
Abstract
Arrhythmogenic cardiomyopathy (ACM) is a hereditary myocardial disease characterized by the replacement of the ventricular myocardium with fibrous fatty deposits. ACM is usually inherited in an autosomal dominant pattern with variable penetrance and expressivity, which is mainly related to ventricular tachyarrhythmia and sudden cardiac death (SCD). Importantly, significant progress has been made in determining the genetic background of ACM due to the development of new techniques for genetic analysis. The exact molecular pathomechanism of ACM, however, is not completely clear and the genotype–phenotype correlations have not been fully elucidated, which are useful to predict the prognosis and treatment of ACM patients. Different gene-targeted and transgenic animal models, human-induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) models, and heterologous expression systems have been developed. Here, this review aims to summarize preclinical ACM models and platforms promoting our understanding of the pathogenesis of ACM and assess their value in elucidating the ACM genotype–phenotype relationship.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Arrhythmogenic cardiomyopathy (ACM), also known as arrhythmogenic right ventricular dysplasia (ARVD) or arrhythmogenic right ventricular cardiomyopathy (ARVC), is an inherited cardiac disease, which is characterized by fibro-fatty replacement of the myocardium, progressive ventricular dysfunction, thrombus formation and an increasingly high risk of sudden cardiac death (SCD) [1,2,3,4]. Despite right ventricular dysfunction being the predominant clinical manifestation, it has been recently proved that left ventricular abnormalities are observed in patients with ACM to some extent and may even be the initial manifestation of the disorder [5,6,7,8,9,10]. Fontaine et al. described ACM in detail for the first time in 1982 [11]. The incidence rate between males and females is about 2:1, especially in young adults and in athletes, probably due to sex hormones, and other factors, such as competitive sports, meteorological factors, intracardiac thrombosis, atrial arrhythmias as well as brady-arrhythmias [2, 8, 12,13,14,15,16,17,18,19,20,21,22].
The definite diagnosis of ACM is established based on major and minor clinical, electrocardiography, and echocardiography and cardiac magnetic resonance (CMR), genetic studies, biopsy, and histology [23, 24] (Fig. 1). Pathogenic variants in ACM-associated genes are the main diagnostic criteria. In 2021, Gasperetti et al. reported that reduced left ventricular ejection fraction (LVEF), advanced atrioventricular block (AVB), prolonged PR interval, longer QRS duration, right ventricular apical involvement, and positive [18] F-FDG PET scan exist in cardiac sarcoidosis (CS) mimicking ACM, whereas larger right ventricular outflow tract (RVOT) dimensions, subtricuspid involvement and T-wave inversions (TWIs) help to diagnose hereditary ACM [25]. In some cases, low specific electrocardiographic abnormalities, difficulties in interpretation imaging to assess right ventricular structure and function, multiple reasons for right ventricular arrhythmias, and the puzzling genetic testing make the diagnosis challenging [1] (Fig. 2). Importantly, there is no curative treatment for this life-threatening disease [26].

Illustrative cardiac MRI images, transthoracic echocardiography, 12-lead ECG with epsilon waves, left-bundle branch block VT with superior axis and 3-D endocardial RV mapping of patients with definite ACM harboring pathogenic desmosomal variants. A. 12-lead ECG of an ACM patient presenting with epsilon waves in right precordial leads harboring a pathogenic DSP variant; B-D. Transthoracic echocardiography in different views presenting RV/RVOT dilatation and regional wall thinning in a patient with ACM harboring a pathogenic PKP2 variant; E. Cardiac MRI presenting fibrosis, dilatation and regional wall thinning of the RV in the same patient; F. Endocardial 3D electro anatomical voltage mapping of the RV showing the typical “C-scar” in the subtricuspid region extending towards the RVOT in a patient with ACM harboring a pathogenic PKP-2 variant being referred for catheter ablation of VT; G.12-lead ECG presenting typical precordial T wave inversions and a ventricular premature beat from the RV in a patient with ACM harboring a pathogenic PKP-2 variant; H. 12-leadECG presenting a sustained ventricular tachycardia with LBBB morphology and superior axis originating in the subtricuspid area of the RV in a patient with ACM harboring a pathogenic PKP2 variant

Diagnosis, pathogenesis, and management of ACM
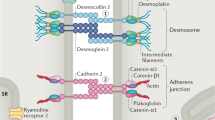
To date, more than 10 ACM-associated genes have been identified [27] (Table 1). Desmosomes in the heart contain five proteins, including junctional plakoglobin encoded by JUP, plakophilin-2 (PKP2) by PKP2 which is the most common genetic cause of ACM, desmoplakin by DSP, desmoglein-2 by DSG2, and desmocollin-2 by DSC2 [27,28,29,30,31,32]. There are also forms of autosomal recessive inheritance. Homozygous mutations in plakoglobin cause Naxos disease, which affects the heart and skin, and symptoms begin in childhood. Other forms of autosomal recessive inheritance are Carvajal syndrome caused by mutations in DSP, which is characterized by woolly hair, palmoplantar keratosis, and heart disease [1], and the homozygous p.Gln554X variant in DSC2, which is prevalent in around 10% of the Hutterite population and causes severe ACM.
In addition to mutations in desmosomal genes, an increasing number of non-desmosomal genes, such as desmin (DES), titin (TTN), lamin A/C (LMNA), Phospholamban (PLN), Transmembrane Protein 43 (TMEM43), and the sodium channel Nav1.5 (SCN5A) have also been reported as causative genes for ACM by using candidate gene sequence methods and linkage analysis, as inherited mutations identified in humans with cardiomyopathy are now modeled in transgenic or knock-in animals [46, 47]. In addition, at least 30–50% of ACM cases are also facilitated by non-genetic factors such as inflammation or exercise (Fig. 2) [48,49,50,51]. However, James et al. reported that only 8 genes including PKP2, DSP, DSG2, DSC2, JUP, TMEM43, PLN, and DES, had definitive or moderate evidence for ACM using the Clinical Genome Resource approach to gene-disease curation [52]. Therefore, understanding its genetic architecture and molecular mechanism, which will need to be fully elucidated by larger studies, may be helpful to improve the treatment of arrhythmias and prevent sudden cardiac death in ACM.
Unfortunately, the exact pathophysiological mechanisms and treatment strategies of ACM are still unclear. Preclinical models such as animal models, heterologous expression systems, and human cardiomyocytes derived from induced pluripotent stem cells (hiPSC-CMs) have been used to model the disease and pave the way to clarify the genotype–phenotype associations and the underlying mechanisms. This review summarizes the role of preclinical models in elucidating the pathophysiological mechanism of ACM, and the association between genotype and phenotype to provide evidence-based personalized management.
Experimental Models of ACM
Presently, roughly half of ACM patients with the clinical phenotype have at least one identifiable genetic variant. This raises the possibility that genetic data could be used to predict disease severity [53]. Over the last 10 years, to gain a clear and in-depth understanding of the pathological mechanisms of ACM, researchers established diverse models in vivo and vitro to investigate the pathogenesis and clinical features of ACM development.
Animal Models
To date, transgenic overexpression of genes and targeted genetic deletion animal models have been established to provide compelling evidence for elucidating the mechanisms of ACM development, which contributed to a better understanding of the pathophysiological processes. Furthermore, due to the developing genetic animal models harboring a clear genetic background, they are very suitable for serving as important tools to help uncover mechanisms that commonly and specifically underlie these disease phenotypes under controlled and standardized conditions.
Spontaneous Animal Models
For many years, people have noticed that boxer dog breeds are susceptible to right ventricular structural abnormalities, fibrofatty replacement, myocarditis, apoptosis, ventricular arrhythmia, and SCD, which means this model constitutes a new and potentially useful research tool that can be used to understand the complex clinical and pathogenic mechanisms of ACM disease, but the underlying diseases that lead to these clinical features have not been fully determined, so a series of related studies have been conducted on dog models. Furthermore, Boxers with ACM had a significant increase in serum cTnI concentration, suggesting that cTnI may be an indicator of stage or severity of ACM [54]. Also, Philip and coworkers identified clinically relevant cardiomyopathy in the common domestic cat having clinical features like right-sided congestive heart failure, supraventricular tachyarrhythmias, ventricular tachycardia, polymorphic ventricular arrhythmias, and right bundle-branch block, closely resembling human ACM [55,56,57]. The clinical features of ACM in dog and cat models can be observed, but they are all spontaneous models, which have certain limitations in studying the pathophysiological mechanism of ACM. Transgenic animals can be used to study the role of specific genes associated with ACM in the pathophysiology of ACM. Therefore, the construction of a transgenic mouse model that mimics the pathological characteristics of ACM through transplantation or gene-editing technology helped to further clarify the research on the pathogenesis of ACM.
Desmosomal Variants in Rodent Models
The cardiac desmosomal cell–cell junction protein complex is made up of five classic proteins. Genetic mutations in the desmosome genes that cause the destabilization/breakdown of the desmosome proteome are the core hallmarks of all genetic-based desmosome-targeted diseases including ACM [58,59,60,61]. Desmosomes are indirectly related to the electrochemical coupling of cardiomyocytes and signal transduction [62, 63]. Currently, the electrophysiological and pathological mechanisms caused by desmosomal protein defects are complex. In addition, electrophysiological abnormalities occur prior to major structural changes such as fibrosis in both the human disease and a murine model as a result of mislocalization and a reduction in expression of connexin-43 (Cx-43), leading to a slowed conduction [64, 65]. Different transgenic, knock-in, and conditional cardiac-specific mice models for the desmosomal genes have been developed to investigate the specific pathophysiological mechanism of ACM (Table 2).
PKP2
One of the most commonly disrupted genes in ACM is PKP2 [1, 115]. Worth noting, even in a structurally normal heart, variants in human PKP2 can cause life-threatening arrhythmias. Surprisingly, Moncayo-Arlandi et al. observed structural but not electrophysiological abnormalities in trained and old PKP2-truncated mice [72]. Of note, the reduced PKP2 expression reduced Na current peak density and also altered channel kinetics as well as significantly reduced conduction velocity (CV) in cultured rat neonatal cardiomyocytes, suggesting that desmosomal proteins also directly interact with ion channels at the intercalated disc [70, 115, 116]. However, the signaling between loss of PKP2 expression and structural cardiomyopathy is still unclear. Cerrone et al. thus investigated the role of ATP/adenosine in PKP2 cKO mice models, finding that an adenosine 2A receptor blocker-Istradefylline mitigated the progression of fibrosis and mechanical failure, while PSB115 (a blocker of the 2B adenosine receptor) had opposite effects [73], indicative of involvement of adenosine receptor signaling. Kim and coworkers, using PKP2 cKO mice 14 days post-tamoxifen (post-TAM) to explore early molecular/cellular events, found that loss of PKP2 disrupted right ventricular-predominant Ca2 + homeostasis prior to cardiomyopathy, which is at least partly mediated by a Cx43-dependent permeability and inhibited by protein kinase C (PKC) inhibitors [74]. In conclusion, the loss of PKP2 led to electrophysiological abnormalities and ultrastructural defects.
