
Abstract
Aortic aneurysm and dissection (AAD) is a cardiovascular disease that poses a severe threat to life and has high morbidity and mortality rates. Clinical and animal-based studies have irrefutably shown that fluoroquinolones, a commonly prescribed antibiotic for treating infections, significantly increase the risk of AAD. Despite this, the precise mechanism by which fluoroquinolones cause AAD remains unclear. Therefore, this study aims to investigate the molecular mechanism and role of Ciprofloxacin definitively—a type of fluoroquinolone antibiotic—in the progression of AAD. Aortic transcriptome data were collected from GEO datasets to detect the genes and pathways expressed differently between healthy donors and AAD patients. Human primary Vascular Smooth Muscle Cells (VSMCs) were isolated from the aorta. After 72 h of exposure to 110ug/ml Ciprofloxacin or 100 nmol/L AngII, either or combined, the senescent cells were identified through SA-β-gal staining. MitoTracker staining was used to examine the morphology of mitochondria in each group. Cellular Reactive Oxygen Species (ROS) levels were measured using MitoSox and DCFH-DA staining. Western blot assay was performed to detect the protein expression level. We conducted an analysis of transcriptome data from both healthy donors and patients with AAD and found that there were significant changes in cellular senescence-related signaling pathways in the latter group. We then isolated and identified human primary VSMCs from healthy donors (control-VSMCs) and patients' (AAD-VSMCs) aortic tissue, respectively. We found that VSMCs from patients exhibited senescent phenotype as compared to control-VSMCs. The higher levels of p21 and p16 and elevated SA-β-gal activity demonstrated this. We also found that pretreatment with Ciprofloxacin promoted angiotensin-II-induced cellular senescence in control-VSMCs. This was evidenced by increased SA-β-gal activity, decreased cell proliferation, and elevation of p21 and p16 protein levels. Additionally, we found that Angiotensin-II (AngII) induced VSMC senescence by promoting ROS generation. We used DCFH-DA and mitoSOX staining to identify that Ciprofloxacin and AngII pretreatment further elevated ROS levels than the vehicle or alone group. Furthermore, JC-1 staining showed that mitochondrial membrane potential significantly declined in the Ciprofloxacin and AngII combination group compared to others. Compared to the other three groups, pretreatment of Ciprofloxacin plus AngII could further induce mitochondrial fission, demonstrated by mitoTracker staining and western blotting assay. Mechanistically, we found that Ciprofloxacin impaired the balance of mitochondrial fission and fusion dynamics in VSMCs by suppressing the phosphorylation of AMPK signaling. This caused mitochondrial dysfunction and ROS generation, thereby elevating AngII-induced cellular senescence. However, treatment with the AMPK activator partially alleviated those effects. Our data indicate that Ciprofloxacin may accelerate AngII-induced VSMC senescence through modulating AMPK/ROS signaling and, subsequently, hasten the progression of AAD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aortic aneurysm and dissection (AAD) is a dangerous medical condition that can have severe consequences. It occurs when the aorta's inner and middle layers separate or when the outer layer ruptures due to problems with the vascular cells and extracellular matrix. Risk factors for AAD include exposure to environmental factors, as well as genetic changes [1,2,3]. Recent studies have shown that fluoroquinolones (FQs), a type of antibiotic used to treat bacterial infections, may increase the risk of developing aortic aneurysm and dissection [4,5,6,7,8,9,10,11]. Ciprofloxacin, one of the most commonly prescribed FQs and the first approved for use in children, may accelerate the progression of this condition. However, the exact mechanism and molecular basis for this effect are still unclear.
Vascular smooth muscle cells (VSMCs) are the central cells in the aortic wall. They maintain the structure of the aortic wall, and when they are affected by environmental factors or mechanical stress, they can change from a contractile to a synthetic phenotype [12,13,14]. Our previous study showed that VSMC senescence contributes to the change in phenotype in abdominal aortic aneurysms [15,16,17]. Similarly, VSMC senescence has also been found in thoracic aortic aneurysm and dissection (TAAD) [18]. We aimed to investigate if ciprofloxacin-induced VSMC senescence increases the risk of AAD. Although the exact mechanism causing cellular senescence is not fully understood, it is known that high levels of reactive oxygen species (ROS) can cause it [19,20,21]. Furthermore, FQs like Ciprofloxacin contribute to bacterial death by causing chromosome fragmentation and ROS accumulation [22,23,24]. However, it is still unclear whether and how Ciprofloxacin influences the mechanism and function linking ROS generation, VSMC senescence, and the development of aortic dissection.
Mitochondria are a vital organelle that plays a critical role in cellular, physiological, and pathophysiological processes. Research has shown that mitochondrial dysfunction could be a significant factor in the development of aortic dissection and aneurysms [19, 25, 26]. The process of mitochondrial dynamics, which is mainly regulated by fission and fusion, has been found to be involved in various human diseases, such as cancer, neurologic, and cardiovascular diseases [27,28,29,30]. Studies have demonstrated that inhibiting mitochondrial fission by using Mdivi-1 can prevent the development of aortic aneurysm and dissection in mice [31,32,33]. There is also strong evidence that disturbing the balance of mitochondrial dynamics can cause an increase in the generation of reactive oxygen species (ROS) and cellular senescence [34,35,36]. It is currently unclear whether and how Ciprofloxacin, which has been shown to induce ROS production in bacteria, regulates mitochondrial dynamics. The AMPK pathway is known to regulate the process of mitochondrial dynamics [37,38,39], and it would be interesting to investigate whether Ciprofloxacin mediates VSMC senescence by suppressing AMPK signaling and thereby increasing susceptibility to aortic aneurysm and dissection.
Materials and Methods
Data Processing and Bioinformatics Analysis.
The gene expression profile of GSE98770 was obtained from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). This dataset includes five normal aorta samples from normal donors and six from patients with TAAD. For the specific data processing of GSE98770, Platform GPL14550 Agilent-028004 SurePrint G3 Human GE 8 × 60 K Microarray was used to probe ID conversion and annotation. The raw data were preprocessed by edgeR in R software. After background correction and data normalization, upregulated and downregulated DEGs were identified in GSE98770, and our validation set with p < 0.05 and |log fold change (FC)|> 1.5. A Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using a clusterprofiler package to elucidate the possible biological role of the selected gene. Benjamini-Hochberg's False Discovery Rate (FDR) was used to correct the results, and p < 0.05 was taken as the significant difference threshold, indicating a significant difference in the KEGG pathway. The GSEA software was downloaded from http://software.broadinstitute.org/gsea/index.jsp. The GSEA analysis was conducted with a cohort GSE98770. Normalized enrichment score (NES) is the primary statistic for examining gene set enrichment results, nominal p value estimates the statistical significance of the enrichment score, and a gene set with nominal p value < 0.05 was considered significantly enriched.
