to enroll in clinical trials [23]. However, the role of these clinical trial design features for patient accrual remains debated [9, 17, 18].
This is the largest cross-sectional study to identify and quantify background, treatment-related, disease-related, and trial design factors that are associated with patient accrual in clinical trials supporting the FDA approval of 170 drugs in 455 anti-cancer indications from 2000 to 2022.
2 Data and Methods
2.1 Sample Identification
All new drug applications and biologic license applications with FDA approval from 1 January 2000 to 1 January 2022 were identified in the Drugs@FDA database. We examined US drug approvals, given that most trials focus on the US and the FDA is typically the first regulatory agency to authorize new drugs [24]. Then, we limited the sample to anti-cancer treatments, excluding non-cancer drugs, supportive cancer agents, diagnostic medicines, and anti-emetics for cancer patients. For these anti-cancer drugs, we then screened the Drugs@FDA database to identify all their original and supplemental indications with FDA approval until 1 January 2022.
2.2 Data Collection
Two independent reviewers collected background-, treatment-, disease-, and trial-related variables from FDA labels, clinicaltrials.gov, associated clinical trial publications, and the Global Burden of Disease study for all identified cancer drugs and supporting clinical trials. The first reviewer (D.T.M.) obtained data from FDA labels, which the second reviewer (T.M.) then cross-checked and extended with data found on clinicaltrials.gov and associated peer-reviewed publications. Full details of the data extraction method have been described elsewhere [25,26,27].
Patient accrual rate The primary endpoint of this study was the patient accrual rate. The patient accrual rate was calculated by the quotient of the total number of recruited patients and the total enrollment period. The patient accrual rate measures the efficiency of patient recruitment [17, 18]. A high accrual rate indicates very efficient patient recruitment, and vice versa.
Background factors We classified the primary clinical trial sponsor (industry vs. government) and obtained the total number of clinical trial sites and the total number of participating countries to describe background-related factors.
Treatment-related factors Drug indications were characterized by their novelty/innovativeness (first-in-indication vs. advance-in-indication vs. addition-to-indication), mechanism of action (cytotoxic chemotherapy vs. targeted therapies vs. immune regulators), biomarkers status, line of therapy (first-line vs. advanced-line), and treatment type (combination therapy vs. monotherapy). Indication novelty was independently assessed by two medical doctors on the basis of the World Health Organization’s Anatomical Therapeutic Chemical code. Adapting Lanthier et al.’s classification of drug innovation and modifying it to indications [28], we considered drugs that were the first in a class to treat a new disease as “first-in-indication” drugs that were not the first in a class to treat a new disease but approved under FDA priority review as “advance-in-indication” and all others as “addition-to-indication”.
Disease-related factors We obtained disability-adjusted life years (DALYs) for the US population in 2019 from the Global Burden of Disease study to describe the burden of each cancer disease [29]. Furthermore, we accessed the FDA’s orphan drug database to link the orphan designation status to each indication. Given the existence of distinct orphan subgroups [26], we stratified orphan indications according to the number of affected US inhabitants into common (> 200,000), rare (200,000–6600), and ultra-rare (< 6600).
Trial design Clinical trials were differentiated by their design (randomized vs. single-arm), phase (phase 1/2 vs. phase 3), masking (open-label vs. double-blind), comparator (active vs. placebo/no treatment), randomization (equal vs. skewed), and crossover (not-specified vs. allowed vs. not allowed).
2.3 Statistical analysis
We used descriptive statistics to present the sample’s baseline characteristics. We conducted distinct analyses for randomized and single-arm trials given their unique characteristics in trial designs and high collinearity with other variables in the dataset. Frequencies were compared using Fisher’s exact test.
First, we conducted a univariable analysis to identify and quantify factors associated with patient accrual. For categorical variables, median accrual rates with interquartile ranges (IQR) were compared using Kruskal–Wallis tests. The association between interval-scaled variables and accrual rates was explored in a univariable regression analysis. The patient accrual rate was specified as the dependent variable.
Thereafter, we conducted a multivariable regression analysis. All collected variables were included in the analysis, except for those identified as collinear in a Pearson correlation matrix, e.g., disease type and orphan designation (Table e1). For the regression analyses, we used Poisson models to account for the left-skewed distribution of the patient accrual rate. For the Poisson regression analysis, we report adjusted rate ratios (aRR) with 95% confidence intervals (95% CI). All regression models account for molecule-clustered standard errors. Furthermore, we conducted an alternative regression model including the FDA approval type (standard approval vs. accelerated approval) as an independent variable.
