
Abstract
The metabolic syndrome describes a clustering of risk factors—visceral obesity, dyslipidaemia, insulin resistance, and salt-sensitive hypertension—that increases mortality related to cardiovascular disease, type 2 diabetes, cancer, and non-alcoholic fatty liver disease. The prevalence of these concurrent comorbidities is ~ 25–30% worldwide, and metabolic syndrome therefore presents a significant global public health burden. Evidence from clinical and preclinical studies indicates that glucocorticoid excess is a key causal feature of metabolic syndrome. This is not increased systemic in circulating cortisol, rather increased bioavailability of active glucocorticoids within tissues. This review examines the role of covert glucocorticoid excess on the hypertension of the metabolic syndrome. Here, the role of the 11β-hydroxysteroid dehydrogenase enzymes, which exert intracrine and paracrine control over glucocorticoid signalling, is examined. 11βHSD1 amplifies glucocorticoid action in cells and contributes to hypertension through direct and indirect effects on the kidney and vasculature. The deactivation of glucocorticoid by 11βHSD2 controls ligand access to glucocorticoid and mineralocorticoid receptors: loss of function promotes salt retention and hypertension. As for hypertension in general, high blood pressure in the metabolic syndrome reflects a complex interaction between multiple systems. The clear association between high dietary salt, glucocorticoid production, and metabolic disorders has major relevance for human health and warrants systematic evaluation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The metabolic syndrome describes a concurrence of interrelated abnormalities, including visceral obesity, dyslipidaemia, insulin resistance, and hypertension. Each of these features independently carries significant cardiovascular risk. In combination, the risk is amplified, and all-cause mortality increases: metabolic syndrome predicts the development of type 2 diabetes, cardiovascular disease, cancer, and non-alcoholic fatty liver disease [1]. Although a single, unifying definition of metabolic syndrome is lacking, the prevalence of these concurrent comorbidities is ~ 25–30% worldwide [2], presenting a significant global public health burden [3].
Metabolic syndrome is more useful as an epidemiologic tool for analysing cardiovascular risk than it is as a clinical entity requiring specialist management above and beyond management of individual components. For example, hypertension is one of the cardinal features of metabolic syndrome, but the origins of high blood pressure are obscure and lost in the complexity of the syndrome. Clearly metabolic syndrome captures a cluster of pathophysiological features that are individually accepted as “pro-hypertensive”: renal dysfunction and sodium retention [4], vascular [5] and microvascular dysfunction [6], activation of the renin-angiotensin-aldosterone system [7], sympathetic overdrive [8], and oxidative stress [9]. These have all been described in metabolic syndrome patients (and in animal models), as they have for uncomplicated hypertension. Indeed, as for uncomplicated hypertension, it is unlikely that any individual component is “causal”, and there is no distinct blood pressure management strategy for metabolic syndrome patients. Lifestyle and nutritional interventions to increase calorific outflow and lower salt intake are advocated, but adherence is poor, and blood pressure control requires early therapeutic intervention [10]. Nevertheless, there are interesting aspects to metabolic syndrome that may offer a route to improve blood pressure control. Glucocorticoids are important regulators of metabolism. Although rare, the systemic glucocorticoid excess of Cushing syndrome displays the same key features as metabolic syndrome [11]. Although circulating cortisol is not elevated in most patients with metabolic syndrome, “glucocorticoid excess” is a complex concept and may instead reflect instead amplification of cellular bioavailability [12•], enhance frequency/amplitude of pulsatile release over the 24-h cycle [13], and/or alter relationship of circadian/ultradian rhythms to external cues [14••].
This review focusses on covert glucocorticoid excess and the role of local glucocorticoid metabolism by the isozymes 11β hydroxysteroid dehydrogenase types 1 and 2 (11βHSD1 and 11βHSD2). 11βHSD1 and 11βHSD2 are products of distinct genes and members of the dehydrogenase/reductase superfamily. Here, the preclinical and clinical data connecting the activity of these enzymes to blood pressure homeostasis is discussed, concluding by addressing the potential therapeutic relevance to the management of patients with the metabolic syndrome.
11βHSDs and Glucocorticoid Signalling
Plasma concentrations of active glucocorticoid (cortisol in humans; corticosterone in rodents) are determined by the balance between synthesis and clearance, and by the high-affinity binding of glucocorticoids to circulating corticosteroid-binding globulin. Glucocorticoids are synthesised in the zona fasiculata of the adrenal cortex in response to ACTH, described as the hypothalamic-pituitary-adrenal axis (HPAA). In peripheral tissues, particularly adipose, liver, skeletal muscle, and kidney, glucocorticoids can be regenerated from inactive 11-keto derivatives (cortisone in humans; 11-dehydrocorticosterone in rodents) by 11βHSD1 (see [12•] for review). Systemic cortisol clearance is primarily mediated by hepatic 5α- and 5β-reductases, with a significant contribution from 11βHSD2 in the distal nephron of the kidney, which converts active glucocorticoids into inactive metabolites (Fig. 1).