Isolation, Culture, and Characterization of VSMCs
Aortic tissue was collected from patients with aortic dissection who underwent surgical repair or healthy donors respectively. Written informed consent was obtained from all study patients. All procedures that involved human samples were approved by the research ethics board of Guangdong Provincial People's Hospital (No. GDREC2018060H). The procedure for isolation and culture VSMCs has been described in our previous study [15,16,17]. In brief, the medial part of aortic tissues was carefully dissected from the adventitia and intima, cut into 1‐2 mm3 pieces, then transferred those pieces into a culture flask(cell culture flask, angled neck; Corning), and incubated for adhesion at 37 °C for 1 h. After attaching to the plate, the medial pieces were gently cultured with Dulbecco's modified Eagle medium (DMEM; Sigma‐Aldrich) containing 10% fetal bovine serum (FBS; Gibco) and 100 µg/mL penicillin and streptomycin (P/S, Thermo Fisher Scientific). Cell cultures were maintained at 37 °C in a humidified 5% CO2 atmosphere. The medial pieces were left undisturbed to prevent detachment for 4 days, and the medium was refreshed approximately every 3 to 4 days. VSMCs migrated out from the medial pieces within 1‐2 weeks. After removing the medial pieces, VSMCs were regularly collected and passaged. All VSMCs in passages 3 − 5 were used in this study. In the current study, we collected three control‐VSMC cell lines from healthy donors and four AAD‐VSMC cell lines from AAD patients (General information of the donors and patients is shown in supplemental Table 1).
CCK8 Assay
Cell viability was detected using CCK-8 assay (CK04; DOJINDO) according to the protocol in this research. After relevant treatment, cells with a concentration of 4 × 103 cells/well were seeded into the 96-well plate and incubated for 24 h. After treatments, 10 μL of CCK-8 solution was added to the culture medium and further incubated for 2 h in the dark at 37 °C. Following that, the absorbance of each well was measured and recorded at 450 nm on a Micro-plate reader. Each experiments were performed in triplicate.
Detection of Intracellular ROS
Use two kinds of fluorescent probes to detect intracellular reactive oxygen species (ROS) production in VSMCs, namely DCFH-DA (D399; Invitrogen) and MitoSOX™ Red (M36008; Invitrogen). Control‐VSMCs were cultured in 6‐well plates and treated with Ciprofloxacin (HY-B0356; MCE) or 100 nM Ang II(HY-13948; MCE) or NAC (MCE, HY-B0215) or AICAR (MCE, HY-13417) alone or combined. For DCFH-DA staining, cells were incubated in the dark with 10 μM DCFH-DA for 15 min at 37 °C. Fluorescence was detected at 488 nm excitation and 525 nm emission. Five different view fields of each sample were photographed, and fluorescence intensity was calculated in three independent experiments using Image J software. For MitoSOX™ Red staining, cells were incubated with 5 μmol/L Mitosox at 37 °C for 15 min in the dark. The fluorescence intensity of control cells normalized the level of mitochondrial ROS.
Immunofluorescent Staining
For immunofluorescent staining, VSMCs were cultured on cover slides in 24‐well plates. After washing with PBS three times, they were fixed in 4% PFA for 15 min, followed by permeabilization with 0.1% Triton X‐100 in PBS for half an hour. 10% BSA blocked cells and then incubated with the following primary antibodies at 4 °C overnight: anti‐calponin (1:100, Abcam, ab46794), anti‐α‐SMA (1:100, Abcam, ab5694), anti‐Smoothelin (1:100, Abcam, ab8969), and anti‐ki67 (1:100, Abcam, ab15580). Next, cells were incubated in the dark with fluorescent-labeled secondary antibodies (1:1000) for 1 h at room temperature. Subsequently, VSMCs were washed with PBS three times and mounted with 4′, 6‐diamidino‐2‐phenylindole (DAPI). Finally, five randomly selected areas of each slide were photographed under a fluorescence microscope.
Senescence‐Associated β‐Galactosidase (SA‐β‐gal) Assay
Use the SA‐β‐gal assay kit to test VSMC senescence according to the manufacturer's protocol (Beyotime, C0602). Control‐VSMCs and AAD‐VSMCs were cultured on 6‐well plates. For intervention groups, control‐VSMCs were treated for 72 h with the vehicle, 110ug/ml Ciprofloxacin, AngII, NAC, and AICAR alone or combined. After washing three times with PBS, cells were fixed for 30 min, then stained with SA‐β‐gal solution at 4 °C overnight (without CO2). Finally, cells were washed with PBS and randomly photographed. The percentage of SA‐β‐gal positive cells was analyzed from five different view fields of each sample in three independent experiments. The percentage of SA‐β‐gal positive cells was calculated to estimate the percentage of senescent cells.
Mitotracker Staining
Use MitoTracker Green FM (Invitrogen, M7514) to examine the mitochondrial morphology of VSMCs. Control‐VSMCs were treated for 72 h with the vehicle, 110ug/ml Ciprofloxacin, AngII, NAC, and AICAR alone or combined. All groups were incubated in the dark for 30 min with DMEM supplemented with 20 nmol/L MitoTracker Green FM. After washing with PBS three times, it was photographed using a confocal microscope.
Western Blotting Assay
The total protein of treated VSMCs was extracted using RIPA (CST, 9806) with Protease/Phosphatase inhibitor (CST, 5872), and the concentration was measured using a bicinchoninic acid (BCA) assay kit (Thermo, 231,227). A 30 µg protein was resolved by 10% Tris‐glycine gel electrophoresis and then transferred onto a PVDF membrane. After blocking with 5% fat‐free milk in TBST, the PVDF membranes were incubated overnight at 4 °C with the following antibodies:anti‐calponin (Abcam, ab46794), anti‐α‐SMA (Abcam, ab5694), anti‐Smoothelin (Abcam, ab8969), anti-OPN (Santa Cruz, sc-21742), anti-p21 (Abcam, ab109199), anti-p16(Abcam, ab108349), anti-pAMPK (CST, 4184), and anti-AMPK (CST, 5832). Membranes were washed three times with TBST, incubated with secondary antibodies (1:3000, CST) at room temperature for 1 h, and then exposed in a dark room. The quantification of Western blotting in three independent experiments was analyzed using Image J software (National Institutes of Health).
Statistical Analysis
Analysis was performed using GraphPad Prism 8 Software. Tested normality of data distribution by using a Shapiro-Wilks test, and statistical significance was determined by independent-sample t test between two groups or analysis of variance (ANOVA) followed by the Bonferroni test between more than two groups. All values are expressed as mean ± SEM. p < 0.05 was considered statistically significant.