Data were stored in Microsoft Excel (Microsoft Corp) and analyzed with Stata software, version 14.2 (StataCorp LLC). Two-tailed p-values below 0.05 were considered significant. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guidelines where applicable.
3 Results
3.1 Sample Overview
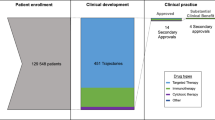
A total of 720 new drugs were granted FDA approval from 2000 to 2022. Of these, 170 drugs were approved for anticancer treatment with a total of 455 FDA-approved indications (Fig. 1).

New cancer drugs and clinical trials included in the analysis. CAR chimeric antigen receptor, FDA US Food and Drug Administration
Of these 455 indications, 292 (64%) were supported by randomized trials and 163 (36%) by single-arm trials. A median of 330 patients (IQR: 128–676) were enrolled per clinical trial with a median accrual rate of 18 patients per month (IQR: 7–36). The majority of trials were sponsored by the pharmaceutical industry (428, 94%). Trials were conducted at a median of 92 sites (IQR: 38–151) across 15 countries (IQR: 8–22). Industry-sponsored trials were conducted at significantly more sites (97 vs. 43, p = 0.034) and countries (16 vs. 2, p < 0.001) than government sponsored trials (Table e2). New drug indications were innovative with the majority being first-in-indication (176 [39%]) or addition-to-indication (211 [46%]). The mechanism of action was cytotoxic chemotherapy for 32 indications (7%), targeted for 273 indications (60%), and immune regulatory for 150 indications (33%). The FDA approved 167 indications (37%) on the basis of a biomarker. Indications were predominantly advanced-line (238 [52%]) monotherapies (298 [65%]) for solid cancers (301 [66%]). The median DALY rate per 100,000 US inhabitants was 117 (IQR: 31–428). The FDA orphan designation was granted to 294 indications, of which 64 (14%) were for common, 205 (45%) for rare, and 25 (5%) for ultra-rare diseases. Randomized and single-arm trials significantly differed in background, treatment-related, and disease-related variables (Table 1). Among the 292 randomized trials, 176 were open-label (60%), 187 (64%) compared the new drug with placebo or no treatment, and 197 (67%) trials used an equal randomization ratio. Crossover was not specified in 153 trials (52%), allowed in 68 trials (23%), and not permitted in 71 trials (24%).
3.2 Randomized trials
Among randomized trials, patient enrollment per month was significantly faster for industry- than government-sponsored trials (median: 32 vs. 14 patients per month, p < 0.001). In the univariable analysis, the patient accrual rate was significantly correlated with the total number of trial sites (Fig. 2a) and countries (Fig. 2b). Slower patient enrollment per month was observed for more innovative indications. Drug indications considered that first-in-indication enrolled fewer patients per month than those considered advance-in-indication and addition-to-indication (median: 22 vs. 33 vs. 32 patients per month, p =0 .005). Accrual was slower for monotherapies than combination treatments (median: 23 vs. 36, p < 0.001). Patient accrual was substantially slower for hematologic than solid cancer trials (median: 15 vs. 33, p < 0.001) and orphan than non-orphan cancer trials (median: 20 vs. 38, p < 0.001). Particularly fewer patients were enrolled in clinical trials for ultra-rare orphans relative to rare, common, and non-orphan cancers (median: 8 vs. 20 vs. 21 vs. 38 patients per month, p < 0.001). Accordingly, accrual rates were significantly associated with the burden of disease measured by DALYs (Fig. 2c). Slower patient enrollment was furthermore observed among phase 1/2 than phase 3 trials (median: 9 vs. 33, p < 0.001) and open-label than double-blind trials (median: 26 vs. 34, p = 0.024). No significant difference in the number of patients enrolled per month was observed for trials with an active comparator, skewed randomization, or trials that allowed cross-over to the new drug.