JUP
To test if reduced desmosomal protein expression causes ACM, Kirchhof et al. for the first time found that heterozygous plakoglobin-deficient mice (plakoglobin+/−) showed an increased right ventricular volume, reduced right ventricular function, and spontaneous ventricular ectopy, which was accelerated by endurance training [79,80,81]. This suggests that plakoglobin deficiency provokes the functional phenotype of ACM. In addition, overexpression of the N-terminal mutants in embryonic mouse hearts can lead to embryonic lethality resembling that seen in JUP- and PKP2-deficient mice [79, 93, 117]. Fabritz et al. confirmed that load-reducing therapy prevents training-induced development of ACM in plako ( ±) mice [82]. Plakoglobin functions as a signaling protein via its ability to modulate the Wnt/β-catenin signaling pathway. However, Li et al. generated cardiac-restricted Jup knockout mice which largely recapitulated the clinical manifestation of human ACM, including ventricular dilation and aneurysm, cardiac fibrosis, cardiac dysfunction and spontaneous ventricular arrhythmias and increased β-catenin at adherens junctions, but there was no abnormality in Wnt/β-catenin signaling [83, 118]. Indeed, they also found cardiomyocyte death in Jup mutant hearts and the cell death was usually confined to the area close to the fibrosis replacement area [83]. Similarly, ablation of plakoglobin caused an increase in β-catenin stabilization associated with activated AKT and an inhibition of glycogen synthase kinase 3β, which may contribute to ACM pathogenesis [84]. They also demonstrated for the first time the dual functions of plakoglobin as a cell adhesion and signaling molecule in the working myocardium [84]. Similarly, the plakoglobin homolog, β-catenin was increased in a plakoglobin hypomorph (PG FN/Δ) hearts [119].
DSG2
In 2009, Pilichou et al. demonstrated for the first time that myocyte necrosis as the key initiator of myocardial injury triggered progressive myocardial damage, including an inflammatory response and massive calcification within the myocardium, followed by injury repair with fibrous tissue replacement, and myocardial atrophy [85]. They were also interested in whether mutation-induced intercalated disc remodeling impacts electrophysiological properties before the onset of cell death and fibrosis. Understanding the electrical remodeling in the early stage of ACM is crucial to elucidate mechanisms for ventricular arrhythmias. Therefore, they employed the same model, finding that a DSG2 variant in a structural component of cardiac desmosomes affects ventricular conduction and arrhythmia susceptibility even prior to the onset of necrosis and replacement fibrosis due to a reduced INa density during the early ACM stages [86]. Inhibiting lectin, galactoside-binding, soluble, 3 (Lgals3) in zebrafish reduced Wnt and TGF-β signaling, increased Hippo/YAP-TAZ signaling, and induced alterations in desmosome integrity and stability [120].
Mutant mice lacking a part of the extracellular adhesive domain of DSG2 developed ventricular dilation leading to cardiac insufficiency and eventually premature death, which are correlated with increased mRNA expression of c-myc, ANF, BNF, CTGF, and GDF15, markers for cardiac stress, remodeling and heart failure [87]. To unravel the sequence of myocardial alterations during ACM onset and progression, histological analyses were performed on the hearts of DSG2 mutant mice from the juvenile to the adult state, suggesting that dying cardiomyocytes with calcification appeared in lesions of all ages and lesions of young mutant mice and the older animals harbored high amounts of CD45 + immune cells [88]. The expression of α1-skeletal muscle actin (Acta1) was upregulated in the myocardium of Dsg2mt/mt and Dsg2cKO/cKO mice. Its early upregulation is related to the impaired mechanical coupling of cardiomyocytes, and the later stage is the production of fibrotic cardiomyopathy and adjacent cardiomyocytes caused by TGFβ [89]. The stabilization of Dsg2 binding by a linking peptide (Dsg2-LP), serving as a novel approach to treat arrhythmia in patients with AC, is efficient to rescue arrhythmia in an AC mouse model, disrupted cohesion induced by siRNA-mediated plakoglobin or Dsg2 depletion as well as Cx-43 mislocalization and conduction irregularities [121].
DSP
Subunit 6 of the cardiac constitutive photomorphogenesis 9 (COP9) signalosome (CSN6), a component of the cardiac desmosome complex, directly interacts with the N-terminus of DSP [110]. Furthermore, hearts from Dsp-cKO mouse model displayed reduced junctional localization of CSN6 and similar protein degradation defects, which is consistent with loss of CSN6 function [110]. Using Cardiac-restricted DSP transgenic mice, Yang et al. identified 4 novel mutations in DSP, like a nonsense mutation in the N terminus of DSP (W233X) leading to haploinsufficiency, two missense mutations in the N terminus of DSP (V30M and Q90R) affecting the normal localization of DSP in vitro probably due to loss of binding to JUP as well as the overexpression of a C-terminal DSP mutation (R2834H) leading to cardiac defects [93]. In addition, studies have linked mutations in the desmosomal protein and desmoplakin to patients primarily exhibiting left ventricular-dominant and biventricular forms of ACM [19, 122, 123]. Homozygous DSP-cKO mice display early ultrastructural defects in desmosomal integrity leading to a cardiomyopathy reminiscent of a biventricular form of ACM, which includes cell death and fibro-fatty replacement within the ventricle leading to biventricular dysfunction, failure and premature death [94]. Exercise and catecholamine stimulation exacerbated ventricular arrhythmias that happened in DSP-cKO mice, whose hearts exhibited right ventricular conduction defects related to loss of connexin 40 expression and electrical wavefront propagation defects associated with loss of Cx43 expression [94].
DSC2
To date, there are few transgenic or mutant DSC2 knock-in mouse models. It has been reported that the graded knockdown of DSC2 in zebrafish embryos can severely disrupt myocardial desmosome structure and contractility. Also, a heterozygous mutation (c.631-2A → G) in DSC2 is a cause of familial ACM11 [111]. However, the G790del mutation in a DSC2 knock-in mouse model showed a slight contractile dysfunction and Ca2+ dysregulation in the left ventricular, which was not relevant to the pathogenesis of ACM, perhaps because G790del in DSC2 alone is insufficient to develop ACM in mice [96]. Other non-genetic factors such as microRNAs may contribute to the down-regulation of desmosomal protein that causes desmosome dysfunction [124]. The overexpression of miR-130a in adult myocardium would promote downregulation of DSC2 and lead to a disease phenotype resembling AC, suggesting that αMHC-tTA/TetO-miR130a mice may serve as a potential model to study ACM [125].
Non-desmosomal Mutations in Rodent Models
Currently, several rare mutations in non-desmosome genes have been found in human ACM patients, including cardiac electrophysiology (encoded by SCN5A, PLN), Z-band proteins (encoded by DES, LDB3, ACTN2), nuclear envelope proteins (encoded by TMEM43, LMNA, LEMD2), or proteins involved in cell–cell or cell to extracellular matrix (ECM) adhesion (encoded by CTNNA3, CDH2, TJP1, ILK, FLNC). However, there are currently a few specific mice or zebrafish models available for ACM-associated non-desmosomal mutations.
PLN
Phospholamban encoded by the PLN gene is present in the sarcoplasmic reticulum membrane, regulating calcium handling by reversibly inhibiting the activity of the sarcoplasmic reticulum calcium ATPase 2 (SERCA2) [126]. Animal models have been established to mimic human ACM. Overexpression of Arginine (Arg) 14 deletion (PLN-R14del) in mice resulted in super inhibition of SERCA, which may be related to cardiac hypertrophy, myocardial fibrosis, and premature death. PLN ablation (PLN-KO) significantly increases cardiac contractile parameters, whereas overexpressing PLN inhibits cardiac function [127, 128]. A mutation in the PLN-R14Del may lead to mis-localization of PLN from SR to the sarcolemma and increased Na/K-ATPase (NKA) activity [129]. PLN-R14Δ/Δ mice accelerated manner in less than 2 months, whereas PLN-R14Δ/+ mice exhibit cardiomyopathy at middle age [99]. Raad et al. found that R14del hearts exhibited increased arrhythmia susceptibility at the early stages of the disease, which provide an electrophysiological basis for the typical mode of SCD in these patients [100].
TMEM43
Transmembrane protein 43 (TMEM43) localized mostly at the nuclear membrane is related to a highly lethal and fully penetrant ACM subtype, which is called ARVD5[MIM:604400] [37, 103, 130,131,132,133]. The Ser358Leu mutation of TMEM43 knock-in mice displays ACM-like phenotypes, such as higher level of left ventricle end-diastolic dimension (LVEDD), lower level of posterior wall thickness in systole (PWTS) as well as cardiac fibrosis and adipogenesis [104]. Of note, TMEM43 S358L mutation up-regulated nuclear factor-κB (NF-κB)-TGFβ signal cascade during ACM cardiac fibrosis, revealing the regulatory mechanism of ACM development [104]. Barthe et al. demonstrated that transgenic mice expressing TMEM43-S358L exhibited myocardial fibrofatty replacement and died at a very young age whereas GSK3β inhibitor or overexpression of calcineurin Aβ1 in TMEM43 mutant mice can improve cardiac function but antifibrotic treatment can’t, suggesting that a new therapeutic approach could be used in ACM5 patients in the future [103]. TMEM43-mutant mice treated with enalapril showed a significantly increased survival rate, and showed increased left ventricular ejection fraction, shortened QRS duration, and decreased left ventricular fibrosis at 4 months of enalapril treatment whereas metoprolol did not show positive effects [105], suggesting that enalapril can preventively treat asymptomatic ACM5 gene carriers.
DES
The prevalence of Des mutations in ACM is higher than previously described, estimated at 2–3%, and Des−/− mice recapitulate most of the pathognomonic features of ACM [46, 134]. The crosstalk between the complement and coagulation systems exacerbated the myocardial injury of ACM mice which was alleviated by using the thrombin inhibitor lepirudin [101]. Des elimination leads to structural and functional abnormalities of the sinoatrial pacemaker complex (SANcl) [101]. As such, clarifying the molecular correlation between coagulation and the complement system may provide potential and new molecular therapeutic targets for ACM to improve clinical outcomes.
Other Factors in Rodent Models
To date, due to low and uncertain ACM penetrance, it is crucial to understand the cardiac remodeling caused by environmental stressors.