Actions of the 11βHSD enzymes. The bioactivity of glucocorticoid is regulated by enzymatic modification of the C11 side chain. In humans, the reduced 11-hydroxy form cortisol (F) is physiologically active at the mineralocorticoid receptor; the oxidised 11-keto form cortisone (E) is inert. The same is true in rodents for active corticosterone (B) and inactive 11-dehydrocorticosterone (A). Interconversion between the oxidised and reduced forms is catalysed by two 11β-hydroxysteroid dehydrogenase (11βHSD) enzymes. 11βHSD1 operates as an NAPDH-dependent reductase, regenerating active glucocorticoids in target tissues. It is co-expressed in the endoplasmic reticulum with hexose-6-phosphate dehydrogenase (H6PDH), which generates NADPH requisite for reductase activity. 11βHSD2 is a high-affinity NAD+-dependent dehydrogenase, inactivating glucocorticoids in vivo. The changes in redox potential that accompany NAD+ metabolism may lock MR-cortisol complexes in an inactive state
The 11βHSD enzymes have conventionally been regarded as regulators of glucocorticoid function at a cell level, but they do exert endocrine influence. Thus, circulating concentration of glucocorticoid is not affected by deletion of 11βHSD2 [15], but the half-life of cortisol is increased in patients with null mutations in the encoding gene, HSD11B2. The effect of 11βHSD1 deletion is also nuanced, appearing as abnormal circadian control of HPAA drive [16]. Micro-dialysis studies suggest that 11βHSDs buffer tissue concentration of glucocorticoid, dissociating this from circulating levels throughout the circadian cycle [17]. Thus, tissue glucocorticoid signalling may be quasi-independent from systemic glucocorticoid status [18]. Mass Spectroscopy imaging is now allowing us to open the black box and peer into tissues: such approaches will significantly advance our understanding of the spatial-temporal regulation of glucocorticoid within tissues and perhaps ultimately cells [19].
Nevertheless, a solely “cellular” view lacks nuance: in humans, 11βHSD1 activity contributes to the postprandial rise in plasma cortisol [20] and in mice 11βHSD2 activity influences the relationship between dietary salt intake and circulating corticosterone [21]. Moreover, gene-targeting strategies in rodents and clinical studies in man clearly demonstrate that these cellular enzymes exert a significant impact on systemic phenotypes, including adiposity and hypertension, discussed below.
Hypertension in Systemic Glucocorticoid Excess
Iatrogenic or endogenous glucocorticoid excess induces hypertension in humans [22], recapitulated in mice models of chronic corticosterone [23•] and ACTH [24] infusion. Suppression of the endogenous diurnal variation causes a loss of nocturnal blood pressure dipping, even when glucocorticoid stays within the physiological range [25•]. The aetiology and treatment of hypertension in Cushing syndrome has been extensively reviewed [26]. Mechanistically, chronic (5-day) infusion of either ACTH or cortisol into healthy men causes antinatriuresis and volume expansion [27]. Studies in mice show activation of sodium reabsorption in the aldosterone-sensitive distal nephron via ENaC [24] and NCC, the thiazide-sensitive cotransporter [25•]. However, conditional deletion of GR in the distal nephron does not blunt the hypertensive response to chronic dexamethasone [28] (a synthetic glucocorticoid) and long-term glucocorticoid excess causes volume contraction rather than expansion. Here, hypertension is maintained by vasoconstriction due to enhanced sympathetic outflow and increased vasopressin [24]: mice with conditional deletion of GR in the vascular endothelium are partially protected against dexamethasone-hypertension [29]. Nevertheless, it is likely that blood pressure control by the kidney is impaired: the combination of glucocorticoid and sympathetic excess induces salt-sensitive hypertension in otherwise healthy rodents due to epigenetic modification of WNK4 kinase that regulates sodium transport in the distal tubule NCC [30••].
Stable hypertension in ACTH or glucocorticoids excess is often associated with electrolyte abnormalities (e.g. hypokalemia) suggestive of aldosterone excess and in mice ACTH induces increased renal transcription of aldosterone-response genes such as sgk1 and that encoding αENaC, scnn1 [31]. As expected, ACTH excess activates gene networks in the adrenal gland that promote steroidogenesis [32], but the effect on circulating aldosterone is transient; the glucocorticoid response is sustained. Thus, GR-mediated pathways are implicated in ACTH-dependent hypertension. MR pathways may come into play if glucocorticoids are sufficiently elevated to breach the 11βHSD2 barrier, as is suggested in human Cushing syndrome [33], or if precursors with mineralocorticoid activity, such as deoxycorticosterone, are elevated to cardiovascular significance [34]. In mice with ACTH excess, both GR and MR antagonism were required to normalise blood pressure [24], and in human ACTH-dependent Cushing, hypertension is often more responsive to mifepristone (RU486) than to MR antagonism [35]. GR antagonism also offers cardiovascular benefits independent of blood pressure control. In a novel rat model of metabolic syndrome, generated by intercross between Dahl-salt-sensitive and Zuker obese rats, RU486 reduced adiposity, 11βHSD1 expression in adipocytes and cardiomyocytes, and reduced cardiac damage without affecting hypertension [36].