Association between patient enrollment per month and study sites, participating countries, and DALYs in cancer trials. a Illustrates the association between patient accrual rates and the total number of registered trial sites; b portrays the association between patient accrual rates and the total number of participating countries; c visualizes the association between patient accrual rates and DALYs (as a rate per 100,000 US inhabitants). The accrual rate, number of registered trial sites, number of participating countries, and DALYs are illustrated on the graphs y and x axis on a logarithmic scale. DALYs disability-adjusted life years, FDA US Food and Drug Administration
Results of the multivariable Poisson regression for randomized trials are presented in the second column of Table 2. Patient enrollment was slower, yet not significant at a 5% level owing to the high collinearity between the number of study sites and countries, for government- than industry-sponsored trials (aRR: 0.80, 95%CI 0.56–1.13, p = 0.209). The accrual rate was significantly associated with the number of trial sites (aRR: 1.001, 95%CI 1.001–1.002, p < 0.001) and the number of participating countries (aRR: 1.02, 95%CI 1.01–1.03, p < 0.001). Patient accrual was negatively associated with advanced-line relative to first-line treatment (aRR: 0.81, 95%CI 0.69–0.95, p = 0.010), monotherapy relative to combination treatments (aRR: 0.80, 95%CI 0.69–0.94, p = 0.007). Patient enrollment was substantially faster in phase 3 than in phase 1 or 2 trials (aRR: 1.64, 95%CI 1.03–2.62, p = 0.037). Patient enrollment was slower for ultra-rare (aRR: 0.30, 95%CI 0.24–0.38, p < 0.001), rare (aRR: 0.73, 95%CI 0.61–0.88, p < 0.001), and common (aRR: 0.88, 95%CI 0.68–1.15, p = 0.361) orphan than non-orphan cancers. Accrual was not associated with trial blinding, crossover, comparator, randomization ratio, mechanism of action, and biomarker status. The alternative regression model shows that accelerated approval was not significantly associated with the accrual rate (Table e3).
3.3 Single-arm trials
In the univariable analysis, faster patient enrollment was observed for industry- than government-sponsored trials (median: 6 vs. 3 patients per month, p = 0.031) (Table 3). However, this association was not significant after adjusting for further variables (aRR: 0.65, 95%CI 0.26–1.64, p = 0.366). In the multivariable model, the number of study sites was positively correlated to accrual rates (aRR: 1.007, 95%CI 1.003–1.012, p = 0.003). Patient enrollment per month was substantially slower for trials in the advanced-line than the first-line setting (aRR: 1.42, 95%CI 1.01–2.00, p = 0.042). Relative to cytotoxic chemotherapy, patient accrual was faster in trials for immune-regulating drugs (aRR 2.35, 95%CI 1.24–4.48, p = 0.009). Relative to non-orphan drugs, trials evaluating new ultra-rare orphan drugs were associated with slower patient accrual (aRR 0.58, 95%CI 0.38–0.90, p = 0.015). In the alternative regression models accelerated approval was not significantly associated with the accrual rate (Table e4).
4 Discussion
This cross-sectional study analyzed factors associated with clinical trial enrollment on the basis of a sample of 170 drugs with FDA approval across 455 cancer indications. We found that several background-, treatment-, and disease-related factors significantly influence the speed of patient enrollment (e.g., the patient accrual rate). Patient accrual is predominantly influenced by the number of participating study sites and countries and the underlying disease incidence and burden (orphan designation status), rather than clinical trial design features.
4.1 Background Factors
Consistent with previous literature [9, 10, 17, 20, 22], this study finds that patient enrollment per month was positively associated with the total number of study sites and participating countries as well as industry sponsorship, especially for randomized trials. These findings indicate that patient enrollment leading to successful trial completion is mainly driven by the financial funding and existing administrative structure to conduct these trials. Of note, industry-sponsorship was not significantly associated with patient accrual, likely owing to collinearity between industry-sponsorship with the number of study sites and participating countries. Pharmaceutical corporations that partner with clinical research organizations and academic and non-academic study sites will, of course, find more eligible patients in a shorter timeframe than government-sponsored clinical trial networks with limited funding that seek to enroll patients at a few study sites in a single country.
In this study, pivotal clinical trials in oncology were conducted at a median of 15 countries globally. However, prior research showed that trials predominantly enrolled patients from high-income countries with only few trials (29%) enrolling patients in low-income countries [30]. Increasing trial participation in low-income countries could expedite patient accrual, allow for faster patient access to new medicines, and test new drugs in a more diverse patient population [31].