Exercise
In addition, mice with Rho-kinase inhibition in the developing heart (SM22α-restricted) spontaneously presented cardiac dilatation and dysfunction, myocardial fibrofatty changes, and ventricular arrhythmias, which further led to premature sudden death, phenotypes consistent with the characteristics of ACM in humans, demonstrating a novel crucial role of Rho-kinase inhibition during cardiac development in the pathogenesis of ACM [106]. Physical exercise has been observed as the common denominator in provoking an arrhythmic phenotype. Physical exercise has been observed to cause arrhythmia phenotype [135]. However, treadmill exercise may restore the transcription levels of most dysregulated genes in cardiomyocytes, reduce cardiomyocyte apoptosis, and induce eccentric cardiac hypertrophy without affecting cardiac dysfunction in the myocyte-specific Dsp haplo-insufficient (Myh6-Cre: DspW/F) mice [109], probably because the exercise protocol used in this study is to gradually increase the workload. The limitation of this study is that it did not involve the effect of treadmill exercise on arrhythmia in elderly mice. These also demonstrate that when studying the effects of physical activity on disease progression and arrhythmia in the ACM model, the type of training plays a critical role. Hammer et al. showed that low-intensity exercise in the PKP2+/- mouse model did not lead to fibrofatty replacements or rearrangement of gap junctions, SCD and an increase in obvious arrhythmias, calcium handling and contractility alterations of isolated myocytes caused by exercise were mostly abolished in these animals, suggesting that the low-intensity exercise seems to be beneficial even in the early, short-term stages [77]. Endurance exercise training caused PKP-2 R735X mutant mice to have a clear RV dysfunction resembling the ACM phenotype, such as impaired global RV systolic function and RV regional wall motion abnormalities and connexin 43 delocalization at intercardiomyocyte gap junctions, suggesting that endurance exercise is a key risk factor for the development of ACM, heart failure, arrhythmias and sudden death [20, 71, 136]. Moreover, Moncayo-Arlandi et al. found that endurance training triggered the ACM phenotype in truncated PKP2 mice [72], whereas endurance exercise accelerated ACM pathogenesis in Tg-DSP (R2834H) mice and this event is associated with perturbed AKT1 and GSK3-β signaling [95].
Inflammation
It has been reported that in addition to exercise, pressure overload and inflammation are associated with ACM [75, 137,138,139,140]. The effects of exercise, pressure overload, and inflammation on the progression of PKP2-related diseases were studied in heterozygous PKP2 knockout mice (PKP2-Hz), showing that PKP2 haploinsufficient mice displayed reduced Ca2 + -handling-related proteins expression, such as CaV1.2, SERCA2a, AnkB, and Casq2. Pressure overload increased levels of fibrosis and impaired electrical conduction rather than structural remodeling, leading to exercise-induced pro-arrhythmic cardiac remodeling [78]. However, whether cardiac electrical remodeling and Ca2+-handling disturbances were directly linked in these models remains uncertain. Patients with ACM have elevated circulating levels of pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α). Thus, inhibiting the complement factor C5a receptor (CD88), blocking GSK3β, activating NF-κB, and employing Bay11-7082 may be the therapeutic options of blunting inflammatory signaling [103, 134, 139, 141, 142]. Moreover, recent advances reveal that extracellular vesicle (EVs) which are secreted by cardiosphere-derived cells (CDCs) also improve cardiac function, reduce cardiac inflammation, and suppress arrhythmogenesis in ACM [91].
miRNAs
In the past few decades, a large number of microRNAs have been proved to play a crucial role in various disease phenotypes, including cardiovascular diseases. Recently, miRNAs have also been shown to be altered in ACM, such as miR-21-5p, and miR-135b, miR-184, miR-130a, miR-217-5p and miR-708-5p, along with miR-499-5p [90, 124, 125, 143, 144]. Yet, further studies will be necessary to reveal the potential pathophysiological roles of miRNA and underlying mechanisms in the development of ACM. Sorbin and SH3 domain-containing 2b (SORBS2) is another potential candidate gene for ACM susceptibility because SORBS2 knockout mouse manifests several key features of ACM, such as right ventricular dilation, right ventricular dysfunction, spontaneous ventricular tachycardia, and premature death [108].
In addition to the cardiac inflammation, patients of ACM show a gradual fibro-adipose replacement of the ventricular myocardium. Yet, there is no pharmacological method available in clinical practice to counteract the replacement of cardiac lipogenesis. The PLN, tumor protein 53 apoptosis effector (PERP), and carnitine palmitoyltransferase 1β (CPT1B) involved in ion channels, apoptosis and adipogenesis play a role in the pathogenesis of ACM [145]. Some studies also demonstrated that inhibition of Wnt/β-catenin signaling can trigger adipogenesis, fibrogenesis, and apoptosis, which are characteristic of human ACM [92, 146,147,148]. It has been reported that the proliferator-activated receptor gamma (PPARγ), activated when Wnt/β-catenin- and Hippo-pathway are impaired, is a key regulator of ACM adipogenesis [92, 149]. Therefore, the PPARγ modulator rosiglitazone or 13-hydroxyoctadecadienoic acid (13HODE) could convert glycolysis into fatty acid metabolism to mimic ACM lipogenesis [150]. Furthermore, ACM patients show high plasma concentration of oxLDL which are major cofactors of adipogenesis. Cardiac adipogenesis and right ventricle systolic impairment are counteracted by atorvastatin treatment in a Pkp2 heterozygous knock-out mice (Pkp2 + / − mice) with a high-fat diet (HFD) [151]. In that study, the authors also demonstrated for the first time, that oxidative stress and oxidized lipid metabolism modulate ACM adipogenic phenotype at the cellular, mouse, and patient levels [151]. Fibrofatty infiltration only appeared in mouse models with DSP mutations, but not in mouse models involving DSG2, DSC2, and JUP [92, 93, 152, 153].
Mutations in Zebrafish Models
Due to the convenience of embryonic morpholine gene knockout and the adaptability of high-throughput inheritance and compound screening, zebrafish ACM models for studying DSC2 and JUP mutations have also been developed [111, 154]. Furthermore, Moriartyet al. knocked down PKP2 in zebrafish through morpholine microinjection, resulting in cardiac oedema, blood pooling, failure of the heart to loop, decreased heart rate and abnormal desmosomes in the heart, suggesting that PKP2 is essential in cardiac development [112]. A more integrative model (zebrafish) is used to investigate the pathophysiological mechanism of ACM, which allowed us to assess the specific effect of genetic variation at the organs and organisms level. Asimaki et al. generated a zebrafish model of ACM carrying a cardiomyocyte-specific expression of the human 2057del2 mutation in the gene encoding plakoglobin to elucidate the underlying mechanisms and discover potential chemical modifiers, and SB216763 showed a remarkable ability to prevent ACM in this model [112]. Giuliodori et al. generated and validated a zebrafish model for DSP-associated AC using a gene knock-down approach, finding that knock-down of zebrafish DSP affects desmosome structure, Wnt/β-catenin, TGF-β/Smad3, and Hippo/YAP-TAZ signaling pathways [113]. Furthermore, GSK3β inhibitor rescues the AC phenotype in the zebrafish model through Wnt/β-catenin signaling, and activation of abnormal Hippo/YAP signaling pathway leads to β-catenin cytoplasmic isolation and JUP nuclear translocation [113, 142, 149, 154]. Moreau et al. established the zebrafish model to validate the effect of the DSC2 p.R132C substitution in‐vivo, indicating that the R132C substitution impairs DSC2 function [114].
These animal models have played a critical role in elucidating the pathophysiological mechanism of ACM and developing targeted therapies. Although many observations are consistent with those of ACM patients, clearly, animal models cannot reproduce the conditions of ACM patients due to the differences in electrophysiological functions between animal and human hearts. Therefore, the main differences in the electrophysiological characteristics of the heart of small animals and humans largely limit the translation of the results to humans. Moreover, animal models often lack research on the molecular level of gene function.
Heterologous Expression Systems
When studying the regulatory function of a gene, normally it is necessary to verify its regulatory effect in different models. Using cell models to study its molecular mechanism is more conducive to the prediction and verification of gene function. Compared with gene knockout animals, gene knockout at the cell level has many advantages, such as lower lethality and faster construction time. Therefore, specific mutant genes identified from ACM patients transfected into cells can be helpful to understand the underlying pathophysiological mechanism and provide unique opportunities to gain insights into different forms of ACM. To date, there are many very meaningful kinds of researches related to ACM at the cellular level (Table 3).
HEK293 Cells
HEK293 cells are widely used in many fields and have become a powerful platform. Importantly, HEK293 cells exhibit high transfection efficiency, fast growth, efficient and flexible metabolism, and have all human post-translational modifications to produce the most similar proteins [170,171,172,173]. Asimaki and colleagues identified a mutation (S39_K40insS) of JUP in an ACM-affected German family, which is a novel autosomal dominant plakoglobin mutation. They reported that HEK293 cells expressing the mutant plakoglobin had a higher proliferation rate and a lower apoptosis rate [158]. However, Christensen et al. did not find that the PKP2 c.419C > T variant has an effect on cell proliferation, indicating that PKP2 c.419C > T lacks a functional effect [159]. Gerull et al. transfected HEK293 cells with constructs expressing human DSC2 WT or mutant DSC2-Q554X, finding that both DSC2-WT and DSC2-Q554X are localized at the cell membrane and remain stable [160]. Desmoplakin knock-down (DP-KD) also impaired the Cx43 membrane localization in neonatal rat ventricular myocytes (NRVCMs), suggesting that DP regulates membrane localization [164]. To document that the current amplitude change is independent of cell-type, Riele et al. transiently transfected HEK293 cells with cDNA coding for SCN5A, demonstrate in that similar to the hiPSC-CMs, the current density generated by a construct containing the SCN5A p.Arg1898His mutation was significantly reduced compared with wild-type SCN5A [174]. Chen et al. identified that the DSG2 gene expression is significantly decreased in a novel nonsense variant in DSG2 (c.710 T > A, p.Leu237Ter) group, suggesting this null variant would decrease the expression of DSG2 gene, and the mutant DSG2 truncated protein was markedly shifted to the cytoplasm, which suggests that the nonsense variant (DSG2 c.710 T > A, p.Leu237Ter) could affect the expression and function of DSG2 protein [161]. More importantly, these results could be repeated in AC16 cell model [161]. Furthermore, Khudiakov et al. observed that GSK3BS9A mutation expression did not result in a decrease of Wnt/β-catenin signaling activity but led to a significant INa density decrease in GSK3BS9A transfected HEK293T cells [162]. HEK293T cells transfected with mutant plasmids led to the truncated DSP mRNA and protein, upregulation of nuclear JUP and downregulation of β-catenin, when compared with WT. Truncation of DSP protein, down-regulation of JUP and up-regulation of β-catenin expression in nuclear but not cytoplasm are observed in the HEK293T cells transfected with DSP c.832delG [163].
HL-1
HL-1(Heart Atrial cells derived from mice) has been extensively characterized and is a valuable model system to address questions of cardiac biology at the cellular & molecular levels with a phenotype similar to adult cardiomyocytes. Garcia-Gras et al. established DP-deficient HL-1 cells and showed that suppression of DP expression led to nuclear localization of the desmosomal protein plakoglobin and a twofold reduction in canonical Wnt/beta-catenin signaling through Tcf/Lef1 transcription factors [92]. Yang et al. also demonstrated that the N-terminal mutants (V30M and Q90R) of DSP failed to localize to the cell membrane in the desomosome-forming cell line (the human tongue squamous cell carcinoma cell line SCC-9) and failed to bind to JUP [93]. HL-1 atrial myocytes maintain the electrophysiological functioning of healthy cardiomyocytes and express near natural levels of connexin43, making them ideal for this particular research [175]. PKP-2 siRNA in HL-1 cardiomyocyte cells decreases connexin43 expression and alters its localization [176]. PKP2 deficient led to decreased INa and NaV1.5 at the site of cell contact in HL-1-derived cells that endogenously express NaV1.5 but have PKP2 deficiency. PKP2 variants that reduce INa could be related to a Brugada syndrome (BrS) phenotype, even without overt structural features characteristic of ACM. Cx43 and Nav1.5 expression decreased and exhibited an abnormal distribution following DSP silencing in HL-1 cells and the DSP suppression also decreased INa and slowed CV, indicating that impaired mechanical coupling affects electrical synchrony in ACM to a great extent [156].