11βHSD1 and Hypertension
11βHSD1 is highly expressed in the key metabolic tissues of liver, adipose, pancreas, and skeletal muscle. The role of 11βHSD1 in metabolism has been extensively studied from a cellular basis in individual tissues through to impact upon an integrated metabolic system [12•]. A consistent finding in obese humans and rodents is that 11βHSD1 activity in subcutaneous adipose more than doubles (e.g. [37, 38]). Increased adipose 11βHSD1 and consequent intracellular glucocorticoid amplification is similarly reported in patients with metabolic syndrome [39]. Transgenic approaches strongly evidence the relationship between adipose 11βHSD1 and metabolic disease: global knockout mice have a favourable metabolic phenotype, even when obese [40] and adipose-specific deletion protects mice against the metabolic consequences of circulating corticosterone excess [23•]. In contrast, transgenic overexpression of the enzyme in adipocytes markedly enhances cellular glucocorticoid, without changing circulating corticosterone levels, and induces a comprehensive metabolic syndrome phenotype [41]. Importantly, these overexpressing mice have the salt-sensitive hypertension and attenuation of the normal sleep-phase dip [42], characteristic of the blood pressure profile in human metabolic syndrome. In the mice, cellular amplification of corticosterone increased production of angiotensinogen by adipocytes, activating the systemic RAAS [42]. Blood pressure was normalised with an angiotensin receptor blocker, and in metabolic syndrome patients, ARBs offer a safe, effective, and well-tolerated means of blood pressure control [10], with added benefit for other aspects of the syndrome [7].
It is of course challenging to ascribe absolute causality of hypertension in a complex disorder, and several studies show that non-adipose 11βHSD1 activity contributes to blood pressure control. Human genetics studies associate the gain of function rs846910 SNP in the HSD11B1 promotor with blood pressure in non-obese people [43,44,45]. This SNP associates with type 2 diabetes but not with the metabolic syndrome [46], and such studies offer limited mechanistic insight. However, 11βHSD1 is expressed in systems with a strong influence on blood pressure homeostasis, including vascular smooth muscle cells. It is well-established that glucocorticoids enhance the vasoconstrictor response to catecholamines, yet global 11βHSD1 knockout did not reduce the contractile response to phenylephrine in either the mesenteric artery (resistance) or thoracic aorta [47]. Recent studies show that 11βHSD1 in perivascular fat, amplified in metabolic syndrome [48•], can influence vascular tone: sympathetic over activation increased 11βHSD1 activity and glucocorticoid amplification in perivascular fat, inducing induced endothelial dysfunction in underlying vessels by activation of MR [49••].
The kidney contributes to long-term blood pressure control through the pressure natriuresis, an integrated tubular-vascular response that stabilises extracellular fluid volume [50]. 11βHSD1 is expressed in the renal vasculature and in proximal and distal convoluted tubules, podocytes, macula densa cells, and the interstitial cells of the medulla [51]. Knockdown of 11βHSD1 activity in the rat renal medulla by targeted siRNA delivery decreased the concentration of corticosterone in the urine [52]. This indicates that 11βHSD1 operates as a reductase in vivo despite the absence of H6PDH expression here. Renal medullary upregulation of 11βHSD1 is critical to the hypertensive response to high salt diet in Dahl salt-sensitive rats, and knockdown by the local injection of siRNA is antihypertensive [52]. The molecular mechanisms connecting renal 11βHSD1 activity in the renal medulla to salt-sensitive blood pressure are not resolved, but it is noted that 11βHSD1 null mice are resistant to the hypertension induced by systemic infusion of corticosterone [23•]. It is plausible that 11βHSD1 regulates tubular sodium reabsorption by generating active glucocorticoid, since the stimulatory effects of moderate glucocorticoid excess are well-defined [53]. However, it is unlikely that the enzyme plays a major role in physiological salt balance, since 11βHSD1 knockout mice adapt perfectly well to dietary sodium restriction [54].
The mechanistic relationship between increased 11βHSD1 activity and disorders of metabolism provided a strong driver for development of pharmacological inhibitors. Preclinical studies showed that 11βHSD1 inhibitors lower systemic blood pressure in obese spontaneously hypertensive rats [55]. A blood pressure-lowering effect of a different inhibitor was also observed in mice [56], but this was an off-target benefit, since a similar antihypertensive action was observed in 11βHSD1 knockout mice. In small clinical trials, selective 11βHSD1 inhibitors caused a modest reduction in blood pressure as a secondary endpoint in patients with either type 2 diabetes or the metabolic syndrome [57•]. This was not statistically significant when assessed as a primary endpoint in obese patients [58].
11βHSD2 and Hypertension
Mineralocorticoid over-activity is often considered a major factor in the hypertension of glucocorticoid excess. Activation of MR by glucocorticoids is normally restricted by the presence in certain cells of 11βHSD2, which convert MR-active glucocorticoids to MR-inactive metabolites. 11βHSD2 is highly expressed in the kidney, defining the aldosterone-sensitive distal nephron, and here, the activity of the enzyme is certainly important for blood pressure control. Congenital or acquired deficiency in 11βHSD2 causes the syndrome of apparent mineralocorticoid excess (AME; OMIM #218030), presenting with salt retention, potassium wasting, and hypertension [51].