4.2 Treatment-Related Factors
Results indicate that the line of therapy, therapy type, and the drug indication’s innovativeness were significantly associated with patient enrollment per month. Similar to prior studies [9, 17], faster patient enrollment was observed in trials for first-line than advanced-line treatments. There is simply a larger eligible patient population that can participate in first-line than advanced-line trials. Furthermore, very innovative drugs are often first tested as monotherapy in advanced-line treatments for common diseases and then “move up” the therapeutic ladder to first-line combination therapies if they continue to show clinical benefit relative to the standard of care [25]. Further, pharmaceutical companies were shown to be inclined to first test and seek approval for cancer drugs with multiple indications in rare diseases such that they receive the orphan designation’s benefits [32]. This commercialization strategy is also referred to as “orphan-first”[33]. These factors could explain the lower patient accrual rates observed for first-in-indication drugs.
4.3 Disease-Related Factors
Consistent with prior studies [9, 10, 17, 20, 22], our findings show that patient enrollment per month is influenced by disease incidence and burden. Patient accrual was particularly low for orphan drugs (median enrollment per month: 20 vs. 38 patients), which are defined as drugs for diseases with a prevalence below 200,000 US inhabitants, or drugs with limited sales potential. However, there are certain orphan subgroups with different disease dynamics [26]. Regarding these orphan subgroups, our results underline patient enrollment in clinical trials significantly differs. Results indicate that patient accrual is substantially more complex for ultra-rare diseases, defined by a prevalence threshold below 6600 US inhabitants equating to 1 in 50,000, than rare or non-orphan drugs (median enrollment per month: 8 vs. 20 vs. 38 patients ). Meanwhile, there was no significant difference between common orphan drugs that received the FDA orphan designation albeit their disease prevalence is beyond 200,000 US inhabitants, and non-orphan drugs (median enrollment per month: 21 vs. 38 patients, aRR 0.88, p = 0.361). These common orphan drugs are often targeted or immune therapies treating biomarker-defined subsets of common diseases [26].
Conducting clinical trials and developing drugs for rare diseases has emerged as “an economically viable strategy” owing to greater financial incentives, lower trial failure rates, and higher drug prices resulting in greater firm valuations and returns for pharmaceutical companies [34, 35]. Although a the share of newly developed orphan drugs has surged over the past decades, orphan drug development has concentrated on few diseases [36]. FDA-approved drugs exist for merely 5% of the more than 7000 rare diseases [36]. Further, clinical trials with new drugs are currently under development only for 15% of these more than 7000 rare diseases [36]. As a result many patients continue to suffer from rare diseases without treatment options—particularly those with ultra-rare diseases. The main challenge in conducting trials for ultra-rare diseases is finding competent investigators that recruit a sufficient number of patients at study sites with an adequate technological infrastructure. These challenges could be overcome by increasing funding for ultra-rare diseases, such that there are more financial resources available to pay clinical specialists that see patients with ultra-rare diseases regularly and provide the highly complex biotechnological infrastructure that is often necessary to administer new treatments for ultra-rare diseases (e.g., gene and cell therapies) across multiple countries. As previously highlighted, increasing the total number of participating study sites and countries will likely also increase patient accrual. Therefore, ultra-rare disease trials must be conducted as an international endeavor across nations where pharmaceutical companies closely collaborate with national healthcare providers and patient organizations in each region. Furthermore, decentralized clinical trials (DCTs) could help to enroll the very limited and geographically disperse patient population that defines these N-of-1 (ultra-rare) diseases [37]. With DCTs, patients are not bound to a single study site, which often demands lengthy travel, in a country to participate in clinical trials for their disease, but could effortlessly enroll in trials with the help of telehealth infrastructure, online consent forms, mail pharmacies, and electronic patient reported outcomes (ePRO) platforms.