Results
VSMCs from Patients with Aortic Dissection Exhibit Cellular Senescence
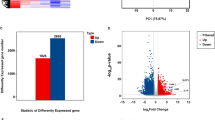
Our previous research has established that vascular smooth muscle cells (VSMCs) from patients with abdominal aortic aneurysm (AAA) or thoracic aortic dissection (TAD) caused by heredity show increased senescent phenotypes [15,16,17, 40]. In this study, we aimed to investigate the potential mechanisms during the process of AAD by analyzing the RNAseq dataset (GSE98770) from the Gene Expression Omnibus (GEO) database to identify differentially expressed genes (DEGs) between normal and patient samples. Our analysis revealed that the "vascular smooth muscle contraction" pathway was downregulated in AAD patients, while pathways related to cell senescence and mitochondrial dynamics were significantly upregulated, including "cellular senescence," "p53 pathway," and "mitochondrial biogenesis" (Fig. 1a-c). We then isolated VSMCs from control donors and AD patients by immunofluorescence staining and found that VSMCs from both groups expressed Calponin, α-SMA, and SM-22, which are widely recognized as contractile VSMC markers. However, all these markers were downregulated in AAD-VSMCs compared to control-VSMCs (Fig. 1d). To compare cellular senescence between control-VSMCs and AAD-VSMCs, we performed an SA-β-gal assay, which showed significantly elevated SA‐β‐gal activity in AAD-VSMCs (Fig. 1e). These results indicate that VSMCs isolated from AAD patients exhibited senescent phenotypes.

VSMCs from patients with aortic dissection exhibit cellular senescence. a Volcano map of GSE98770. b KEGG enrichment analysis of GSE98770. c GSEA enrichment analysis of GSE98770. d Immunofluorescence staining and quantitative analysis of Calponin, α‐SMA, and SM-22 in control-VSMCs and AAD-VSMCs. e Representative SA-β-gal staining images and quantitative analysis of control-VSMCs and AAD-VSMCs. All data were obtained from at least three independent experiments and each error bar represents the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Ciprofloxacin Accelerates AngII-Induced VSMC Senescence
Various clinical studies and animal experiments have indicated that fluoroquinolones (FQs) antibiotics increase the risk of aortic aneurysm disease (AAD) [4,5,6,7,8,9,10,11]. Ciprofloxacin, which belongs to the FQs antibiotics, has been proven to inhibit smooth muscle cell proliferation and aggravate apoptosis or inflammatory. The reduced proliferative capacity is one of the main biological indexes during cellular senescence [4, 41, 42]. This leads to the question of whether Ciprofloxacin accelerates the progression of aortic aneurysm and dissection by inducing vascular smooth muscle cell (VSMC) senescence.
To address this query, first we used human VSMC proteomics data after treated with CPFX which was published [41]. Our analysis revealed that significantly change proteins were enriched in pathways including “smooth muscle contraction,” “cellular response to reactive oxygen species,” and “cellular senescence”(Fig. 2a-b), which was similar to AAD patients. Then, we tested the effects of Ciprofloxacin in the range of 0–160 ug/ml on VSMC viability by cck8 assay. It was observed that Ciprofloxacin was most effective in inhibiting proliferation at a dose of 110ug/ml for 72 h. Moreover, when combined with AngII, it further decreased proliferation compared to the AngII alone group in Figure S1. Furthermore, compared to a blank group, pretreatment of Ciprofloxacin or AngII induced VSMC senescence, identified by SA-β-gal activity, while the combination group was more severe (Fig. 2c). Similar results were obtained from western blotting: Ciprofloxacin or AngII elevated the expression of p21 and p16, and the union group was higher than the single (Fig. 2d). We then used ki-67 staining to assess the proliferation ability of VSMCs in each group. Results showed that both Ciprofloxacin and AngII reduced the level of ki-67, and the combination of them was lower than treated alone (Fig. 2e). Additionally, in our previous studies, we observed that the progress of VSMC senescence was always accompanied by its phenotype transition, such as contractile status switching into synthetic [15,16,17, 40]. Thus, we detected the expression of contractile markers, including Calponin and α-SMA, and synthetic marker-OPN. Consistent with that, compared to vehicle control, alone or combination groups are all characterized by lower levels of contractile markers and higher levels of synthetic markers (Fig. 2f). These findings suggest that Ciprofloxacin induces cellular senescence in VSMCs and has a synergistic effect with AngII further to promote the senescent phenotype in the combination group.

Ciprofloxacin accelerates AngII-induced VSMCs senescence. a Volcano map of control and CPFX-treated VSMC. b Enrichment analysis significantly change proteins after CPFX treated in human VSMC. c Representative SA-β-gal staining images and quantitative analysis of vehicle, CPFX, AngII, or CPFX + AngII treated control-VSMCs. d Western blotting and quantitative analysis of p21 and p16 expression in vehicle, CPFX-VSMCs, AngII-VSMCs, and CPFX + AngII-VSMCs. e Representative images and quantitative analysis of Ki67 immunostaining in vehicle, CPFX-VSMCs, AngII-VSMCs, and CPFX + AngII-VSMCs. f Western blotting and quantitative analysis of Calponin, α‐SMA and OPN expression in vehicle, CPFX-VSMCs, AngII-VSMCs, and CPFX + AngII-VSMCs. All data were obtained from at least three independent experiments and each error bar represents the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Ciprofloxacin Induces Cellular Senescence of VSMCs via ROS Generation
Studies in the past few decades have shown that certain antibiotics, such as fluoroquinolones, work by increasing the level of reactive oxidative stress (ROS) in bacteria [22,23,24]. It has also been found that the increase in ROS generation can contribute to cellular senescence [19,20,21]. Therefore, we investigated the ROS generation in each group using MitoSOX and DHE staining. The results showed that all groups with Ciprofloxacin, AngII, and ciprofloxacin and AngII combinations had additive ROS generation, with the combination group having the highest (Fig. 3a-b). The researchers also used N-acetylcysteine (NAC), an inhibitor of ROS, to treat VSMCs with Ciprofloxacin and AngII and found that intervening with NAC reduced the ROS level induced by Ciprofloxacin and AngII (Fig. 3c). Additionally, the SA-β-gal assay demonstrated that NAC alleviated VSMC senescence caused by ciprofloxacin + AngII (Fig. 3d). Notably, ciprofloxacin + AngII combined with NAC lowered p21 expression (Fig. 3e). The above results suggest that Ciprofloxacin regulates AngII-induced VSMC senescence via ROS generation.