Renal 11βHSD2 activity is regulated by glucocorticoids. It is downregulated following adrenalectomy and restored by corticosterone replacement [59]. Such regulation of 11βHSD2 expression would defend against glucocorticoid-driven sodium retention during periods of physiological glucocorticoid excess. However, recent data from our lab indicate that high doses of dexamethasone actually reduce renal Hsd11b2 expression [51]. Studies in obese humans also find impaired renal 11βHSD2 activity [60] due to downregulation of gene expression [61]. Renal 11βHSD2 activity is the primary source of 11-dehydrocorticosterone, the substrate for 11βHSD1, and this supply role for peripheral glucocorticoid amplification is metabolically significant [62••]. Thus, metabolic disorders are caught between Scylla and Charybdis: reducing 11βHSD2 activity is metabolically beneficial but increases susceptibility to salt-sensitive hypertension, as demonstrated in the 11βHSD2 knockout rat [15] and discussed below.
In common with other Mendelian disorders, high blood pressure in AME is thought to originate in the kidney [63]. Renal transplant reverses AME in humans [64]; selective deletion of 11βHSD2 in the renal tubule induces key features of AME, including salt-sensitive hypertension [65••]. However, this view is too simplistic. In the global 11βHSD2 knockout mouse, increased vascular tone, reflecting either a defect in endothelial NO production [47] or enhanced sympathetic-induced vasoconstriction [66], maintains hypertension even when sodium balance is restored: activation of the MR target protein ENaC, increases vascular stiffness in obesity [67]. In the CNS, 11βHSD2 is expressed in a subset of neurons in the nucleus of the solitary tract. These neurons are activated by dietary salt restriction. Conditional deletion of 11βHSD2 in the nucleus of the solitary tract does not change blood pressure per se [68] but induces a powerful phenotypic switch from salt-resistant to salt-sensitivity BP so that even modest increases in dietary salt intake cause hypertension [69••]. The switch to salt sensitivity is amplified by an abnormal salt appetite: under free-choice regimens, CNS-knockout mice ingested ~ 3 times more salt than controls [69••].
Mechanistically, null mutations in HSD11B2, or inhibition of the enzyme by glycerrhetinic acid such as found in liquorice [70], would permit activation of MR by cortisol (or corticosterone in rodent models), causing sodium retention [71] due to enhanced reabsorption in the distal nephron via ENaC [66, 72]. Hypertension develops, because HPAA activity is not subject to negative feedback through volume/electrolyte status. However, it is likely that 11βHSD2 in the distal nephron does not act merely as a guardian of renal MR. For example, high salt diet caused moderate glucocorticoid excess in hsd11b2 heterozygote null mice, and salt-sensitive hypertension was prevented by GR blockade with RU486 rather than by MR blockade with spironolactone [21, 73]. The relationship between 11βHSD2, GR, and MR appears to be more complex than previously thought, at least in the kidney. Here, GR translocation to the cytoplasm is strongly influenced by aldosterone, rather than by physiological levels of corticosterone [74]. It may be that 11βHSD2 also determines the function of GR in “aldosterone-sensitive” cell types. Indeed, studies in a colonic cell line suggest that GR occupancy is a pre-requisite for aldosterone-MR signalling [75]. MR and GR share many of the post-receptor signalling pathways, and a molecular framework for corticosteroid regulation of distal nephron sodium transport—and the role of 11βHSD2 within this framework—is currently being elucidated in our laboratory and in others.
The physiological ramifications of MR/GR interaction are not clear, but it is likely that aldosterone and glucocorticoids normally have mutually reinforcing roles. In a collecting duct cell line, for example, aldosterone activation of MR controls sodium transport during circadian cycles [76]. If aldosterone rises, as seen during salt restriction, activation of GR by aldosterone maximises sodium transport via ENaC. Furthermore, ultradian fluctuations in circulating corticosterone, amplified by renal 11βHSD1 activity, mean that local glucocorticoid may periodically exceed the enzymatic capacity of 11βHSD2. Thus, at key times of the day, or after meals, sodium transport may be physiologically regulated by glucocorticoid. Clearly, this has implications for blood pressure regulation in the metabolic syndrome, where local glucocorticoid excess may underpin enhanced sodium reabsorption in the distal nephron. High-fat feeding to mice recapitulates key features of metabolic syndrome. Impaired sodium excretion and salt-sensitive hypertension reflect activation of furosemide-sensitive NKCC2 [77] and NCC [78], rather than ENaC [79].
Conclusions
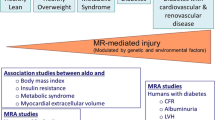
Conditions associated with increased circulating or intracellular glucocorticoids are common and often associated with hypertension. The metabolic syndrome exemplifies the complexity of glucocorticoid-dependent hypertension: clinical investigation and studies in experimental models demonstrate impairment of all the major homeostatic systems controlling blood pressure (Fig. 2). A unifying factor is that hypertension in the metabolic syndrome is commonly salt-sensitive. This presents a major challenge for clinical management, since salt intake is habitually high, and adherence to salt-restricted diets is notoriously poor [80•]. Moreover, high salt diet itself alters the dynamic regulation of the HPAA. In mice, this manifests as an amplified diurnal peak and enhanced 24-h excretion of corticosterone and metabolites, consistent with enhanced production [21]. In Dahl salt-sensitive rats, high salt diet does not enhance circulating corticosterone but does increase activity of 11βHSD1 in adipocytes [81]. In humans, a direct relationship between salt intake and glucocorticoid production is suggested, also involving peripheral metabolism [82]. Importantly, this relationship between salt intake and glucocorticoid production predicted metabolic syndrome status [83••]. This study was observational, and the association between dietary salt intake, cortisol production, and metabolic disease cannot be regarded as causal. Nevertheless, these relationships have implications for human health and disease and warrant systematic evaluation.