Primary Cells
What’s more, compared with cell lines, primary cells retain more biological characteristics of the original tissue, such as growth and senescence, so better cellular diseases models can be established through primary cells. Oxford et al. decreased PKP2 expression in NRVCMs using RNA silencing technology, showing that loss of PKP2 expression led to a decrease in total Cx43 content, and Cx43 had a significant redistribution in the intracellular space, which demonstrated that there is a molecular crosstalk between desmosomal and gap junction proteins [165]. The two missense mutations of DSC2 (p.E102K mutation and p.I345T mutation) in the N-terminal domain affect the normal localization of DSC2 on neonatal rat cardiomyocytes and HL-1 cells [166]. Mutations R79x and 179 fs of PKP2 did not alter the localization of endogenous PKP2, DP or Cx43, and the mutation R79x expression significantly reduces HSP90 levels, which leads to facilitated activation of myocyte apoptotic pathways [167]. Moreover, Wei et al. observed hypoxia/serum depletion stimulation induced significantly elevation of intracellular and extracellular HSP70 in NRCMs, indicating that elevated HSP70 is a feature of heart failure caused by ACM [168]. Epicardial explants after PKP2 knockdown obtained from neonatal rat hearts increased abundance of α-smooth muscle actin-positive cells, cell migration speed, abundance of cell proliferation markers, and abundance of lipid markers, suggesting that a group of non-excitable cardiac resident cells express desmosome molecules and rely on PKP2 expression in vitro [169]. Pathobiological features seen in patients with ACM can be observed in NRVMs expressed 2057del2 plakoglobin, which was reversed or prevented by SB216763 which is a suppressor of the disease phenotype [154]. NRVCMs with 2157del2 in JUP exhibits several features, such as abnormal redistribution of intercalated disk proteins, the release of inflammatory cytokines, myocyte apoptosis, which can be prevented by Bay 11–7082 (a small-molecule inhibitor of NF-κB signaling) [141]. Asimaki et al. transfected adenovirus into normal NRVCMs to express 2057del2 mutation plakoglobin, showing increased myocyte apoptosis, decreased immunoreactive signal for Cx43 at cell–cell junctions as well as diminished immunoreactive signal for plakoglobin at cell–cell junctions and abundant signals in cell nuclei, which recapitulates cardiac characteristics of ACM patients [112].
From the above review, heterologous expression systems expressing disease-specific mutations, such as HEK293 cells, HL-1 cells etc., and transgenic animal models have greatly facilitated our understanding of the pathogenic mechanisms associated with ACM. In fact, it is true that all published animal or cell models of ACM as well as the elucidation of the involved mechanisms and the applicability of experimental data have inherent limitations in studying human diseases, which have hampered the exploration of potential therapies for the management of human ACM. Similarly, human non-cardiac cell lines (such as HEK-293 or CHO cells) are not ideal to model the heart as they are different from cardiomyocytes in many aspects such as sarcomere tissue, metabolism and electrophysiology, etc. [177]. In this regard, cardiomyocytes derived from induced pluripotent stem cells have advantages over animal or human non-cardiac cells.
Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs)
The use of hiPSC-CMs is extremely versatile in studying basic and profound mechanisms of cardiomyopathies such as familial dilated cardiomyopathy (DCM), familial hypertrophic cardiomyopathy (HCM), catecholaminergic polymorphic ventricular tachycardia (CPVT), long QT syndrome (LQTS), short QT syndrome (SQTS), ACM and BrS [154, 178,179,180,181,182,183,184,185,186,187,188]. Compared with animal models and heterologous expression systems, one of the most important advantages of hiPSC-CMs is that it closely matches the genes of patients with specific diseases. The second advantage is the possibility to generate patient-specific cardiomyocytes for patient-specific investigation including mechanistic and therapeutic studies [183, 189]. Therefore, hiPSC-CMs are a significant preclinical model system for studying the genetic basis of human cardiovascular diseases (Table 4). hiPSC-CMs have been used to better understand the occurrence of arrhythmias [192, 196]. Several research groups have successfully generated iPSC-CMs from patients with hereditary cardiac ion channel diseases, such as the LQTS and BrS [182, 197,198,199]. hiPSC-CMs retain the patient’s genetic information and exclude the influence of environmental factors. Additionally, ACM patient-specific hiPSC-CMs that can model disease-specific abnormalities have been proved to recapitulate key characteristics observed in human disease [200].
Desmosomal Variants in hiPSC-CMs
In 2012, Ma et al. for the first time generated iPSC-derived cardiomyocytes from a patient with a clinical diagnosis of ACM and demonstrated significant phenotypes such as reduced gene expression of PKP2 and JUP, reduced immunofluorescence signals for these desmosomal proteins, and increased potential for adipocytic change [192]. hiPSC-CMs from 2 ACM patients with PKP2 mutations were established, displaying similar results, such as a significant decrease in the expression of PKP2 and reduced densities of PKP2, the associated desmosomal protein plakoglobin as well as the gap-junction protein Cx-43, which was related to upregulation of the proadipogenic transcription factor PPAR-γ, whereas elevated estradiol levels decreased apoptosis and lipid accumulation of CMs in an in-vitro ACM model [22, 150, 191].
Sex Hormones Study in hiPSC-CMs
It is known that sex hormones regulate metabolic homeostasis of various cell types and the occurrence of arrhythmias [154, 201], especially testosterone which regulates adipogenesis of fat cells and is also associated with a high incidence of cardiovascular disease [202, 203]. Akdis et al. reported that in male ACM patients, increased serum testosterone levels were independently linked to major arrhythmic cardiovascular events (MACE), while in female MACE patients, estradiol levels were reduced, suggesting that testosterone worsened and estradiol improved cardiomyocyte apoptosis and lipogenesis in an induced pluripotent stem cell-derived ACM model [22]. Currently, researchers have successfully established the iPS cell line HUBUi001-A from a patient carrying the DSP heterozygous variants (c.104G > T p.G35V; c.5617C > T p.R1873C) and an iPSC cell line with a pathogenic heterozygous variant in PKP2 (c.1799delA) from a patient affected by ACM [204, 205].
Ion Channel Dysfunction in hiPSC-CMs
Some researches related to arrhythmias in ACM have been conducted, mainly focusing on the electrophysiological properties of hiPSC-CMs. Riele et al. revealed reduced peak sodium current and reduced abundance of NaV1.5 and N-Cadherin clusters at the intercalated disc, indicating that Nav1.5 and adhesion molecules are in a functional complex, and Nav1.5 dysfunction may contribute to ACM [174]. El-Battrawy et al. reported that ACM-hiPSC-CMs carrying a DSG2 gene missense variant (G to A substitution at nucleotide p.Gly638Arg) showed an abnormal action potential with reduced APA and Vmax, multiple ion channel current dysfunctions such as reduced INa, Ito, ISK, IKATP, INCX, and enhanced IKr, and also showed that ion channels were more sensitive to adrenergic stimulation, suggesting that multiple ion channel dysfunction and increased sensitivity to adrenergic stimulation are associated with arrhythmias in ACM patients [193]. Buljubasic et al. demonstrated that both NDPK-B and SK4 expressions were elevated in ACM-hiPSC-CMs with the same mutation and that recombinant NDPK-B enhanced ISK4, cell automaticity and arrhythmic events, whereas protein histidine phosphatase 1 (PHP-1), a counter actor of NDPK-B, prevented the NDPK-B effect, suggesting possible involvement of NDPKB/SK4 in arrhythmogenesis of ACM with DSG2 mutations [206]. Moreover, hiPSC-CM derived from the DSC2 patient showed that reduced Ca2+ current density and increased K+ current density led to a shortened action potential duration (APD), which may be used to elucidate the abnormal repolarization dynamics in ACM patients [114].
hiPSC-CMs with the p.S358L variant in TMEM43 also showed contractile dysfunction, which partially recovered after GSK3β inhibition [103]. There are no reports indicating that variants in the OBSCN gene cause ACM. Thus, Chen et al. generated iPSC-CMs isolated from an ACM patient carrying a variant in the OBSCN gene, showing that the calcium current increased, the structure of mutant OBSCN protein and its anchor Ank1.5 protein structure was disordered and the expression of both proteins together with other desmosomal proteins was reduced, whereas the adipogenesis pathway-related proteins such as PPARγ, C/EBPα, and FABP4 were increased, which may explain the fibrofatty replacement of the myocardium and calcium channel-related myocardial contraction abnormalities in ACM patients [195]. Additionally, Hawthorne et al. established a novel hiPSC-CM model derived from ACM patients with a c.2358delA variant in DSG2, confirming that DSG2Mut CMs harbored decreased DSG2 expression and disrupted protein localization, whereas the expression or localization of other key desmosome components did not change significantly [190]. At the same time, the APD and the time to reach peak calcium in DSG2Mut CMs were shortened, and the Ca2+ handling and expression of immune cytokines were altered [190]. Similarly, hiPSC-CMs from a patient with a PKP2 deficit showed drastically reduced INa, whereas transfection of wild-type PKP2 can restore these deficits [155]. Khudiakov et al. generated the hiPSC-CMs model of ACM with PKP2 genetic variants c.354delT and p.Lys859Arg to study its molecular and functional role, displaying that after inhibiting GSK3β, sodium current was restored through Wnt/β-catenin-independent mechanisms [162, 207].
hiPSCs derived from patients with specific variants can be frozen, stored, and used as an in vitro model of ACM, providing structure and function-based data for the development of new therapeutic applications. The pathogenic role of a variant can be confirmed by gene editing. It was demonstrated that repairing the gene variant could revert the disease-phenotype in the DSC2 hiPSC‐CMs to normal state, and in addition, sotalol improved electrical activity as well as mechanical function, and flecainide normalized the frequency of spontaneous Ca2+ transients and the occurrence of Ca2+ sparks, which provide a rationale for their therapeutic application in ACM [114]. The same gene, even sometimes the same genetic variation can also cause totally different clinical characteristics, indicating that the disease entity results from multiple disease-causing factors. In addition to the genetic causes, a host of other factors, such as age, environment, genetic background, complications together with epigenetic factors will also affect the occurrence, progression, and prognosis of ACM [208].