Ciprofloxacin induces cellular senescence of VSMCs via ROS generation. a Representative MitoSox staining images and quantitative fluorescence analysis of vehicle, CPFX, AngII, or CPFX + AngII treated control-VSMCs. b Representative DHE staining and quantitative fluorescence analysis of vehicle, CPFX, AngII, or CPFX + AngII treated control-VSMCs. c Representative MitoSox staining images and quantitative fluorescence analysis of vehicle, CPFX, AngII, or CPFX + AngII treated control-VSMCs. d Representative SA-β-gal staining images and quantitative analysis of vehicle, CPFX + AngII, NAC, and CPFX + AngII + NAC treated control-VSMCs. e Western blotting and quantitative analysis of p21 expression in vehicle, CPFX + AngII, NAC, and CPFX + AngII + NAC treated control-VSMCs. All data were obtained from at least three independent experiments and each error bar represents the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Ciprofloxacin Promotes ROS Generation by Impairing Mitochondrial Function
It has been widely reported that there is a close association between mitochondrial dysfunction, excessive ROS generation, and cellular senescence [27,28,29]. Therefore, we examined the mitochondrial function in four groups: vehicle control, ciprofloxacin-treated, AngII-treated, and a combination of Ciprofloxacin and AngII. To determine the ΔΨm in the mitochondria, we used the JC-1 signal, which displays the red color (aggregates, high potential) and the green color (monomers, low potential). We found a significant reduction in the red/green fluorescence ratio in all three treatment groups compared to the vehicle control (Fig. 4a). We also tested the mitochondria morphology in each group, which is an essential measure of normal mitochondrial function. Mitotracker staining revealed that mitochondrial length decreased among all three treatment groups, with the combination group exhibiting the shortest length (Fig. 4b). Western blotting showed that, compared to the vehicle control, the mitochondrial fission protein p-Drp-1 ser616 level increased in all three treatment groups. In contrast, the level of mitochondrial fusion proteins Mitofusin 1 (Mfn1) and Mitofusin 2 (Mfn2) significantly decreased, particularly in the Ciprofloxacin and AngII combination group (Fig. 4c). Furthermore, we tested mtROS level by MitoSOX staining and found that Mdivi-1, an inhibitor of mitochondrial fission, could significantly reduce ROS generation after being treated with Ciprofloxacin and AngII (Fig. 4d). These data indicate that Ciprofloxacin further promotes AngII-induced mitochondrial fission and its dysfunction, which is related to excessive ROS generation in VSMCs.

Ciprofloxacin promotes ROS generation by impairing mitochondrial function. a Representative JC-1 staining images in vehicle, AngII, CPFX, and CPFX + AngII treated control-VSMCs. b Representative MitoTracker staining images in vehicle, AngII, CPFX, and CPFX + AngII treated control-VSMCs. c Western blotting and quantitative analysis of Mfn1, Mfn2, p-Drp1, and total Drp1 in vehicle, AngII, CPFX, and CPFX + AngII treated control-VSMCs. d Representative histograms of flow cytometric analysis of MitoSOX fluorescence in vehicle, CPFX + AngII, CPFX + AngII + Mdivi-1 treated control-VSMCs. All data were obtained from at least three independent experiments and each error bar represents the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Ciprofloxacin Partially Mediates VSMC Senescence by AMPK Signaling
It is well known that the AMPK signaling pathway plays a crucial role in regulating the dynamics of mitochondria and the generation of reactive oxygen species (ROS) [37,38,39]. Through bioinformatics analysis, we found that the AMPK signaling pathway and cellular senescence process were significantly enriched in the gene sets of healthy donors and patients with AAD, respectively, as shown in Fig. 1b. This led us to investigate whether Ciprofloxacin accelerates the senescence of VSMCs induced by AngII by regulating the AMPK signaling pathway. To test this hypothesis, we assessed the levels of total and phosphorylated AMPK proteins in all groups using western blotting. Our results showed that both Ciprofloxacin and AngII could inhibit AMPK phosphorylation, which was further decreased when they were combined (Fig. 5a). In Fig. 5b, we observed that treating cells with AICAR partially alleviated SA-β-gal activity when compared to the group treated with both Ciprofloxacin and AngII, while AICAR alone did not show any significant changes compared to the vehicle group. Additionally, ROS generation was significantly reduced after treatment with AICAR (Fig. 5c). Moreover, treatment with AICAR combined with Ciprofloxacin and AngII restored AMPK phosphorylation and reduced p21 expression to a similar level as the vehicle group (Fig. 5d). Our findings suggest that Ciprofloxacin mediates ROS generation and cellular senescence in VSMCs by suppressing the AMPK signaling pathway.

Ciprofloxacin partially mediates VSMC senescence by AMPK signaling. a Western blotting and quantitative analysis of AMPK and p-AMPK in vehicle, AngII, CPFX, and CPFX + AngII treated control-VSMCs. b Representative SA-β-gal staining images and quantitative analysis of vehicle, AngII, CPFX + AngII, AICAR, CPFX + AngII + AICAR treated control-VSMCs. c Representative MitoSox staining images and quantitative analysis of vehicle, AngII, CPFX + AngII, AICAR, CPFX + AngII + AICAR treated control-VSMCs. d Western blotting and quantitative analysis of p-AMPK and p21 expression in vehicle, AngII, CPFX + AngII, AICAR, CPFX + AngII + AICAR treated control-VSMCs. All data were obtained from at least three independent experiments and each error bar represents the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
In this study, it was found that vascular smooth muscle cells (VSMCs) from patients with aortic dissection exhibited a senescent phenotype compared to those from healthy donors. Additionally, Ciprofloxacin was found to increase AngII-induced VSMC senescence in vitro significantly. However, activating the AMPK signaling pathway was found to partially alleviate cellular senescence caused by ciprofloxacin treatment by reducing ROS generation. The study suggests that administering Ciprofloxacin can interfere with AMPK/ROS signaling, leading to senescence in VSMCs. Furthermore, co-administration of AngII can expedite the senescence process, which may increase susceptibility to aortic dissection formation or rupture.
Aortic dissection is defined as a tear within the intimal layer of the aorta, causing separation of the intimal and medial layers of the aortic wall. The incidence of aortic dissection has been recorded to be 3 to 20 per 100,000 population annually. Acute aortic dissection or its rupture is associated with an overall high mortality rate of 24% in the first 24 h, 29% at 48 h, 44% at day 7, and 50% after 2 weeks [1,2,3]. The aorta can be affected by a broad spectrum of risk factors, including acute aggravated factors and chronic acquired and/or genetic conditions, which result in increased aortic wall stress and weakened aortic structure. Recent clinical or animal studies have indicated that the use of fluoroquinolones (FQs) can increase the risk of aortic dissection incidence [4,5,6,7,8,9,10,11]. However, the mechanism and molecular basis of this effect are yet to be elucidated.
According to current consensus, the weakening of the aortic wall in aortic aneurysm and dissection (AAD) is characterized by the loss of vascular smooth muscle cells (VSMCs) and degradation of extracellular matrix (ECM) [43, 44]. As the predominant cell type in the middle layer, VSMCs are known to demonstrate plasticity, which allows them to adapt to stress and maintain cellular phenotypic homeostasis [12,13,14]. However, VSMC senescence has been identified as a critical factor in the early stages of AAD, leading to imbalanced phenotypic switching. In AAD mouse models induced by β-aminopropionitrile, single-cell RNA sequencing has shown that cellular senescence and death-related pathways were significantly upregulated in VSMCs during the early stage of AAD [18]. Additionally, sustained ADP/P2ry12 signaling activation contributes to VSMC senescence, leading to AAD [45]. In an AngII-induced mouse model, MKL1 cooperates with p38MAPK to promote vascular senescence [46]. To determine whether AAD-VSMCs were senescent and underwent phenotypic switching, we performed SA-β-gal staining and western blot assay on primary VSMCs from AAD patients and healthy donors. Our results showed that the positive rate of SA-β-gal staining was significantly higher in the AAD group. Moreover, AAD-VSMCs expressed more p21, p16, and OPN, while the contraction protein level of α-SMA and Calponin was lower than that of control-VSMCs. These findings suggest that VSMCs from AAD patients exhibit a senescent phenotype. Furthermore, proteomic data from ciprofloxacin-treated VSMC profiled that the different expressed proteins were enriched in pathways including “cellular senescence” and “smooth muscle contraction”, which was similar to the changes between donors and AAD patients. Based on the data from bulk RNA-seq, proteomics, and our experimental evidence, we found ciprofloxacin-induced VSMC contractile dysfunction via supposed ciprofloxacin-induced VSMC dysfunction by elevating cellular senescence. Of note, AngII is one of the most prevailing drugs used to challenge the AAD mice model and its circulating level always increases in those patients with heart failure, hypertension, and atherosclerosis due to renin-angiotensin system dysregulation [47,48,49]. In our study, we first demonstrated that ciprofloxacin exhibited a synergistic effect with AngII which indicated that ciprofloxacin used in individuals with high circulating AngII may increase their susceptibility to AAD or other cardiovascular disease. This is consistent with a previous independent report showing that combining ciprofloxacin with oxy-LDL to treat monocytes raised the level of myristic acid and primed atherosclerosis [50].