Mechanisms contributing to systemic arterial hypertension in the metabolic syndrome. Hypertension is salt-sensitive and reflects renal, vascular, and central mechanisms. The concept that hypertension is driven by sodium retention arising from renal mineralocorticoid actions is not the whole story, and glucocorticoid receptor blockade is likely to be beneficial. (ASDN aldosterone-sensitive distal nephron)
References
Papers of particular interest, published recently, have been highlighted as: •Of importance •• Of major importance
McCullough AJ. Epidemiology of the metabolic syndrome in the USA. J Dig Dis. 2011;12:333–40.
O'Neill S, O'Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16:1–12.
Sperling LS, Mechanick JI, Neeland IJ, Herrick CJ, Despres JP, Ndumele CE, et al. The cardiometabolic health alliance: working toward a new care model for the metabolic syndrome. J Am Coll Cardiol. 2015;66:1050–67.
Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116:991–1006.
Limberg JK, Morgan BJ, Sebranek JJ, Proctor LT, Walker BJ, Eldridge MW, et al. Altered neurovascular control of the resting circulation in human metabolic syndrome. J Physiol. 2012;590:6109–19.
Tune JD, Goodwill AG, Sassoon DJ, Mather KJ. Cardiovascular consequences of metabolic syndrome. Transl Res. 2017;183:57–70.
Yamada S. Pleiotropic effects of ARB in metabolic syndrome. Curr Vasc Pharmacol. 2011;9:158–61.
Grassi G, Seravalle G, Quarti-Trevano F, Scopelliti F, Dell'Oro R, Bolla G, et al. Excessive sympathetic activation in heart failure with obesity and metabolic syndrome: characteristics and mechanisms. Hypertension. 2007;49:535–41.
Youn JY, Siu KL, Lob HE, Itani H, Harrison DG, Cai H. Role of vascular oxidative stress in obesity and metabolic syndrome. Diabetes. 2014;63:2344–55.
Owen JG, Reisin E. Anti-hypertensive drug treatment of patients with and the metabolic syndrome and obesity: a review of evidence, meta-analysis, post hoc and guidelines publications. Curr Hypertens Rep. 2015;17:558.
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367:1605–17.
• Chapman K, Holmes M, Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93:1139–206. A comprehensive review of the 11BHSDs, ranging from basic biological concepts through to therapeutic implications.
Baudrand R, Campino C, Carvajal CA, Olivieri O, Guidi G, Faccini G, et al. Increased urinary glucocorticoid metabolites are associated with metabolic syndrome, hypoadiponectinemia, insulin resistance and beta cell dysfunction. Steroids. 2011;76:1575–81.
•• Henley DE, Lightman SL. Cardio-metabolic consequences of glucocorticoid replacement: relevance of ultradian signalling. Clin Endocrinol. 2014;80:621–8. Often overlooked, this review emphasizes that the physiology of glucocorticoid signalling is dynamic and abnormalities in rhythm will increase cardiovascular risk.
Mullins LJ, Kenyon CJ, Bailey MA, Conway BR, Diaz ME, Mullins JJ. Mineralocorticoid excess or glucocorticoid insufficiency: renal and metabolic phenotypes in a rat Hsd11b2 knockout model. Hypertension. 2015;66:667–73.
Harris HJ, Kotelevtsev Y, Mullins JJ, Seckl JR, Holmes MC. Intracellular regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase (11beta-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: analysis of 11beta-HSD-1-deficient mice. Endocrinology. 2001;142:114–20.
Usa K, Singh RJ, Netzel BC, Liu Y, Raff H, Liang M. Renal interstitial corticosterone and 11-dehydrocorticosterone in conscious rats. Am J Physiol Renal Physiol. 2007;293:F186–92.
Kilgour AH, Semple S, Marshall I, Andrews P, Andrew R, Walker BR. 11beta-Hydroxysteroid dehydrogenase activity in the brain does not contribute to systemic interconversion of cortisol and cortisone in healthy men. J Clin Endocrinol Metab. 2015;100:483–9.
Andrew R, Homer NZ. Mass spectrometry and its evolving role in assessing tissue specific steroid metabolism. Biochem Soc Trans. 2016;44:645–51.
Stimson RH, Mohd-Shukri NA, Bolton JL, Andrew R, Reynolds RM, Walker BR. The postprandial rise in plasma cortisol in men is mediated by macronutrient-specific stimulation of adrenal and extra-adrenal cortisol production. J Clin Endocrinol Metab. 2014;99:160–8.
Craigie E, Evans LC, Mullins JJ, Bailey MA. Failure to downregulate the epithelial sodium channel causes salt sensitivity in Hsd11b2 heterozygote mice. Hypertension. 2012;60:684–90.
Goodwin JE, Geller DS. Glucocorticoid-induced hypertension. Pediatr Nephrol. 2012;27:1059–66.