Limitations of hiPSC-CMs Models
hiPSC-CMs exhibit a relatively immature phenotype and are more depolarized than adult ventricular cardiomyocytes in a resting state, which will affect the dynamics of voltage-gated ion channels and change the excitability [209]. In typical monolayer culture, they will not exhibit an elongated morphology or form fully organized insertion discs, which may affect the level and spatial distribution of desmosomal protein expression. Now more chemical, genetic, and biomechanical approaches are developed to promote the maturation of cardiomyocytes, such as the incorporation of CMs into 3D tissue constructs, bioelectrical stimulation, mechanical stretch, biochemical stimulation, and long-term culture [210,211,212,213,214]. For example, Liu et al. first reported that PGC-1α activator ZLN005 promotes the maturation of mitochondrial biology and energy metabolism, enhances structural maturity including sarcomere length, increases CX43 expression, enhances electrical activity, improves Ca2+ handling and electrophysiological characteristics to promote the maturation of hPSC-CMs [215]. Furthermore, Miki et al. identified that ERRγ agonists make hiPSC-CM be larger in size, possess longer sarcomere length, present transverse tubules, and enhance metabolic function, contraction and electrical properties, which is consistent with the characteristics of the mature neonatal CMs and contributes to disease modeling and regenerative medicine [216]. The combinations of hiPSC-derived CMs, cardiac fibroblasts (CFs), and cardiac endothelial cells can also promote the maturation of scaffold-free three-dimensional microtissues (MTs) [217]. A better hiPSC-CM platform that is more similar to native cardiomyocytes will be available in the near future.
Conclusions and Future Perspectives
ACM is a genetic disease of the myocardium, which is characterized by ventricular arrhythmias and SCD, right ventricular dysfunction, and subsequent progressive heart failure. To date, some animals such as mice, dogs, and cats and cell models have been used to study clinical features of ACM. Each model has its unique advantages and limitations (Table 5). Transgenic animals are increasingly used in in-vivo experiments to examine the effects of gene functions, their regulation, or genetic changes on disease development. The health of many genetically modified animals has been affected by genetic modification, regardless of the procedures performed on them. Therefore, some researchers have proposed non-animal transgenic methods, such as cell transfection technology. However, cell transfection technology has several disadvantages such as low cell transfection efficiency and high cell death rate, especially the primary cultured cells. Some diseases lack appropriate animal models or in vitro models. Therefore, hiPSC-CMs become a valuable model for studying the pathological mechanism in ACM. Although hiPSC-CMs are relatively less mature than isolated adult ventricular cardiomyocytes, such as disorganized sarcomeric filament, lack of t-tubular network, polygonal shapes, and rhythmic automaticity, more useful approaches have been studied to promote the maturity of hiPSC-CMs. There is no doubt that the advantages of this cell type outweigh the disadvantages, e. g. generating patient-specific cells, editing the genome of healthy and disease cells to insert or correct mutations/variants.
Moreover, genome editing tools including clustered regularly interspaced short palindromic repeats (CRISPR/Cas9), transcription activator-like effector nucleases (TALEN) etc. have largely facilitated the applications of hiPSC-CMs by altering gene expression and correct genetic variation [218,219,220,221,222]. Hence, hiPSC-CMs combined with gene-editing technology have become a powerful tool for studying the pathophysiological mechanism of ACM and a substitute for animal models. In general, hiPSC-CMs are more widely used in preclinical research and regenerative medicine. Currently, different methods have been adopted to study the pathogenesis of ACM, including in vitro cell and tissue models and in vivo models. However, it is still challenging to fully reproduce the clinicopathological features of ACM in a laboratory environment. Therefore, more work needs to be done to promote the innovation of advanced ACM models to reproduce these subtle physiological effects to uncover the pathology and clinical findings of ACM.
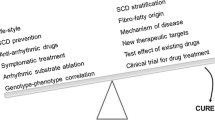
Currently, the source of the correlation between genetic variation and phenotype in ACM can be the direct, major, and mixed impact of variation. For the complex traits present in ACM, it is still challenging to identify all the causal variants and clarify their underlying mechanisms. Male and multiple gene variants, which are important factors, affect the prognosis of ACM [8, 21]. Current effective treatments for ACM include lifestyle changes, traditional pharmacological therapy, catheter ablation, ICD, and heart transplantation. However, every treatment has its limitations. Numerous approaches such as using cardiac stem cells to regenerate cardiomyocytes or collecting cardiac progenitor cells to produce beneficial factors are rapidly entering clinical trials to solve various forms of cardiomyopathy [223]. WES and whole-genome sequencing (WGS), multi-omics technology and hiPSC-CMs are new tools for studying the genetics of ACM, which provide a new platform for unraveling the complex molecular interactions of ACM and clinical management. Therefore, an in-depth understanding of gene mutation-phenotype association and individualized treatment are the prerequisites for achieving precision medicine in ACM.
Data Availability
Not applicable
Code Availability
Not applicable
References
Corrado, D., Link, M. S., & Calkins, H. (2017). Arrhythmogenic right ventricular cardiomyopathy. New England Journal of Medicine, 376(1), 61–72.
Priori, S. G., Blomstrom-Lundqvist, C., Mazzanti, A., et al. (2015). 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). European Heart Journal, 36(41), 2793–2867.
Dalal, D., Nasir, K., Bomma, C., et al. (2005). Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation, 112(25), 3823–3832.
Akdis, D., Chen, K., Saguner, A. M., et al. (2019). Clinical characteristics of patients with a right ventricular thrombus in arrhythmogenic right ventricular cardiomyopathy. Thrombosis and Haemostasis, 119(8), 1373–1378.
Sultan, F. A., Ahmed, M. A., Miller, J., & Selvanayagam, J. B. (2017). Arrhythmogenic right ventricular cardiomyopathy with biventricular involvement and heart failure in a 9-year old girl. J Saudi Heart Assoc., 29(2), 139–142.
Suzuki, H., Sumiyoshi, M., Kawai, S., et al. (2000). Arrhythmogenic right ventricular cardiomyopathy with an initial manifestation of severe left ventricular impairment and normal contraction of the right ventricle. Japanese Circulation Journal, 64(3), 209–213.
Sen-Chowdhry, S., Syrris, P., Prasad, S. K., et al. (2008). Left-dominant arrhythmogenic cardiomyopathy: An under-recognized clinical entity. Journal of the American College of Cardiology, 52(25), 2175–2187.
Bhonsale, A., Groeneweg, J. A., James, C. A., et al. (2015). Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. European Heart Journal, 36(14), 847–855.
Burke, A. P., Farb, A., Tashko, G., & Virmani, R. (1998). Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: Are they different diseases? Circulation, 97(16), 1571–1580.
Casella, M., Gasperetti, A., Sicuso, R., et al. (2020). Characteristics of patients with arrhythmogenic left ventricular cardiomyopathy: combining genetic and histopathologic findings. Circulation: Arrhythmia and Electrophysiology, 13(12), e009005.
Marcus, F. I., Fontaine, G. H., Guiraudon, G., et al. (1982). Right ventricular dysplasia: A report of 24 adult cases. Circulation, 65(2), 384–398.
Ruwald, A. C., Marcus, F., Estes, N. A., 3rd., et al. (2015). Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: Results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. European Heart Journal, 36(27), 1735–1743.
Chung, F. P., Li, H. R., Chong, E., et al. (2013). Seasonal variation in the frequency of sudden cardiac death and ventricular tachyarrhythmia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: The effect of meteorological factors. Heart Rhythm, 10(12), 1859–1866.
Wu, L., Yao, Y., Chen, G., et al. (2014). Intracardiac thrombosis in patients with arrhythmogenic right ventricular cardiomyopathy. Journal of Cardiovascular Electrophysiology, 25(12), 1359–1362.
Camm, C. F., James, C. A., Tichnell, C., et al. (2013). Prevalence of atrial arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm, 10(11), 1661–1668.
Burghouwt, D. E., Kammeraad, J. A., Knops, P., du Plessis, F. A., & de Groot, N. M. (2015). Bradyarrhythmias: First presentation of arrhythmogenic right ventricular cardiomyopathy? Journal of Clinical Medical Research, 7(4), 278–281.
Costa, S., Cerrone, M., Saguner, A. M., Brunckhorst, C., Delmar, M., & Duru, F. (2021). Arrhythmogenic cardiomyopathy: An in-depth look at molecular mechanisms and clinical correlates. Trends in Cardiovascular Medicine, 31(7), 395–402.
Corrado, D., Basso, C., Rizzoli, G., Schiavon, M., & Thiene, G. (2003). Does sports activity enhance the risk of sudden death in adolescents and young adults? Journal of the American College of Cardiology, 42(11), 1959–1963.
Corrado, D., & Thiene, G. (2006). Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Clinical impact of molecular genetic studies. Circulation, 113(13), 1634–1637.
James, C. A., Bhonsale, A., Tichnell, C., et al. (2013). Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Journal of the American College of Cardiology, 62(14), 1290–1297.
Rigato, I., Bauce, B., Rampazzo, A., et al. (2013). Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circulation: Cardiovascular Genetics, 6(6), 533–542.
Akdis, D., Saguner, A. M., Shah, K., et al. (2017). Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. European Heart Journal, 38(19), 1498–1508.
Corrado, D., Basso, C., & Judge, D. P. (2017). Arrhythmogenic cardiomyopathy. Circulation Research, 121(7), 784–802.
Towbin, J. A., McKenna, W. J., Abrams, D. J., et al. (2019). 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm, 16(11), e301–e372.
Gasperetti, A., Rossi, V. A., Chiodini, A., et al. (2021). Differentiating hereditary arrhythmogenic right ventricular cardiomyopathy from cardiac sarcoidosis fulfilling 2010 ARVC Task Force Criteria. Heart Rhythm, 18(2), 231–238.
Gandjbakhch, E., Redheuil, A., Pousset, F., Charron, P., & Frank, R. (2018). Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. Journal of the American College of Cardiology, 72(7), 784–804.
Ohno, S. (2016). The genetic background of arrhythmogenic right ventricular cardiomyopathy. Journal of Arrhythmia, 32(5), 398–403.
Rampazzo, A., Nava, A., Malacrida, S., et al. (2002). Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. American Journal of Human Genetics, 71(5), 1200–1206.
Gerull, B., Heuser, A., Wichter, T., et al. (2004). Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nature Genetics, 36(11), 1162–1164.
Pilichou, K., Nava, A., Basso, C., et al. (2006). Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation, 113(9), 1171–1179.
Syrris, P., Ward, D., Evans, A., et al. (2006). Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. American Journal of Human Genetics, 79(5), 978–984.
McKoy, G., Protonotarios, N., Crosby, A., et al. (2000). Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet, 355(9221), 2119–2124.
Klauke, B., Kossmann, S., Gaertner, A., et al. (2010). De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Human Molecular Genetics, 19(23), 4595–4607.
Taylor, M., Graw, S., Sinagra, G., et al. (2011). Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation, 124(8), 876–885.
Hall, C. L., Gurha, P., Sabater-Molina, M., et al. (2020). RNA sequencing-based transcriptome profiling of cardiac tissue implicates novel putative disease mechanisms in FLNC-associated arrhythmogenic cardiomyopathy. International Journal of Cardiology, 302, 124–130.