4.4 Trial Design
Apart from the clinical trial phase, trial design features were not associated with faster patient accrual. These findings are consistent with previous studies [9, 17, 18]. A study of 747 trials supported by the National Clinical Trials Network found that trial blinding and comparator were not associated with patient accrual [9]. A study of 69 pivotal FDA approval trials for metastatic solid tumors (2006–2017) found no difference in patient accrual rates for trials with and without crossover [18]. Accordingly, a study of 194 pivotal FDA approval trials for solid and hematologic cancers could not find any association between patient accrual rates and crossover, trial blinding, primary endpoint, and randomization [17]. Although pharmaceutical companies and researchers believe that these clinical trial design features encourage patients to enroll in clinical trials [23], to date no evidence supports this optimistic belief. The use of crossover in clinical trials remains debated. “Although crossover is essential for studies that test the timing or sequence of therapies, […] crossover confounds interpretation of overall survival” [18]. For trials permitting crossover, pharmaceutical companies are frequently using crossover as the main reason why a surrogate endpoint, such as progression-free survival or tumor response, yet not the clinical endpoint, e.g., overall survival, showed a significant result [38]. Scholars increasingly argue in favor of clinical endpoints, e.g., overall survival and quality of life. These patient-centered outcomes are only rarely assessed and reported by cancer trials [39]. However, there is no evidence these endpoints are associated with faster patient enrollment [17], likely given that endpoints are only rarely discussed with and understood by patients [40]. In theory, a skewed randomization ratio favorably increases patients’ likelihood to be allocated to the treatment arm (e.g., 2:1, 3:2, 3:1) and should thereby entice enrollment. In practice, there is no evidence that skewed randomization ratios encourage patients to enroll in clinical trials. Perhaps the positive effect of the favorable randomization ratio on patient accrual is alleviated by the greater number of patients required to conduct trials with skewed randomization.
4.5 Limitations
There are certain limitations underlying our analysis. First, we only evaluated clinical trials that lead to the FDA approval of new cancer drugs. Thereby, our sample is biased to only successful trials with sufficient patient accrual. However, Jenei et al. showed that patient accrual results and their interpretation are consistent between successful FDA approval trials and other cancer trials [17]. Furthermore, with this uniquely large dataset of predominantly industry-sponsored trials, our study extends the existing body of research examining barriers to patient accrual in government-sponsored trials. Future research could examine factors associated with completed relative to withdrawn/terminated clinical trials. Second, our analysis focused on trial-level factors affecting patient accrual. However, previous studies highlighted that there are multiple patient-level factors, including socio-economic status, geographic location, age, sex, race, attending physician, and others, that influence patients’ decision to enroll in clinical trials [2, 7, 12,13,14, 16]. Finally, results and policy implications derived from our sample of cancer trials should be confirmed for other therapeutic areas.
5 Conclusions
Insufficient patient enrollment remains the leading cause of early trial termination. Prior studies highlighted patient and physician barriers to clinical trial participation. On the basis of a uniquely large dataset of 170 drugs with FDA approval in 455 cancer indications, we examined trial-level factors associated with patient enrollment per month. In this study, disease incidence and disease burden alongside the number of study sites and participating countries were the main drivers of patient enrollment. More financial incentives for rare disease trials and closer international collaborations could expedite drug development and encourage patient enrollment for (ultra-)orphan drugs. There is no evidence that trial designs that are commonly believed to entice patient enrollment, including skewed randomization, crossover, or open-label masking, result in faster accrual.
References
Comis RL, Miller JD, Aldigé CR, Krebs L, Stoval E. Public attitudes toward participation in cancer clinical trials. J Clin Oncol. 2003;21(5):830–5.
Murthy VH, Krumholz HM, Gross CP. Participation in cancer clinical trials: race-, sex-, and age-based disparities. J Am Med Assoc. 2004;291(22):2720–6.
American Cancer Society Cancer Action Network. Barriers to patient enrollment in therapeutic clinical trials to cancer—a landscape report [Internet]. 2018 [cited 2023 May 30]. Available from: https://www.fightcancer.org/sites/default/files/National%20Documents/Clinical-Trials-Landscape-Report.pdf. Accessed 30 Jul 2023.
Cheng SK, Dietrich MS, Dilts DM. A sense of urgency: evaluating the link between clinical trial development time and the accrual performance of cancer therapy evaluation program (NCI-CTEP) sponsored studies. Clin Cancer Res. 2010;16(22):5557–63.
Zhang E, DuBois SG. Early termination of oncology clinical trials in the United States. Cancer Med. 2023;12(5):5517–25.
Williams RJ, Tse T, DiPiazza K, Zarin DA. Terminated trials in the ClinicalTrialsgov results database: evaluation of availability of primary outcome data and reasons for termination. PLoS ONE. 2015;10(5):e0127242.
Walsh E, Sheridan A. Factors affecting patient participation in clinical trials in Ireland: a narrative review. Contemp Clin Trials Commun. 2016;3:23–31.