• Morgan SA, McCabe EL, Gathercole LL, Hassan-Smith ZK, Larner DP, Bujalska IJ, et al. 11beta-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci U S A. 2014;111:E2482–91. Interesting study that reveals the contribution of tissue regeneration of glucocorticoid to blood pressure homeostasis.
Bailey MA, Mullins JJ, Kenyon CJ. Mineralocorticoid and glucocorticoid receptors stimulate epithelial sodium channel activity in a mouse model of Cushing syndrome. Hypertension. 2009;54:890–6.
• Ivy JR, Oosthuyzen W, Peltz TS, Howarth AR, Hunter RW, Dhaun N, et al. Glucocorticoids induce nondipping blood pressure by activating the thiazide-sensitive cotransporter. Hypertension. 2016;67:1029–37. This study shows that flattening the diurnal glucocorticoid rhythm within the physiological range exerts significant effects of renal function and blood pressure.
Isidori AM, Graziadio C, Paragliola RM, Cozzolino A, Ambrogio AG, Colao A, et al. The hypertension of Cushing’s syndrome: controversies in the pathophysiology and focus on cardiovascular complications. J Hypertens. 2015;33:44–60.
Connell JM, Whitworth JA, Davies DL, Lever AF, Richards AM, Fraser R. Effects of ACTH and cortisol administration on blood pressure, electrolyte metabolism, atrial natriuretic peptide and renal function in normal man. J Hypertens. 1987;5:425–33.
Goodwin JE, Zhang J, Velazquez H, Geller DS. The glucocorticoid receptor in the distal nephron is not necessary for the development or maintenance of dexamethasone-induced hypertension. Biochem Biophys Res Commun. 2010;394:266–71.
Goodwin JE, Zhang J, Gonzalez D, Albinsson S, Geller DS. Knockout of the vascular endothelial glucocorticoid receptor abrogates dexamethasone-induced hypertension. J Hypertens. 2011;29:1347–56.
•• Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, et al. Epigenetic modulation of the renal beta-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17:573–80. The study shows, with mechanistic insight, the impact on blood pressure of concomitant HPAA and sympathetic activation. It has high clinical relevance.
Dunbar DR, Khaled H, Evans LC, Al-Dujaili EA, Mullins LJ, Mullins JJ, et al. Transcriptional and physiological responses to chronic ACTH treatment by the mouse kidney. Physiol Genomics. 2010;40:158–66.
Menzies RI, Zhao X, Mullins LJ, Mullins JJ, Cairns C, Wrobel N, et al. Transcription controls growth, cell kinetics and cholesterol supply to sustain ACTH responses. Endocr Connect. 2017;6:446–57.
Stewart PM, Walker BR, Holder G, O’Halloran D, Shackleton CH. 11 beta-Hydroxysteroid dehydrogenase activity in Cushing’s syndrome: explaining the mineralocorticoid excess state of the ectopic adrenocorticotropin syndrome. J Clin Endocrinol Metab. 1995;80:3617–20.
Mullins LJ, Peter A, Wrobel N, McNeilly JR, McNeilly AS, Al-Dujaili EA, et al. Cyp11b1 null mouse, a model of congenital adrenal hyperplasia. J Biol Chem. 2009;284:3925–34.
Nieman LK, Chrousos GP, Kellner C, Spitz IM, Nisula BC, Cutler GB, et al. Successful treatment of Cushing’s syndrome with the glucocorticoid antagonist RU 486. J Clin Endocrinol Metab. 1985;61:536–40.
Takeshita Y, Watanabe S, Hattori T, Nagasawa K, Matsuura N, Takahashi K, et al. Blockade of glucocorticoid receptors with RU486 attenuates cardiac damage and adipose tissue inflammation in a rat model of metabolic syndrome. Hypertens Res. 2015;38:741–50.
Livingstone DE, Jones GC, Smith K, Jamieson PM, Andrew R, Kenyon CJ, et al. Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology. 2000;141:560–3.
Rask E, Olsson T, Soderberg S, Andrew R, Livingstone DE, Johnson O, et al. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab. 2001;86:1418–21.
Rask E, Walker BR, Soderberg S, Livingstone DE, Eliasson M, Johnson O, et al. Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11beta-hydroxysteroid dehydrogenase type 1 activity. J Clin Endocrinol Metab. 2002;87:3330–6.
Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A. 1997;94:14924–9.
Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–70.
Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, et al. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90.
Franks PW, Knowler WC, Nair S, Koska J, Lee YH, Lindsay RS, et al. Interaction between an 11betaHSD1 gene variant and birth era modifies the risk of hypertension in Pima Indians. Hypertension. 2004;44:681–8.
He J, Gu D, Kelly TN, Hixson JE, Rao DC, Jaquish CE, et al. Genetic variants in the renin-angiotensin-aldosterone system and blood pressure responses to potassium intake. J Hypertens. 2011;29:1719–30.
Morales MA, Carvajal CA, Ortiz E, Mosso LM, Artigas RA, Owen GI, et al. Possible pathogenetic role of 11 beta-hydroxysteroid dehydrogenase type 1 (11betaHSD1) gene polymorphisms in arterial hypertension. Rev Med Chil. 2008;136:701–10.