Quarta, G., Syrris, P., Ashworth, M., et al. (2012). Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. European Heart Journal, 33(9), 1128–1136.
Merner, N. D., Hodgkinson, K. A., Haywood, A. F., et al. (2008). Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. American Journal of Human Genetics, 82(4), 809–821.
Good, J. M., Fellmann, F., Bhuiyan, Z. A., Rotman, S., Pruvot, E., & Schlapfer, J. (2020). ACTN2 variant associated with a cardiac phenotype suggestive of left-dominant arrhythmogenic cardiomyopathy. HeartRhythm Case Reports, 6(1), 15–19.
Lopez-Ayala, J. M., Ortiz-Genga, M., Gomez-Milanes, I., et al. (2015). A mutation in the Z-line Cypher/ZASP protein is associated with arrhythmogenic right ventricular cardiomyopathy. Clinical Genetics, 88(2), 172–176.
van der Zwaag, P. A., van Rijsingen, I. A., Asimaki, A., et al. (2012). Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. European Journal of Heart Failure, 14(11), 1199–1207.
Erkapic, D., Neumann, T., Schmitt, J., et al. (2008). Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace, 10(7), 884–887.
Beffagna, G., Occhi, G., Nava, A., et al. (2005). Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovascular Research, 65(2), 366–373.
Mayosi, B. M., Fish, M., Shaboodien, G., et al. (2017). Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circulation: Cardiovascular Genetics, 10(2), e001605.
De Bortoli, M., Postma, A. V., Poloni, G., et al. (2018). Whole-exome sequencing identifies pathogenic variants in TJP1 gene associated with arrhythmogenic cardiomyopathy. Circulation: Genomic and Precision Medicine, 11(10), e002123.
van Hengel, J., Calore, M., Bauce, B., et al. (2013). Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. European Heart Journal, 34(3), 201–210.
James, C. A., Syrris, P., van Tintelen, J. P., & Calkins, H. (2020). The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. European Heart Journal, 41(14), 1393–1400.
James, C. A., & Calkins, H. (2019). Arrhythmogenic Right Ventricular Cardiomyopathy: Progress Toward Personalized Management. Annual Review of Medicine, 70, 1–18.
Marcus, F. I., McKenna, W. J., Sherrill, D., et al. (2010). Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. European Heart Journal, 31(7), 806–814.
Mestroni, L., Maisch, B., McKenna, W. J., et al. (1999). Guidelines for the study of familial dilated cardiomyopathies. Collaborative research group of the european human and capital mobility project on familial dilated cardiomyopathy. European Heart Journal, 20(2), 93–102.
Lubos, N., van der Gaag, S., Gercek, M., Kant, S., Leube, R. E., & Krusche, C. A. (2020). Inflammation shapes pathogenesis of murine arrhythmogenic cardiomyopathy. Basic Research in Cardiology, 115(4), 42.
Prior, D., & La Gerche, A. (2020). Exercise and arrhythmogenic right ventricular cardiomyopathy. Heart, Lung & Circulation, 29(4), 547–555.
James, C. A., Jongbloed, J. D. H., Hershberger, R. E., et al. (2021). International evidence based reappraisal of genes associated with arrhythmogenic right ventricular cardiomyopathy using the clinical genome resource framework. Circ Genom Precis Med., 14(3), e003273.
Ackerman, M. J., Priori, S. G., Willems, S., et al. (2011). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace, 13(8), 1077–1109.
Baumwart, R. D., Orvalho, J., & Meurs, K. M. (2007). Evaluation of serum cardiac troponin I concentration in Boxers with arrhythmogenic right ventricular cardiomyopathy. American Journal of Veterinary Research, 68(5), 524–528.
Fox, P. R., Maron, B. J., Basso, C., Liu, S. K., & Thiene, G. (2000). Spontaneously occurring arrhythmogenic right ventricular cardiomyopathy in the domestic cat: A new animal model similar to the human disease. Circulation, 102(15), 1863–1870.
Ciaramella, P., Basso, C., Di Loria, A., & Piantedosi, D. (2009). Arrhythmogenic right ventricular cardiomyopathy associated with severe left ventricular involvement in a cat. Journal of Veterinary Cardiology, 11(1), 41–45.
Harvey, A. M., Battersby, I. A., Faena, M., Fews, D., Darke, P. G., & Ferasin, L. (2005). Arrhythmogenic right ventricular cardiomyopathy in two cats. Journal of Small Animal Practice, 46(3), 151–156.
Najor, N. A. (2018). Desmosomes in human disease. Annual Review of Pathology: Mechanisms of Disease, 13, 51–70.
Vimalanathan, A. K., Ehler, E., & Gehmlich, K. (2018). Genetics of and pathogenic mechanisms in arrhythmogenic right ventricular cardiomyopathy. Biophysical Reviews, 10(4), 973–982.
Dusek, R. L., & Attardi, L. D. (2011). Desmosomes: New perpetrators in tumour suppression. Nature Reviews Cancer, 11(5), 317–323.
Vermij, S. H., Abriel, H., & van Veen, T. A. (2017). Refining the molecular organization of the cardiac intercalated disc. Cardiovascular Research, 113(3), 259–275.
Zhao, G., Qiu, Y., Zhang, H. M., & Yang, D. (2019). Intercalated discs: Cellular adhesion and signaling in heart health and diseases. Heart Failure Reviews, 24(1), 115–132.
Cerrone, M., & Delmar, M. (2014). Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends in Cardiovascular Medicine, 24(5), 184–190.
Gomes, J., Finlay, M., Ahmed, A. K., et al. (2012). Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. European Heart Journal, 33(15), 1942–1953.
Akdis, D., Brunckhorst, C., Duru, F., & Saguner, A. M. (2016). Arrhythmogenic cardiomyopathy: Electrical and structural phenotypes. Arrhythmia & Electrophysiology Review, 5(2), 90–101.
Basso, C., Fox, P. R., Meurs, K. M., et al. (2004). Arrhythmogenic right ventricular cardiomyopathy causing sudden cardiac death in boxer dogs: A new animal model of human disease. Circulation, 109(9), 1180–1185.
Spier, A. W., & Meurs, K. M. (2004). Assessment of heart rate variability in Boxers with arrhythmogenic right ventricular cardiomyopathy. Journal of the American Veterinary Medical Association, 224(4), 534–537.
Spier, A. W., & Meurs, K. M. (2004). Evaluation of spontaneous variability in the frequency of ventricular arrhythmias in Boxers with arrhythmogenic right ventricular cardiomyopathy. Journal of the American Veterinary Medical Association, 224(4), 538–541.
Meurs, K. M., Spier, A. W., Miller, M. W., Lehmkuhl, L., & Towbin, J. A. (1999). Familial ventricular arrhythmias in boxers. Journal of Veterinary Internal Medicine, 13(5), 437–439.
Cerrone, M., Noorman, M., Lin, X., et al. (2012). Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovascular Research, 95(4), 460–468.
Cruz, F. M., Sanz-Rosa, D., Roche-Molina, M., et al. (2015). Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. Journal of the American College of Cardiology, 65(14), 1438–1450.
Moncayo-Arlandi, J., Guasch, E., Sanz-de la Garza, M., et al. (2016). Molecular disturbance underlies to arrhythmogenic cardiomyopathy induced by transgene content, age and exercise in a truncated PKP2 mouse model. Human Molecular Genetics, 25(17), 3676–3688.
Cerrone, M., van Opbergen, C. J. M., Malkani, K., et al. (2018). Blockade of the adenosine 2A receptor mitigates the cardiomyopathy induced by loss of plakophilin-2 expression. Frontiers in Physiology, 9, 1750.
Kim, J. C., Perez-Hernandez, M., Alvarado, F. J., et al. (2019). Disruption of Ca(2+)i homeostasis and connexin 43 hemichannel function in the right ventricle precedes overt arrhythmogenic cardiomyopathy in plakophilin-2-deficient mice. Circulation, 140(12), 1015–1030.
Perez-Hernandez, M., Marron-Linares, G. M., Schlamp, F., et al. (2020). Transcriptomic coupling of PKP2 with inflammatory and immune pathways endogenous to adult cardiac myocytes. Frontiers in Physiology, 11, 623190.
Cerrone, M., Montnach, J., Lin, X., et al. (2017). Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nature Communications, 8(1), 106.
Hammer, K. P., Mustroph, J., Stauber, T., Birchmeier, W., Wagner, S., & Maier, L. S. (2021). Beneficial effect of voluntary physical exercise in Plakophilin2 transgenic mice. PLoS ONE, 16(6), e0252649.
van Opbergen, C. J. M., Noorman, M., Pfenniger, A., et al. (2019). Plakophilin-2 haploinsufficiency causes calcium handling deficits and modulates the cardiac response towards stress. International Journal of Molecular Sciences, 20(17), 4076.
Bierkamp, C., McLaughlin, K. J., Schwarz, H., Huber, O., & Kemler, R. (1996). Embryonic heart and skin defects in mice lacking plakoglobin. Developmental Biology, 180(2), 780–785.
Ruiz, P., Brinkmann, V., Ledermann, B., et al. (1996). Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. Journal of Cell Biology, 135(1), 215–225.
Kirchhof, P., Fabritz, L., Zwiener, M., et al. (2006). Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation, 114(17), 1799–1806.
Fabritz, L., Hoogendijk, M. G., Scicluna, B. P., et al. (2011). Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. Journal of the American College of Cardiology, 57(6), 740–750.
Li, D., Liu, Y., Maruyama, M., et al. (2011). Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Human Molecular Genetics, 20(23), 4582–4596.
Li, J., Swope, D., Raess, N., Cheng, L., Muller, E. J., & Radice, G. L. (2011). Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of {beta}-catenin signaling. Molecular and Cellular Biology, 31(6), 1134–1144.
Pilichou, K., Remme, C. A., Basso, C., et al. (2009). Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. Journal of Experimental Medicine, 206(8), 1787–1802.
Rizzo, S., Lodder, E. M., Verkerk, A. O., et al. (2012). Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovascular Research, 95(4), 409–418.
Krusche, C. A., Holthofer, B., Hofe, V., et al. (2011). Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Research in Cardiology, 106(4), 617–633.
Kant, S., Krull, P., Eisner, S., Leube, R. E., & Krusche, C. A. (2012). Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell and Tissue Research, 348(2), 249–259.
Kant, S., Freytag, B., Herzog, A., et al. (2019). Desmoglein 2 mutation provokes skeletal muscle actin expression and accumulation at intercalated discs in murine hearts. Journal of Cell Science, 132(5), jcs199612.
Calore, M., Lorenzon, A., Vitiello, L., et al. (2019). A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of Wnt signalling and miRNA dysregulation. Cardiovascular Research, 115(4), 739–751.
Lin, Y. N., Mesquita, T., Sanchez, L., et al. (2021). Extracellular vesicles from immortalized cardiosphere-derived cells attenuate arrhythmogenic cardiomyopathy in desmoglein-2 mutant mice. European Heart Journal, 42(35), 3558–3571.