Besides, in the past years, there was plenty of evidence verified that, beyond its anti-infection effect, ciprofloxacin treatment induced intercellular ROS generation and lead to DNA, protein, and lipid damage in various types of cells and resulted in organ dysfunction, including primary human aortic endothelial cells, primary human mammary epithelial cells, human gut epithelial cells, and human tenocytes. The half-maximal inhibitory concentration values for ciprofloxacin varied from 40 to 200 μg/ml, depending on the cell line sources or tissues [4, 42, 50]. Intriguingly, in recent years, ciprofloxacin has been extensively studied for its anticancer effects both in vitro and in vivo. It has been demonstrated that ciprofloxacin inhibits the cellular proliferation of melanoma cells B16F10, triple-negative breast cancer cells MDA-MB − 231, pancreatic cancer cells Panc − 1, non-small cell lung cancer cells A549, hepatocellular carcinoma cells HepG2, and glioblastoma A-172 cell, and similarly, the lethal concentration 50 (LC50) varies from 40 ng/ml to 200 μg/ml [51,52,53,54,55]. And in our study, we used CCK8 assay to test the effect of ciprofloxacin on human primary VSMC and found the IC50 value was 110ug/ml. Those data indicated that tissue or cells under disparate disease conditions showed tissue-specific or cell-specific changes after ciprofloxacin stimuli, and further, although ciprofloxacin may be used to inhibit the development of a wide range of tumors, it cannot specifically recognize tumor cells and normal tissue of which targets topoisomerases of all eukaryotes and carry out cellular damage. For instance, while higher concentration of ciprofloxacin effectively eliminates tumors, it might elevate the risk of AAD.
More specifically, as mitochondrial dynamics and function are closely related to cellular senescence [34,35,36], it has been found that the other second generation of fluoroquinolones, enrofloxacin, could induce ROS generation and promote mitochondrial fragmentation via regulating DRP1 in aquatic animals [56]. However, the relationship between ciprofloxacin and mitochondrial fragmentation remains unclear. Mitochondria are dynamic organelles that undergo fission and fusion. The fusion dynamics are mainly mediated by Mfn1/2 and optic atrophy protein 1 (OPA1), while the fission process, also known as fragmentation, is regulated by Drp1 and fission-1 (Fis1) [49]. Recent studies have highlighted that excessive fission is involved in AngII-induced aortic aneurysm and dissection(AAD), and several studies have confirmed that targeting mitochondrial fission is a potential therapeutic for AAD [27,28,29]. In this study, we performed the combined use of Ciprofloxacin and AngII worsened the decrease in mitochondrial membrane potential (MMP, ΔΨm) and the increase in ROS generation compared to each treatment alone. Our findings suggest that Ciprofloxacin and AngII work together to induce VSMC senescence further. It has been proven that AMPK signaling regulates mitochondrial dynamics [37,38,39], but it is unclear if Ciprofloxacin accelerates AngII-induced abnormal mitochondrial dynamics and VSMCs senescence via mediating the AMPK/ROS signaling pathway. We found that the AMPK signaling was inhibited in the AAD group compared to normal VSMCs. Additionally, we identified that ciprofloxacin treatment restrained the phosphorylation of AMPK, with a more severe effect in the combined treatment group than in the single treatment group, consistent with the above results. We also discovered that AICAR, an AMPK activator, partially rescued ciprofloxacin-induced mitochondrial fission and ROS generation, thus alleviating VSMC senescence in vitro.
Our study indicates that Ciprofloxacin can worsen AngII-triggered VSMC senescence by affecting the AMPK/ROS signaling pathway. This finding uncovers a new mechanism that explains the harmful effects of Ciprofloxacin on VSMCs. The study also suggests that individuals who have diseases with increased levels of circulating AngII should be cautious while using FQs as it may increase the risk of aortic dilation, dissection, and rupture.
Conclusion
Our study shows that Ciprofloxacin, which belongs to the fluoroquinolone antibiotics, can significantly increase the senescence of human primary vascular smooth muscle cells induced by Angiotensin II (AngII) in vitro. We also found that Ciprofloxacin accelerates AngII-induced senescence by affecting the AMPK/ROS pathway. These findings suggest that fluoroquinolones should not be recommended for people with high levels of circulating AngII. Additionally, targeting the AMPK signaling pathway may help reduce the risk of aortic aneurysm and dissection caused by fluoroquinolones-induced vascular smooth muscle cell senescence.
Data Availability
The data that support the findings of this study are openly available in[the Gene Expression Omnibus (GEO) database] at[GSE98770].