Devang N, Satyamoorthy K, Rai PS, Nandini M, Rao S, Phani NM, et al. Association of HSD11B1 gene polymorphisms with type 2 diabetes and metabolic syndrome in South Indian population. Diabetes Res Clin Pract. 2017;131:142–8.
Hadoke PW, Christy C, Kotelevtsev YV, Williams BC, Kenyon CJ, Seckl JR, et al. Endothelial cell dysfunction in mice after transgenic knockout of type 2, but not type 1, 11beta-hydroxysteroid dehydrogenase. Circulation. 2001;104:2832–7.
• Siegel-Axel DI, Haring HU. Perivascular adipose tissue: an unique fat compartment relevant for the cardiometabolic syndrome. Rev Endocr Metab Disord. 2016;17:51–60. This recent review highlights the importance of perivascular fat in the cardiovascular complications of metabolic disorders.
•• Victorio JA, Clerici SP, Palacios R, Alonso MJ, Vassallo DV, Jaffe IZ, et al. Spironolactone prevents endothelial nitric oxide synthase uncoupling and vascular dysfunction induced by beta-adrenergic overstimulation: role of perivascular adipose tissue. Hypertension. 2016;68:726–35. This paper shows that corticosteroids are involved in intracrine signalling between adipocytes and the vasculature, which can contribute to vascular dysfunction.
Ivy JR, Bailey MA. Pressure natriuresis and the renal control of arterial blood pressure. J Physiol. 2014;592:3955–67.
Hunter RW, Bailey MA. Glucocorticoids and 11beta-hydroxysteroid dehydrogenases: mechanisms for hypertension. Curr Opin Pharmacol. 2015;21:105–14.
Liu Y, Singh RJ, Usa K, Netzel BC, Liang M. Renal medullary 11 beta-hydroxysteroid dehydrogenase type 1 in Dahl salt-sensitive hypertension. Physiol Genomics. 2008;36:52–8.
Hunter RW, Ivy JR, Bailey MA. Glucocorticoids and renal Na+ transport: implications for hypertension and salt sensitivity. J Physiol. 2014;592:1731–44.
Christensen TH, Bailey MA, Kenyon CJ, Jensen BL, Hunter RW. Sodium homeostasis is preserved in a global 11beta-hydroxysteroid dehydrogenase type 1 knockout mouse model. Exp Physiol. 2015;100:1362–78.
Schnackenberg CG, Costell MH, Krosky DJ, Cui J, Wu CW, Hong VS, et al. Chronic inhibition of 11 beta -hydroxysteroid dehydrogenase type 1 activity decreases hypertension, insulin resistance, and hypertriglyceridemia in metabolic syndrome. Biomed Res Int. 2013;2013:427640.
Bauman DR, Whitehead A, Contino LC, Cui J, Garcia-Calvo M, Gu X, et al. Evaluation of selective inhibitors of 11beta-HSD1 for the treatment of hypertension. Bioorg Med Chem Lett. 2013;23:3650–3.
• Feig PU, Shah S, Hermanowski-Vosatka A, Plotkin D, Springer MS, Donahue S, et al. Effects of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes Metab. 2011;13:498–504. This study shows modest reductions in blood pressure with 11HSD1 inhibitors, suggesting that they may be a useful additional antihypertensive strategy in metabolic disorders.
Shah S, Hermanowski-Vosatka A, Gibson K, Ruck RA, Jia G, Zhang J, et al. Efficacy and safety of the selective 11beta-HSD-1 inhibitors MK-0736 and MK-0916 in overweight and obese patients with hypertension. J Am Soc Hypertens. 2011;5:166–76.
Zallocchi ML, Matkovic L, Calvo JC, Damasco MC. Adrenal gland involvement in the regulation of renal 11beta-hydroxysteroid dehydrogenase 2. J Cell Biochem. 2004;92:591–602.
Wirix AJ, Finken MJ, Von Rosenstiel-Jadoul IA, Heijboer AC, Nauta J, Groothoff JW, Chinapaw MJ and Kist-Van Holthe JE. Is there an association between cortisol and hypertension in overweight or obese children?. J Clin Res Pediatr Endocrinol. 2017. https://doi.org/10.4274/jcrpe.4802.
Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Heintze U, Janke J, et al. Regulation of 11beta-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res. 2004;12:9–17.
•• Harno E, Cottrell EC, Keevil BG, DeSchoolmeester J, Bohlooly YM, Andersen H, et al. 11-Dehydrocorticosterone causes metabolic syndrome, which is prevented when 11beta-HSD1 is knocked out in livers of male mice. Endocrinology. 2013;154:3599–609. This study highlights the concept that covert glucocorticoid excess underlies the metabolic syndrome.
Mullins LJ, Bailey MA, Mullins JJ. Hypertension, kidney, and transgenics: a fresh perspective. Physiol Rev. 2006;86:709–46.
Razzaghy-Azar M, Yau M, Khattab A, New MI. Apparent mineralocorticoid excess and the long term treatment of genetic hypertension. J Steroid Biochem Mol Biol. 2017;165:145–50.
•• Ueda K, Nishimoto M, Hirohama D, Ayuzawa N, Kawarazaki W, Watanabe A, et al. Renal dysfunction induced by kidney-specific gene deletion of Hsd11b2 as a primary cause of salt-dependent hypertension. Hypertension. 2017;70:111–8. This study uses a conditional deletion strategy to support the concept that Apparent Mineralocorticoid Excess is a syndrome of renal dysfunction.