Garcia-Gras, E., Lombardi, R., Giocondo, M. J., et al. (2006). Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. The Journal of Clinical Investigation, 116(7), 2012–2021.
Yang, Z., Bowles, N. E., Scherer, S. E., et al. (2006). Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation Research, 99(6), 646–655.
Lyon, R. C., Mezzano, V., Wright, A. T., et al. (2014). Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Human Molecular Genetics, 23(5), 1134–1150.
Martherus, R., Jain, R., Takagi, K., et al. (2016). Accelerated cardiac remodeling in desmoplakin transgenic mice in response to endurance exercise is associated with perturbed Wnt/beta-catenin signaling. American Journal of Physiology. Heart and Circulatory Physiology, 310(2), H174-187.
Hamada, Y., Yamamoto, T., Nakamura, Y., et al. (2020). G790del mutation in DSC2 alone is insufficient to develop the pathogenesis of ARVC in a mouse model. Biochemistry and Biophysics Reports, 21, 100711.
Brodehl, A., Belke, D. D., Garnett, L., et al. (2017). Transgenic mice overexpressing desmocollin-2 (DSC2) develop cardiomyopathy associated with myocardial inflammation and fibrotic remodeling. PLoS ONE, 12(3), e0174019.
Haghighi, K., Kolokathis, F., Gramolini, A. O., et al. (2006). A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America, 103(5), 1388–1393.
Eijgenraam, T. R., Boukens, B. J., Boogerd, C. J., et al. (2020). The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy. Scientific Reports, 10(1), 9819.
Raad, N., Bittihn, P., Cacheux, M., et al. (2021). Arrhythmia mechanism and dynamics in a humanized mouse model of inherited cardiomyopathy caused by phospholamban R14del mutation. Circulation, 144(6), 441–454.
Ren, J., Tsilafakis, K., Chen, L., et al. (2021). Crosstalk between coagulation and complement activation promotes cardiac dysfunction in arrhythmogenic right ventricular cardiomyopathy. Theranostics., 11(12), 5939–5954.
Wang, Y., Li, C., Shi, L., et al. (2020). Integrin beta1D deficiency-mediated RyR2 dysfunction contributes to catecholamine-sensitive ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy. Circulation, 141(18), 1477–1493.
Padron-Barthe, L., Villalba-Orero, M., Gomez-Salinero, J. M., et al. (2019). Severe cardiac dysfunction and death caused by arrhythmogenic right ventricular cardiomyopathy type 5 are improved by inhibition of glycogen synthase kinase-3beta. Circulation, 140(14), 1188–1204.
Zheng, G., Jiang, C., Li, Y., et al. (2019). TMEM43-S358L mutation enhances NF-kappaB-TGFbeta signal cascade in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Protein & Cell, 10(2), 104–119.
Dominguez, F., Lalaguna, L., Lopez-Olaneta, M., et al. (2021). Early preventive treatment with Enalapril improves cardiac function and delays mortality in mice with arrhythmogenic right ventricular cardiomyopathy type 5. Circulation: Heart Failure, 14(9), e007616.
Ellawindy, A., Satoh, K., Sunamura, S., et al. (2015). Rho-kinase inhibition during early cardiac development causes arrhythmogenic right ventricular cardiomyopathy in mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(10), 2172–2184.
Asano, Y., Takashima, S., Asakura, M., et al. (2004). Lamr1 functional retroposon causes right ventricular dysplasia in mice. Nature Genetics, 36(2), 123–130.
Ding, Y., Yang, J., Chen, P., et al. (2020). Knockout of SORBS2 protein disrupts the structural integrity of intercalated disc and manifests features of arrhythmogenic cardiomyopathy. Journal of the American Heart Association, 9(17), e017055.
Cheedipudi, S. M., Hu, J., Fan, S., et al. (2020). Exercise restores dysregulated gene expression in a mouse model of arrhythmogenic cardiomyopathy. Cardiovascular Research, 116(6), 1199–1213.
Liang, Y., Lyon, R. C., Pellman, J., et al. (2021). Desmosomal COP9 regulates proteome degradation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Journal of Clinical Investigation, 131(11), e137689.
Heuser, A., Plovie, E. R., Ellinor, P. T., et al. (2006). Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. American Journal of Human Genetics, 79(6), 1081–1088.
Moriarty, M. A., Ryan, R., Lalor, P., Dockery, P., Byrnes, L., & Grealy, M. (2012). Loss of plakophilin 2 disrupts heart development in zebrafish. International Journal of Developmental Biology, 56(9), 711–718.
Giuliodori, A., Beffagna, G., Marchetto, G., et al. (2018). Loss of cardiac Wnt/beta-catenin signalling in desmoplakin-deficient AC8 zebrafish models is rescuable by genetic and pharmacological intervention. Cardiovascular Research, 114(8), 1082–1097.
Moreau, A., Reisqs, J. B., Delanoe-Ayari, H., et al. (2021). Deciphering DSC2 arrhythmogenic cardiomyopathy electrical instability: From ion channels to ECG and tailored drug therapy. Clinical and Translational Medicine, 11(3), e319.
Delmar, M., & McKenna, W. J. (2010). The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circulation Research, 107(6), 700–714.
Sato, P. Y., Musa, H., Coombs, W., et al. (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circulation Research, 105(6), 523–526.
Grossmann, K. S., Grund, C., Huelsken, J., et al. (2004). Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. Journal of Cell Biology, 167(1), 149–160.
Haq, S., Michael, A., Andreucci, M., et al. (2003). Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci U S A., 100(8), 4610–4615.
Swope, D., Li, J., Muller, E. J., & Radice, G. L. (2012). Analysis of a Jup hypomorphic allele reveals a critical threshold for postnatal viability. Genesis, 50(10), 717–727.
Cason, M., Celeghin, R., Marinas, M. B., et al. (2021). Novel pathogenic role for galectin-3 in early disease stages of arrhythmogenic cardiomyopathy. Heart Rhythm, 18(8), 1394–1403.
Schinner, C., Erber, B. M., & Yeruva, S. (2020). Stabilization of desmoglein-2 binding rescues arrhythmia in arrhythmogenic cardiomyopathy. JCI Insight, 5(9), e130141.
Norman, M., Simpson, M., Mogensen, J., et al. (2005). Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation, 112(5), 636–642.
Te Riele, A. S., Bhonsale, A., Burt, J. R., Zimmerman, S. L., & Tandri, H. (2012). Genotype-specific pattern of LV involvement in ARVD/C. JACC: Cardiovascular Imaging, 5(8), 849–851.
Bueno Marinas, M., Celeghin, R., Cason, M., Thiene, G., Basso, C., & Pilichou, K. (2020). The role of MicroRNAs in arrhythmogenic cardiomyopathy: Biomarkers or innocent bystanders of disease progression? International Journal of Molecular Science, 21(17), 6434.
Mazurek, S. R., Calway, T., Harmon, C., Farrell, P., & Kim, G. H. (2017). MicroRNA-130a regulation of desmocollin 2 in a novel model of arrhythmogenic cardiomyopathy. Microrna, 6(2), 143–150.
MacLennan, D. H., & Kranias, E. G. (2003). Phospholamban: A crucial regulator of cardiac contractility. Nature Reviews Molecular Cell Biology, 4(7), 566–577.
Luo, W., Wolska, B. M., Grupp, I. L., et al. (1996). Phospholamban gene dosage effects in the mammalian heart. Circulation Research, 78(5), 839–847.
Kadambi, V. J., Ponniah, S., Harrer, J. M., et al. (1996). Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. The Journal of Clinical Investigation, 97(2), 533–539.
Haghighi, K., Pritchard, T., Bossuyt, J., et al. (2012). The human phospholamban Arg14-deletion mutant localizes to plasma membrane and interacts with the Na/K-ATPase. Journal of Molecular and Cellular Cardiology, 52(3), 773–782.
Baskin, B., Skinner, J. R., Sanatani, S., et al. (2013). TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Human Genetics, 132(11), 1245–1252.
Christensen, A. H., Andersen, C. B., Tybjaerg-Hansen, A., Haunso, S., & Svendsen, J. H. (2011). Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clinical Genetics, 80(3), 256–264.
Haywood, A. F., Merner, N. D., Hodgkinson, K. A., et al. (2013). Recurrent missense mutations in TMEM43 (ARVD5) due to founder effects cause arrhythmogenic cardiomyopathies in the UK and Canada. European Heart Journal, 34(13), 1002–1011.
Milting, H., Klauke, B., Christensen, A. H., et al. (2015). The TMEM43 Newfoundland mutation p.S358L causing ARVC-5 was imported from Europe and increases the stiffness of the cell nucleus. European Heart Journal, 36(14), 872–881.
Mavroidis, M., Davos, C. H., Psarras, S., et al. (2015). Complement system modulation as a target for treatment of arrhythmogenic cardiomyopathy. Basic Research in Cardiology, 110(3), 27.
Costa, S., Gasperetti, A., Medeiros-Domingo, A., et al. (2020). Familial arrhythmogenic cardiomyopathy: Clinical determinants of phenotype discordance and the impact of endurance sports. Journal of Clinical Medicine, 9(11), 3781.
Goff, Z. D., & Calkins, H. (2019). Sudden death related cardiomyopathies - Arrhythmogenic right ventricular cardiomyopathy, arrhythmogenic cardiomyopathy, and exercise-induced cardiomyopathy. Progress in Cardiovascular Diseases, 62(3), 217–226.
Austin, K. M., Trembley, M. A., Chandler, S. F., et al. (2019). Molecular mechanisms of arrhythmogenic cardiomyopathy. Nature Reviews Cardiology, 16(9), 519–537.
Basso, C., Thiene, G., Corrado, D., Angelini, A., Nava, A., & Valente, M. (1996). Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation, 94(5), 983–991.
Asimaki, A., Tandri, H., Duffy, E. R., et al. (2011). Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circulation: Arrhythmia and Electrophysiology, 4(5), 743–752.
Haugaa, K. H., Haland, T. F., Leren, I. S., Saberniak, J., & Edvardsen, T. (2016). Arrhythmogenic right ventricular cardiomyopathy, clinical manifestations, and diagnosis. Europace, 18(7), 965–972.
Chelko, S. P., Asimaki, A., Lowenthal, J., et al. (2019). Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation, 140(18), 1491–1505.
Chelko, S. P., Asimaki, A., Andersen, P., et al. (2016). Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight, 1(5), e85923.
Zhang, H., Liu, S., Dong, T., et al. (2016). Profiling of differentially expressed microRNAs in arrhythmogenic right ventricular cardiomyopathy. Science and Reports, 6, 28101.
Gurha, P., Chen, X., Lombardi, R., Willerson, J. T., & Marian, A. J. (2016). Knockdown of plakophilin 2 downregulates miR-184 through CpG hypermethylation and suppression of the E2F1 pathway and leads to enhanced adipogenesis in vitro. Circulation Research, 119(6), 731–750.
Akdis, D., Medeiros-Domingo, A., Gaertner-Rommel, A., et al. (2016). Myocardial expression profiles of candidate molecules in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia compared to those with dilated cardiomyopathy and healthy controls. Heart Rhythm, 13(3), 731–741.