References
Bossone, E., LaBounty, T. M., & Eagle, K. A. (2018). Acute aortic syndromes: Diagnosis and management, an update. European Heart Journal, 39(9), 739–749d. https://doi.org/10.1093/eurheartj/ehx319
Zhu, Y., Lingala, B., Baiocchi, M., Tao, J. J., Toro, A. V., Khoo, J. W., Williams, K. M., Traboulsi, A. A., Hammond, H. C., Lee, A. M., Hiesinger, W., Boyd, J., Oyer, P. E., Stinson, E. B., Reitz, B. A., Mitchell, R. S., Miller, D. C., Fischbein, M. P., & Woo, Y. J. (2020). Type A aortic dissection-experience over 5 decades: JACC historical breakthroughs in perspective. Journal of the American College of Cardiology, 76(14), 1703–1713. https://doi.org/10.1016/j.jacc.2020.07.061
Yang, K., Ren, J., Li, X., Wang, Z., Xue, L., Cui, S., Sang, W., Xu, T., Zhang, J., Yu, J., Liu, Z., Shang, H., Pang, J., Huang, X., Chen, Y., & Xu, F. (2020). Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype. European Heart Journal, 41(26), 2442–2453. https://doi.org/10.1093/eurheartj/ehaa352
LeMaire, S. A., Zhang, L., Luo, W., Ren, P., Azares, A. R., Wang, Y., Zhang, C., Coselli, J. S., & Shen, Y. H. (2018). Effect of ciprofloxacin on susceptibility to aortic dissection and rupture in mice. JAMA Surgery, 153(9), e181804. https://doi.org/10.1001/jamasurg.2018.1804
Chen, S. W., Chan, Y. H., Chien-Chia, Wu. V., Cheng, Y. T., Chen, D. Y., Lin, C. P., Hung, K. C., Chang, S. H., Chu, P. H., & Chou, A. H. (2021). Effects of fluoroquinolones on outcomes of patients with aortic dissection or aneurysm. Journal of the American College of Cardiology, 77(15), 1875–1887. https://doi.org/10.1016/j.jacc.2021.02.047
Newton, E. R., Akerman, A. W., Strassle, P. D., & Kibbe, M. R. (2021). Association of fluoroquinolone use with short-term risk of development of aortic aneurysm. JAMA Surgery, 156(3), 264–272. https://doi.org/10.1001/jamasurg.2020.6165
Lai, C. C., Lu, C. T., Kao, K. C., Lu, M. C., Ko, W. C., & Hsueh, P. R. (2021). Association of fluoroquinolones use with the risk of aortic aneurysm or aortic dissection: Facts and myths. Journal of Microbiology, Immunology, and Infection, 54(2), 182–184. https://doi.org/10.1016/j.jmii.2021.03.002
LeMaire, S. A., Zhang, L., Zhang, N. S., Luo, W., Barrish, J. P., Zhang, Q., Coselli, J. S., & Shen, Y. H. (2022). Ciprofloxacin accelerates aortic enlargement and promotes dissection and rupture in Marfan mice. Journal of Thoracic and Cardiovascular Surgery, 163(3), e215–e226. https://doi.org/10.1016/j.jtcvs.2020.09.069
Inada, K., Koga, M., Yamada, A., Dohgu, S., & Yamauchi, A. (2022). Moxifloxacin induces aortic aneurysm and dissection by increasing osteopontin in mice. Biochemical and Biophysical Research Communication. https://doi.org/10.1016/j.bbrc.2022.08.080
Chen, Y. Y., Yang, S. F., Yeh, H. W., Yeh, Y. T., Huang, J. Y., Tsao, S. L., & Yeh, C. B. (2022). Association between aortic aneurysm and aortic dissection with fluoroquinolones use in patients with urinary tract infections: A population-based cohort study. Journal of the American Heart Association, 11(6), e023267. https://doi.org/10.1161/jaha.121.023267
Son, N., Choi, E., Chung, S. Y., Han, S. Y., & Kim, B. (2022). Risk of aortic aneurysm and aortic dissection with the use of fluoroquinolones in Korea: A nested case-control study. BMC Cardiovascular Disorders, 22(1), 44. https://doi.org/10.1186/s12872-022-02488-x
Petsophonsakul, P., Furmanik, M., Forsythe, R., Dweck, M., Schurink, G. W., Natour, E., Reutelingsperger, C., Jacobs, M., Mees, B., & Schurgers, L. (2019). Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arteriosclerosis, Thrombosis, and Vascular Biology, 39(7), 1351–1368. https://doi.org/10.1161/atvbaha.119.312787
Clément, M., Chappell, J., Raffort, J., Lareyre, F., Vandestienne, M., Taylor, A. L., Finigan, A., Harrison, J., Bennett, M. R., Bruneval, P., Taleb, S., Jørgensen, H. F., & Mallat, Z. (2019). Vascular smooth muscle cell plasticity and autophagy in dissecting aortic aneurysms. Arteriosclerosis, Thrombosis, and Vascular Biology, 39(6), 1149–1159. https://doi.org/10.1161/atvbaha.118.311727
Chen, P. Y., Qin, L., Li, G., Malagon-Lopez, J., Wang, Z., Bergaya, S., Gujja, S., Caulk, A. W., Murtada, S. I., Zhang, X., Zhuang, Z. W., Rao, D. A., Wang, G., Tobiasova, Z., Jiang, B., Montgomery, R. R., Sun, L., Sun, H., Fisher, E. A., … Simons, M. (2020). Smooth muscle cell reprogramming in aortic aneurysms. Cell Stem Cell, 26(4), 542-557.e11. https://doi.org/10.1016/j.stem.2020.02.013
Tao, W., Hong, Y., He, H., Han, Q., Mao, M., Hu, B., Zhang, H., Huang, X., You, W., Liang, X., Zhang, Y., & Li, X. (2021). MicroRNA-199a-5p aggravates angiotensin II-induced vascular smooth muscle cell senescence by targeting Sirtuin-1 in abdominal aortic aneurysm. Journal of Cellular and Molecular Medicine, 25(13), 6056–6069. https://doi.org/10.1111/jcmm.16485
Zhang, Y., Huang, X., Sun, T., Shi, L., Liu, B., Hong, Y., Fu, Q. L., Zhang, Y., & Li, X. (2023). MicroRNA-19b-3p dysfunction of mesenchymal stem cell-derived exosomes from patients with abdominal aortic aneurysm impairs therapeutic efficacy. Journal of Nanobiotechnology, 21(1), 135. https://doi.org/10.1186/s12951-023-01894-3
Ouyang, Y., Hong, Y., Mai, C., Yang, H., Wu, Z., Gao, X., Zeng, W., Deng, X., Liu, B., Zhang, Y., Fu, Q., Huang, X., Liu, J., & Li, X. (2024). Transcriptome analysis reveals therapeutic potential of NAMPT in protecting against abdominal aortic aneurysm in human and mouse. Bioactive Materials, 34, 17–36. https://doi.org/10.1016/j.bioactmat.2023.11.020
Liu, X., Chen, W., Zhu, G., Yang, H., Li, W., Luo, M., Shu, C., & Zhou, Z. (2022). Single-cell RNA sequencing identifies an Il1rn(+)/Trem1(+) macrophage subpopulation as a cellular target for mitigating the progression of thoracic aortic aneurysm and dissection. Cell Discovery, 8(1), 11. https://doi.org/10.1038/s41421-021-00362-2
Huang, X., Zhang, H., Liang, X., Hong, Y., Mao, M., Han, Q., He, H., Tao, W., Jiang, G., Zhang, Y., & Li, X. (2019). Adipose-derived mesenchymal stem cells isolated from patients with abdominal aortic aneurysm exhibit senescence phenomena. Oxidative Medicine and Cellular Longevity, 2019, 5049. https://doi.org/10.1155/2019/1305049