Bailey MA, Paterson JM, Hadoke PW, Wrobel N, Bellamy CO, Brownstein DG, et al. A switch in the mechanism of hypertension in the syndrome of apparent mineralocorticoid excess. J Am Soc Nephrol. 2008;19:47–58.
Martinez-Lemus LA, Aroor AR, Ramirez-Perez FI, Jia G, Habibi J, DeMarco VG, et al. Amiloride improves endothelial function and reduces vascular stiffness in female mice fed a western diet. Front Physiol. 2017;8:456.
Wyrwoll C, Keith M, Noble J, Stevenson PL, Bombail V, Crombie S, et al. Fetal brain 11beta-hydroxysteroid dehydrogenase type 2 selectively determines programming of adult depressive-like behaviors and cognitive function, but not anxiety behaviors in male mice. Psychoneuroendocrinology. 2015;59:59–70.
•• Evans LC, Ivy JR, Wyrwoll C, McNairn JA, Menzies RI, Christensen TH, et al. Conditional deletion of Hsd11b2 in the brain causes salt appetite and hypertension. Circulation. 2016;133:1360–70. This study shows that abnormal aldosterone signalling in the neucleus of the solitary tract is capable of inducing a switch from salt-resistsnce to salt-sensitive hypertension.
Flores-Robles BJ, Sandoval AR, Dardon JD and Blas CA. Lethal liquorice lollies (liquorice abuse causing pseudohyperaldosteronism). BMJ Case Rep 2013. https://doi.org/10.1136/bcr-2013-201007.
Stewart PM, Corrie JE, Shackleton CH, Edwards CR. Syndrome of apparent mineralocorticoid excess. A defect in the cortisol-cortisone shuttle. J Clin Invest. 1988;82:340–9.
Bailey MA, Unwin RJ, Shirley DG. In vivo inhibition of renal 11beta-hydroxysteroid dehydrogenase in the rat stimulates collecting duct sodium reabsorption. Clin Sci (Lond). 2001;101:195–8.
Bailey MA, Craigie E, Livingstone DEW, Kotelevtsev YV, Al-Dujaili EAS, Kenyon CJ, et al. Hsd11b2 haploinsufficiency in mice causes salt sensitivity of blood pressure. Hypertension. 2011;57:515–20.
Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, et al. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol. 2010;299:F1473–85.
Bergann T, Fromm A, Borden SA, Fromm M, Schulzke JD. Glucocorticoid receptor is indispensable for physiological responses to aldosterone in epithelial Na+ channel induction via the mineralocorticoid receptor in a human colonic cell line. Eur J Cell Biol. 2011;90:432–9.
Gaeggeler HP, Gonzalez-Rodriguez E, Jaeger NF, Loffing-Cueni D, Norregaard R, Loffing J, et al. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol. 2005;16:878–91.
Davies M, Fraser SA, Galic S, Choy SW, Katerelos M, Gleich K, et al. Novel mechanisms of Na+ retention in obesity: phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am J Physiol Renal Physiol. 2014;307:F96–F106.
Davies MR, Gleich K, Katerelos M, Lee M, Mount PF, Power DA. The thiazide-sensitive co-transporter promotes the development of sodium retention in mice with diet-induced obesity. Kidney Blood Press Res. 2015;40:509–19.
Nizar JM, Dong W, McClellan RB, Labarca M, Zhou Y, Wong J, et al. Na+-sensitive elevation in blood pressure is ENaC independent in diet-induced obesity and insulin resistance. Am J Physiol Renal Physiol. 2016;310:F812–20.
• Franco V, Oparil S. Salt sensitivity, a determinant of blood pressure, cardiovascular disease and survival. J Am Coll Nutr. 2006;25:247S–55S. Insightful and comprehensive review of the impact of high salt intake on cardiovascular risk.
Usukura M, Zhu A, Yoneda T, Karashima S, Yagi K, Yamagishi M, et al. Effects of a high-salt diet on adipocyte glucocorticoid receptor and 11-beta hydroxysteroid dehydrogenase 1 in salt-sensitive hypertensive rats. Steroids. 2009;74:978–82.
Lewicka S, Nowicki M, Vecsei P. Effect of sodium restriction on urinary excretion of cortisol and its metabolites in humans. Steroids. 1998;63:401–5.
•• Baudrand R, Campino C, Carvajal CA, Olivieri O, Guidi G, Faccini G, et al. High sodium intake is associated with increased glucocorticoid production, insulin resistance and metabolic syndrome. Clin Endocrinol. 2014;80:677–84. This study suggests that salt-induced production of cortisol is predicative of salt-sensitivity and metabolic syndrome.
Acknowledgments
Research in the author’s laboratory was funded by The British Heart Foundation and Kidney Research UK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Hypertension and Metabolic Syndrome
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bailey, M.A. 11β-Hydroxysteroid Dehydrogenases and Hypertension in the Metabolic Syndrome. Curr Hypertens Rep 19, 100 (2017). https://doi.org/10.1007/s11906-017-0797-z
Published:
DOI: https://doi.org/10.1007/s11906-017-0797-z