Unger JM, Cook E, Tai E, Bleyer A. The role of clinical trial participation in cancer research: barriers, evidence, and strategies. Am Soc Clin Oncol Educ Book. 2016;35:185–98.
Bennette CS, Ramsey SD, McDermott CL, Carlson JJ, Basu A, Veenstra DL. Predicting low accrual in the National Cancer Institute’s Cooperative Group clinical trials. J Natl Cancer Inst. 2016;108(2):djv324.
Hauck CL, Kelechi TJ, Cartmell KB, Mueller M. Trial-level factors affecting accrual and completion of oncology clinical trials: a systematic review. Contemp Clin Trials Commun. 2021;24: 100843.
You KH, Lwin Z, Ahern E, Wyld D, Roberts N. Factors that influence clinical trial participation by patients with cancer in Australia: a scoping review protocol. Br Med J Open. 2022;12(4): e057675.
Unger JM, Hershman DL, Till C, Minasian LM, Osarogiagbon RU, Fleury ME, et al. “When offered to participate”: a systematic review and meta-analysis of patient agreement to participate in cancer clinical trials. JNCI J Natl Cancer Inst. 2021;113(3):244–57.
Ford JG, Howerton MW, Lai GY, Gary TL, Bolen S, Gibbons MC, et al. Barriers to recruiting underrepresented populations to cancer clinical trials: a systematic review. Cancer. 2008;112(2):228–42.
Townsley CA, Selby R, Siu LL. Systematic review of barriers to the recruitment of older patients with cancer onto clinical trials. J Clin Oncol. 2005;23(13):3112–24.
Fayter D, McDaid C, Eastwood A. A systematic review highlights threats to validity in studies of barriers to cancer trial participation. J Clin Epidemiol. 2007;60(10):990.e1-990.e33.
Unger JM, Vaidya R, Hershman DL, Minasian LM, Fleury ME. Systematic review and meta-analysis of the magnitude of structural, clinical, and physician and patient barriers to cancer clinical trial participation. JNCI J Natl Cancer Inst. 2019;111(3):245–55.
Jenei K, Haslam A, Olivier T, Miljkovíc M, Prasad V. What drives cancer clinical trial accrual? An empirical analysis of studies leading to FDA authorisation (2015–2020). BMJ Open. 2022;12(10): e064458.
Chen EY, Prasad V. Crossover is not associated with faster trial accrual. Ann Oncol. 2018;29(3):776–7.
Khunger M, Rakshit S, Hernandez AV, Pasupuleti V, Glass K, Galsky MD, et al. Premature clinical trial discontinuation in the era of immune checkpoint inhibitors. Oncologist. 2018;23(12):1494–9.
Tate WR, Cranmer LD. Development and validation of a clinical trial accrual predictive regression model at a single NCI-designated comprehensive cancer center. J Natl Compr Canc Netw. 2016;14(5):561–9.
Schroen AT, Petroni GR, Wang H, Thielen MJ, Gray R, Benedetti J, et al. Achieving sufficient accrual to address the primary endpoint in phase III clinical trials from U.S. Cooperative Oncology Groups. Clin Cancer Res. 2012;18(1):256–62.
Carlisle B, Kimmelman J, Ramsay T, MacKinnon N. Unsuccessful trial accrual and human subjects protections: an empirical analysis of recently closed trials. Clin Trials Lond Engl. 2015;12(1):77–83.
Newington L, Metcalfe A. Factors influencing recruitment to research: qualitative study of the experiences and perceptions of research teams. BMC Med Res Methodol. 2014;14(1):10.
Lythgoe MP, Desai A, Gyawali B, Savage P, Krell J, Warner JL, et al. Cancer therapy approval timings, review speed, and publication of pivotal registration trials in the US and Europe, 2010–2019. JAMA Netw Open. 2022;5(6): e2216183.
Michaeli DT, Michaeli T. Overall Survival, Progression-free survival, and tumor response benefit supporting initial FDA approval and indication extension of new cancer drugs, 2003–2021. J Clin Oncol. 2022;40(35):4095–106.
Michaeli T, Jürges H, Michaeli DT. FDA approval, clinical trial evidence, efficacy, epidemiology, and price for non-orphan and ultra-rare, rare, and common orphan cancer drug indications: cross sectional analysis. Br Med J. 2023;281: e073242.