Lagnado, A., Leslie, J., Ruchaud-Sparagano, M. H., Victorelli, S., Hirsova, P., Ogrodnik, M., Collins, A. L., Vizioli, M. G., Habiballa, L., Saretzki, G., Evans, S. A., Salmonowicz, H., Hruby, A., Geh, D., Pavelko, K. D., Dolan, D., Reeves, H. L., Grellscheid, S., Wilson, C. H., … Passos, J. F. (2021). Neutrophils induce paracrine telomere dysfunction and senescence in ROS-dependent manner. Embo Journal, 40(9), 6048. https://doi.org/10.15252/embj.2020106048
Lin, J., & Epel, E. (2022). Stress and telomere shortening: Insights from cellular mechanisms. Ageing Research Review, 73, 101507. https://doi.org/10.1016/j.arr.2021.101507
Drlica, K., & Zhao, X. (2021). Bacterial death from treatment with fluoroquinolones and other lethal stressors. Expert Review of Anti-Infective Therapy, 19(5), 601–618. https://doi.org/10.1080/14787210.2021.1840353
Badawy, S., Yang, Y., Liu, Y., Marawan, M. A., Ares, I., Martinez, M. A., Martínez-Larrañaga, M. R., Wang, X., Anadón, A., & Martínez, M. (2021). Toxicity induced by ciprofloxacin and enrofloxacin: Oxidative stress and metabolism. Critical Reviews in Toxicology, 51(9), 754–787. https://doi.org/10.1080/10408444.2021.2024496
Salimiaghdam, N., Singh, L., Schneider, K., Chwa, M., Atilano, S. R., Nalbandian, A., Limb, G. A., & Kenney, M. C. (2022). Effects of fluoroquinolones and tetracyclines on mitochondria of human retinal MIO-M1 cells. Experiment Eye Research, 214, 108857. https://doi.org/10.1016/j.exer.2021.108857
Li, Y., Ren, P., Dawson, A., Vasquez, H. G., Ageedi, W., Zhang, C., Luo, W., Chen, R., Li, Y., Kim, S., Lu, H. S., Cassis, L. A., Coselli, J. S., Daugherty, A., Shen, Y. H., & LeMaire, S. A. (2020). Single-cell transcriptome analysis reveals dynamic cell populations and differential gene expression patterns in control and aneurysmal human aortic tissue. Circulation, 142(14), 1374–1388. https://doi.org/10.1161/circulationaha.120.046528
Zhong, X., Wu, Q., Wang, Z., Zhang, M., Zheng, S., Shi, F., Chen, Y., Che, Y., Yuan, S., & Xing, K. (2022). Iron deficiency exacerbates aortic medial degeneration by inducing excessive mitochondrial fission. Food & Function, 13(14), 7666–7683. https://doi.org/10.1039/d2fo01084d
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annual Review Pathology, 15, 235–259. https://doi.org/10.1146/annurev-pathmechdis-012419-032711
Longo, M., Meroni, M., Paolini, E., Macchi, C., & Dongiovanni, P. (2021). Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): New perspectives for a fairy-tale ending? Metabolism, 117, 154708. https://doi.org/10.1016/j.metabol.2021.154708
Jin, J. Y., Wei, X. X., Zhi, X. L., Wang, X. H., & Meng, D. (2021). Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacologica Sinica, 42(5), 655–664. https://doi.org/10.1038/s41401-020-00518-y
Kraus, F., Roy, K., Pucadyil, T. J., & Ryan, M. T. (2021). Function and regulation of the divisome for mitochondrial fission. Nature, 590(7844), 57–66. https://doi.org/10.1038/s41586-021-03214-x
Abudupataer, M., Zhu, S., Yan, S., Xu, K., Zhang, J., Luo, S., Ma, W., Alam, M. F., Tang, Y., Huang, H., Chen, N., Wang, L., Yan, G., Li, J., Lai, H., Wang, C., Zhu, K., & Zhang, W. (2021). Aorta smooth muscle-on-a-chip reveals impaired mitochondrial dynamics as a therapeutic target for aortic aneurysm in bicuspid aortic valve disease. Elife. https://doi.org/10.7554/eLife.69310
Cooper, H. A., Cicalese, S., Preston, K. J., Kawai, T., Okuno, K., Choi, E. T., Kasahara, S., Uchida, H. A., Otaka, N., Scalia, R., Rizzo, V., & Eguchi, S. (2021). Targeting mitochondrial fission as a potential therapeutic for abdominal aortic aneurysm. Cardiovascular Research, 117(3), 971–982. https://doi.org/10.1093/cvr/cvaa133
Ma, D., Zheng, B., Liu, H. L., Zhao, Y. B., Liu, X., Zhang, X. H., Li, Q., Shi, W. B., Suzuki, T., & Wen, J. K. (2020). Klf5 down-regulation induces vascular senescence through eIF5a depletion and mitochondrial fission. PLoS Biology, 18(8), e3000808. https://doi.org/10.1371/journal.pbio.3000808
Giacomello, M., Pyakurel, A., Glytsou, C., & Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nature Reviews Molecular Cell Biology, 21(4), 204–224. https://doi.org/10.1038/s41580-020-0210-7
Yu, B., Ma, J., Li, J., Wang, D., Wang, Z., & Wang, S. (2020). Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nature Communications, 11(1), 2549. https://doi.org/10.1038/s41467-020-16312-7
Yamamoto-Imoto, H., Minami, S., Shioda, T., Yamashita, Y., Sakai, S., Maeda, S., Yamamoto, T., Oki, S., Takashima, M., Yamamuro, T., Yanagawa, K., Edahiro, R., Iwatani, M., So, M., Tokumura, A., Abe, T., Imamura, R., Nonomura, N., Okada, Y., … Yoshimori, T. (2022). Age-associated decline of MondoA drives cellular senescence through impaired autophagy and mitochondrial homeostasis. Cell Reports, 38(9), 110444. https://doi.org/10.1016/j.celrep.2022.110444
Wang, Y., An, H., Liu, T., Qin, C., Sesaki, H., Guo, S., Radovick, S., Hussain, M., Maheshwari, A., Wondisford, F. E., O’Rourke, B., & He, L. (2019). Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Reports, 29(6), 1511-1523.e5. https://doi.org/10.1016/j.celrep.2019.09.070
Hu, Y., Chen, H., Zhang, L., Lin, X., Li, X., Zhuang, H., Fan, H., Meng, T., He, Z., Huang, H., Gong, Q., Zhu, D., Xu, Y., He, P., Li, L., & Feng, D. (2021). The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy, 17(5), 1142–1156. https://doi.org/10.1080/15548627.2020.1749490
Hsu, C. C., Zhang, X., Wang, G., Zhang, W., Cai, Z., Pan, B. S., Gu, H., Xu, C., Jin, G., Xu, X., Manne, R. K., Jin, Y., Yan, W., Shao, J., Chen, T., Lin, E., Ketkar, A., Eoff, R., Xu, Z. G., … Lin, H. K. (2021). Inositol serves as a natural inhibitor of mitochondrial fission by directly targeting AMPK. Molecular Cell, 81(18), 3803-3819.e7. https://doi.org/10.1016/j.molcel.2021.08.025
You, W., Hong, Y., He, H., Huang, X., Tao, W., Liang, X., Zhang, Y., & Li, X. (2019). TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging (Albany NY), 11(11), 3574–3584. https://doi.org/10.18632/aging.101998