Michaeli DT, Michaeli T. Cancer drug prices in the United States: efficacy, innovation, clinical trial evidence, and epidemiology. Value Health. 2023;26(11):1590–600.
Lanthier M, Miller KL, Nardinelli C, Woodcock J. An improved approach to measuring drug innovation finds steady rates of first-in-class pharmaceuticals, 1987–2011. Health Aff (Millwood). 2013;32(8):1433–9.
Institute for Health Metrics and Evaluation (IHME). GBD Results [Internet]. Seattle, WA: IHME, University of Washington; 2020. Available from: https://vizhub.healthdata.org/gbd-results/. Accessed 30 Jul 2023.
Rubagumya F, Hopman WM, Gyawali B, Mukherji D, Hammad N, Pramesh CS, et al. Participation of lower and upper middle-income countries in clinical trials led by high-income countries. JAMA Netw Open. 2022;5(8): e2227252.
Wilson BE, Sullivan R, Peto R, Abubakar B, Booth C, Werutsky G, et al. Global cancer drug development—a report from the 2022 accelerating anticancer agent development and validation meeting. JCO Glob Oncol. 2023;9: e2300294.
Michaeli DT, Mills M, Michaeli T, Miracolo A, Kanavos P. Initial and supplementary indication approval of new targeted cancer drugs by the FDA, EMA, Health Canada, and TGA. Invest New Drugs. 2022;40:798–809.
Michaeli T, Michaeli DT. Partial orphan cancer drugs: US Food and Drug Administration approval, clinical benefit, trials, epidemiology, price, beneficiaries, and spending. Value Health. 2024;27(4):449–57.
Meekings KN, Williams CSM, Arrowsmith JE. Orphan drug development: an economically viable strategy for biopharma R&D. Drug Discov Today. 2012;17(13):660–4.
Michaeli DT, Yagmur HB, Achmadeev T, Michaeli T. Value drivers of development stage biopharma companies. Eur J Health Econ. 2022;23(8):1287–96.
Fermaglich LJ, Miller KL. A comprehensive study of the rare diseases and conditions targeted by orphan drug designations and approvals over the forty years of the Orphan Drug Act. Orphanet J Rare Dis. 2023;18(1):163.
Van Norman GA. Decentralized clinical trials. JACC Basic Transl Sci. 2021;6(4):384–7.
Prasad V. Double-crossed: why crossover in clinical trials may be distorting medical science. J Natl Compr Canc Netw. 2013;11(5):625–7.
Gloy V, Schmitt AM, Düblin P, Hirt J, Axfors C, Kuk H, et al. The evidence base of US Food and Drug Administration approvals of novel cancer therapies from 2000 to 2020. Int J Cancer. 2023;152(12):2474–84.
Boudewyns V, Southwell BG, DeFrank JT, Ferriola-Bruckenstein K, Halpern MT, O’Donoghue AC, et al. Patients’ understanding of oncology clinical endpoints: a literature review. Patient Educ Couns. 2020;103(9):1724–35.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. T.M. was supported by the DKFZ clinician scientist program. D.T.M. was supported by the Federal Ministry of Education and Research (BMBF) Young Scientist Award and the German Cancer Society’s (DKG) Young Talent Award.
Conflict of Interest Disclosures:
D.T.M., T.M., S.A., and J.C.M. declare that they have no conflicts of interest that might be relevant to the contents of this manuscript.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Participate
Not applicable.
Data Sharing Statement
All data used in this study were in the public domain. All data relevant to the study are included in the article or uploaded as supplementary information.
Code Availability
Not applicable.
Author Contributions
D.T.M. and T.M. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analyses. D.T.M. developed the study concept and design, drafted the manuscript, performed statistical analyses, and supervised the study. All authors carried out acquisition, analysis, or interpretation of data, critically revised the manuscript for important intellectual content, and provided administrative, technical, or material support.
Additional Contributions
The authors are grateful for comments and suggestions received by the editor of the journal and the anonymous referees. The authors thank Priv.-Doz. Dr. med. Tobias Boch, MD, Heidelberg University, for his medical advice. None of the additional contributors received any compensation.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Michaeli, D.T., Michaeli, T., Albers, S. et al. Patient Enrollment per Month (Accrual) in Clinical Trials Leading to the FDA Approval of New Cancer Drugs. Targ Oncol 19, 797–809 (2024). https://doi.org/10.1007/s11523-024-01081-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-024-01081-w