Chen, Z., Wu, J., Wang, W., Tang, X., Zhou, L., Lv, Y., & Zheng, Y. (2023). Investigation of the pathogenic mechanism of ciprofloxacin in aortic aneurysm and dissection by an integrated proteomics and network pharmacology strategy. Journal of Clinical Medicine, 12(4), 1270. https://doi.org/10.3390/jcm12041270
Xiang, B., Abudupataer, M., Liu, G., Zhou, X., Liu, D., Zhu, S., Ming, Y., Yin, X., Yan, S., Sun, Y., Lai, H., Wang, C., Li, J., & Zhu, K. (2023). Ciprofloxacin exacerbates dysfunction of smooth muscle cells in a microphysiological model of thoracic aortic aneurysm. JCI Insight. https://doi.org/10.1172/jci.insight.161729
Lu, H., Du, W., Ren, L., Hamblin, M. H., Becker, R. C., Chen, Y. E., & Fan, Y. (2021). Vascular smooth muscle cells in aortic aneurysm: from genetics to mechanisms. Journal of the American Heart Association, 10(24), e023601. https://doi.org/10.1161/jaha.121.023601
Zhang, W. M., Liu, Y., Li, T. T., Piao, C. M., Liu, O., Liu, J. L., Qi, Y. F., Jia, L. X., & Du, J. (2016). Sustained activation of ADP/P2ry12 signaling induces SMC senescence contributing to thoracic aortic aneurysm/dissection. Journal of Molecular and Cellular Cardiology, 99, 76–86. https://doi.org/10.1016/j.yjmcc.2016.08.008
Gao, P., Gao, P., Zhao, J., Shan, S., Luo, W., Slivano, O. J., Zhang, W., Tabuchi, A., LeMaire, S. A., Maegdefessel, L., Shen, Y. H., Miano, J. M., Singer, H. A., & Long, X. (2021). MKL1 cooperates with p38MAPK to promote vascular senescence, inflammation, and abdominal aortic aneurysm. Redox Biology, 41, 101903. https://doi.org/10.1016/j.redox.2021.01903
STARSurg Collaborative, Drake, T. M., Cheung, L. K., Gaba, F., Glasbey, J., Griffiths, N., Helliwell, R. J., Huq, T., Khaw, R., Mayes, J., & Khan, S. (2018). Association between peri-operative angiotensin-converting enzyme inhibitors and angiotensin-2 receptor blockers and acute kidney injury in major elective non-cardiac surgery: a multicentre, prospective cohort study. Anaesthesia, 73(10), 1214–1222. https://doi.org/10.1111/anae.14349
Jiang, S., Chen, G., Yang, Z., Wang, D., Lu, Y., Zhu, L., & Wang, X. (2021). Testosterone attenuates hypoxia-induced hypertension by affecting NRF1-mediated transcriptional regulation of ET-1 and ACE. Hypertension Research, 44(11), 1395–1405. https://doi.org/10.1038/s41440-021-00703-4
Kukida, M., Sawada, H., Daugherty, A., & Lu, H. S. (2020). Megalin: A bridge connecting kidney, the renin-angiotensin system, and atherosclerosis. Pharmacological Research, 151, 104537. https://doi.org/10.1016/j.phrs.2019.104537
Gao, S., & Hu, J. (2021). Mitochondrial fusion: The machineries in and out. Trends in Cell Biology, 31(1), 62–74. https://doi.org/10.1016/j.tcb.2020.09.008
Binesh, A., Devaraj, S. N., & Halagowder, D. (2020). Monocytes treated with ciprofloxacin and oxyLDL express myristate, priming atherosclerosis. Journal of Biochemical and Molecular Toxicology, 34(3), e22442. https://doi.org/10.1002/jbt.22442
Yadav, V., Varshney, P., Sultana, S., Yadav, J., & Saini, N. (2015). Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC Cancer, 15, 581. https://doi.org/10.1186/s12885-015-1560-y
Beberok, A., Wrześniok, D., Rok, J., Rzepka, Z., Respondek, M., & Buszman, E. (2018). Ciprofloxacin triggers the apoptosis of human triple-negative breast cancer MDA-MB-231 cells via the p53/Bax/Bcl-2 signaling pathway. International Journal of Oncology, 52(5), 1727–1737. https://doi.org/10.3892/ijo.2018.4310
Jaber, D. F., Jallad, M. N., & Abdelnoor, A. M. (2017). The effect of ciprofloxacin on the growth of B16F10 melanoma cells. Journal of Cancer Research and Therapeutics, 13(6), 956–960. https://doi.org/10.4103/0973-1482.180610
Beberok, A., Wrześniok, D., Minecka, A., Rok, J., Delijewski, M., Rzepka, Z., Respondek, M., & Buszman, E. (2018). Ciprofloxacin-mediated induction of S-phase cell cycle arrest and apoptosis in COLO829 melanoma cells. Pharmacological Reports, 70(1), 6–13. https://doi.org/10.1016/j.pharep.2017.07.007
Suaifan, G., Mohammed, A. A. M., & Alkhawaja, B. A. (2022). Fluoroquinolones’ biological activities against laboratory microbes and cancer cell lines. Molecules. https://doi.org/10.3390/molecules27051658
Wei, X. L., Xu, Y. C., Tan, X. Y., Lv, W. H., Zhang, D. G., He, Y., & Luo, Z. (2023). Enrofloxacin (ENR) exposure induces lipotoxicity by promoting mitochondrial fragmentation via dephosphorylation of DRP1 at S627 site. Chemosphere, 340, 139892. https://doi.org/10.1016/j.chemosphere.2023.139892
Funding
The study work was supported by grants from Intergovernmental key projects of national key research and development program(2023YFE0114300 to Xin Li), National Natural Science Grant of China (No. 82072225, 82272246, 81270290, 8150056 and 81500567 to Xin Li), the Science and Technology Foundation of Guangzhou (No. 202206010044 to Xin Li), the High-level Hospital Construction Project of Guangdong Provincial People’s Hospital (No. DFJHBF202104 to Xin Li), NSFC Incubation Project of Guangdong Provincial People's Hospital (NO.KY0120220050 to Xiaoran Huang), and Guangdong Medical Science and Technology Research Fund Project (A2021067 to Weifeng Li).
Author information
Authors and Affiliations
Contributions
XL and XH designed the research. WZ, YL, SH, JZ, and CM performed experiments. YL and XH finished the transcriptome data analysis. BH and WL collected clinical samples. LS and BL analyzed and interpreted data. WZ, YL, SH, WL, XH, and XL discussed the data and drafted the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Ethical Approval
The entire or partial content of the paper has yet to be submitted or published elsewhere. The paper will not be submitted to another location until the journal editing department completes the program.
Additional information
Handling Editor: Daniel Conklin.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, W., Liang, Y., Huang, S. et al. Ciprofloxacin Accelerates Angiotensin-II-Induced Vascular Smooth Muscle Cells Senescence Through Modulating AMPK/ROS pathway in Aortic Aneurysm and Dissection. Cardiovasc Toxicol 24, 889–903 (2024). https://doi.org/10.1007/s12012-024-09892-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-024-09892-z