Ross, S. E., Hemati, N., Longo, K. A., et al. (2000). Inhibition of adipogenesis by Wnt signaling. Science, 289(5481), 950–953.
Chen, S., Guttridge, D. C., You, Z., et al. (2001). Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. Journal of Cell Biology, 152(1), 87–96.
Longo, K. A., Kennell, J. A., Ochocinska, M. J., Ross, S. E., Wright, W. S., & MacDougald, O. A. (2002). Wnt signaling protects 3T3-L1 preadipocytes from apoptosis through induction of insulin-like growth factors. Journal of Biological Chemistry, 277(41), 38239–38244.
Chen, S. N., Gurha, P., Lombardi, R., Ruggiero, A., Willerson, J. T., & Marian, A. J. (2014). The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circulation Research, 114(3), 454–468.
Kim, C., Wong, J., Wen, J., et al. (2013). Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature, 494(7435), 105–110.
Sommariva, E., Stadiotti, I., Casella, M., et al. (2021). Oxidized LDL-dependent pathway as new pathogenic trigger in arrhythmogenic cardiomyopathy. EMBO Molecular Medicine, 13(9), e14365.
Padron-Barthe, L., Dominguez, F., Garcia-Pavia, P., & Lara-Pezzi, E. (2017). Animal models of arrhythmogenic right ventricular cardiomyopathy: What have we learned and where do we go? Insight for therapeutics. Basic Research in Cardiology, 112(5), 50.
Sen-Chowdhry, S., Syrris, P., & McKenna, W. J. (2005). Genetics of right ventricular cardiomyopathy. Journal of Cardiovascular Electrophysiology, 16(8), 927–935.
Asimaki, A., Kapoor, S., Plovie, E., et al. (2014). Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Science Translational Medicine, 6(240), 240ra274.
Cerrone, M., Lin, X., Zhang, M., et al. (2014). Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation, 129(10), 1092–1103.
Zhang, Q., Deng, C., Rao, F., et al. (2013). Silencing of desmoplakin decreases connexin43/Nav1.5 expression and sodium current in HL1 cardiomyocytes. Molecular Medicine Reports, 8(3), 780–786.
Gerull, B., & Brodehl, A. (2020). Genetic animal models for arrhythmogenic cardiomyopathy. Frontiers in Physiology, 11, 624.
Asimaki, A., Syrris, P., Wichter, T., Matthias, P., Saffitz, J. E., & McKenna, W. J. (2007). A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. American Journal of Human Genetics, 81(5), 964–973.
Christensen, A. H., Kamstrup, P. R., Gandjbakhch, E., et al. (2016). Plakophilin-2 c.419C>T and risk of heart failure and arrhythmias in the general population. European Journal of Human Genetics, 24(5), 732–738.
Gerull, B., Kirchner, F., Chong, J. X., et al. (2013). Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circulation: Cardiovascular Genetics, 6(4), 327–336.
Chen, P., Li, Z., Yu, B., Ma, F., Li, X., & Wang, D. W. (2020). Distal myopathy induced arrhythmogenic right ventricular cardiomyopathy in a pedigree carrying novel DSG2 null variant. International Journal of Cardiology, 298, 25–31.
Khudiakov, A., Zaytseva, A., Perepelina, K., et al. (2020). Sodium current abnormalities and deregulation of Wnt/beta-catenin signaling in iPSC-derived cardiomyocytes generated from patient with arrhythmogenic cardiomyopathy harboring compound genetic variants in plakophilin 2 gene. Biochimica et Biophysica Acta, Molecular Basis of Disease, 1866(11), 165915.
Lin, X., Ma, Y., Cai, Z., et al. (2020). Next-generation sequencing identified novel Desmoplakin frame-shift variant in patients with Arrhythmogenic cardiomyopathy. BMC Cardiovascular Disorders, 20(1), 74.
Patel, D. M., Dubash, A. D., Kreitzer, G., & Green, K. J. (2014). Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin-EB1 interactions. Journal of Cell Biology, 206(6), 779–797.
Oxford, E. M., Musa, H., Maass, K., Coombs, W., Taffet, S. M., & Delmar, M. (2007). Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circulation Research, 101(7), 703–711.
Beffagna, G., De Bortoli, M., Nava, A., et al. (2007). Missense mutations in desmocollin-2 N-terminus, associated with arrhythmogenic right ventricular cardiomyopathy, affect intracellular localization of desmocollin-2 in vitro. BMC Medical Genetics, 8, 65.
Joshi-Mukherjee, R., Coombs, W., Musa, H., Oxford, E., Taffet, S., & Delmar, M. (2008). Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVC)-related plakophilin-2 (PKP2) mutations. Heart Rhythm, 5(12), 1715–1723.
Wei, Y. J., Huang, Y. X., Shen, Y., et al. (2009). Proteomic analysis reveals significant elevation of heat shock protein 70 in patients with chronic heart failure due to arrhythmogenic right ventricular cardiomyopathy. Molecular and Cellular Biochemistry, 332(1–2), 103–111.
Matthes, S. A., Taffet, S., & Delmar, M. (2011). Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Communication & Adhesion, 18(4), 73–84.
Dalton, A. C., & Barton, W. A. (2014). Over-expression of secreted proteins from mammalian cell lines. Protein Science, 23(5), 517–525.
Swiech, K., Picanco-Castro, V., & Covas, D. T. (2012). Human cells: New platform for recombinant therapeutic protein production. Protein Expression and Purification, 84(1), 147–153.
Dumont, J., Euwart, D., Mei, B., Estes, S., & Kshirsagar, R. (2016). Human cell lines for biopharmaceutical manufacturing: History, status, and future perspectives. Critical Reviews in Biotechnology, 36(6), 1110–1122.
Roman, R., Miret, J., Scalia, F., Casablancas, A., Lecina, M., & Cairo, J. J. (2016). Enhancing heterologous protein expression and secretion in HEK293 cells by means of combination of CMV promoter and IFNalpha2 signal peptide. Journal of Biotechnology, 239, 57–60.
Te Riele, A. S., Agullo-Pascual, E., James, C. A., et al. (2017). Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovascular Research, 113(1), 102–111.
Claycomb, W. C., Lanson, N. A., Jr., Stallworth, B. S., et al. (1998). HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proceedings of the National Academy of Sciences of the United States of America, 95(6), 2979–2984.
Fidler, L. M., Wilson, G. J., Liu, F., et al. (2009). Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. Journal of Cellular and Molecular Medicine, 13(10), 4219–4228.
Pourrier, M., & Fedida, D. (2020). The emergence of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-hiPSCCMs) as a platform to model arrhythmogenic diseases. International Journal of Molecular Sciences, 21(2), 657.
Itzhaki, I., Maizels, L., Huber, I., et al. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature, 471(7337), 225–229.
Wei, H., Wu, J., & Liu, Z. (2018). Studying KCNQ1 mutation and drug response in type 1 long QT syndrome using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Methods in Molecular Biology, 1684, 7–28.
Itzhaki, I., Maizels, L., Huber, I., et al. (2012). Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. Journal of the American College of Cardiology, 60(11), 990–1000.
Davis, R. P., Casini, S., van den Berg, C. W., et al. (2012). Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation, 125(25), 3079–3091.
Liang, P., Sallam, K., Wu, H., et al. (2016). Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of brugada syndrome. Journal of the American College of Cardiology, 68(19), 2086–2096.
Fan, X., Yang, G., Kowitz, J., et al. (2021). Preclinical short QT syndrome models: Studying the phenotype and drug-screening. Europace, 24(3), 481–493.
El-Battrawy, I., Albers, S., Cyganek, L., et al. (2019). A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. Europace, 21(9), 1410–1421.
El-Battrawy, I., Lan, H., Cyganek, L., et al. (2018). Modeling short QT syndrome using human-induced pluripotent stem cell- derived cardiomyocytes. Journal of the American Heart Association, 7(7), e007394.
Huang, M., Fan, X., Yang, Z., et al. (2021). Alpha 1-adrenoceptor signalling contributes to toxic effects of catecholamine on electrical properties in cardiomyocytes. Europace, 23(7), 1137–1148.
El-Battrawy, I., Zhao, Z., Lan, H., et al. (2018). Ion channel dysfunctions in dilated cardiomyopathy in limb-girdle muscular dystrophy. Circulation: Genomic and Precision Medicine, 11(3), e001893.
Fan, X., Yang, G., Kowitz, J., Akin, I., Zhou, X., & El-Battrawy, I. (2022). Takotsubo syndrome: Translational implications and pathomechanisms. International Journal of Molecular Sciences, 23(4), 1951.
Schwartz, P. J., Gnecchi, M., Dagradi, F., et al. (2019). From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome Type 2. European Heart Journal, 40(23), 1832–1836.
Hawthorne, R. N., Blazeski, A., Lowenthal, J., et al. (2021). Altered electrical, biomolecular, and immunologic phenotypes in a novel patient-derived stem cell model of desmoglein-2 mutant ARVC. Journal of Clinical Medicine, 10(14), 3061.
Caspi, O., Huber, I., Gepstein, A., et al. (2013). Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circulation: Cardiovascular Genetics, 6(6), 557–568.
Ma, D., Wei, H., Lu, J., et al. (2013). Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. European Heart Journal, 34(15), 1122–1133.
El-Battrawy, I., Zhao, Z., Lan, H., et al. (2018). Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace, 20(FI1), f46–f56.
Gusev, K., Khudiakov, A., Zaytseva, A., et al. (2020). Impact of the DSP-H1684R genetic variant on ion channels activity in iPSC-derived cardiomyocytes. Cellular Physiology and Biochemistry, 54(4), 696–706.
Chen, P., Xiao, Y., Wang, Y., et al. (2020). Intracellular calcium current disorder and disease phenotype in OBSCN mutant iPSC-based cardiomyocytes in arrhythmogenic right ventricular cardiomyopathy. Theranostics, 10(24), 11215–11229.
Wen, J. Y., Wei, C. Y., Shah, K., Wong, J., Wang, C., & Chen, H. S. (2015). Maturation-based model of arrhythmogenic right ventricular dysplasia using patient-specific induced pluripotent stem cells. Circulation Journal, 79(7), 1402–1408.
Yazawa, M., Hsueh, B., Jia, X., et al. (2011). Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature, 471(7337), 230–234.
Egashira, T., Yuasa, S., Suzuki, T., et al. (2012). Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovascular Research, 95(4), 419–429.
Moretti, A., Bellin, M., Welling, A., et al. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. New England Journal of Medicine, 363(15), 1397–1409.
Ross, S. B., Fraser, S. T., & Semsarian, C. (2016). Induced pluripotent stem cells in the inherited cardiomyopathies: From disease mechanisms to novel therapies. Trends in Cardiovascular Medicine, 26(8), 663–672.
Costa, S., Saguner, A. M., Gasperetti, A., Akdis, D., Brunckhorst, C., & Duru, F. (2021). The link between sex hormones and susceptibility to cardiac arrhythmias: From molecular basis to clinical implications. Frontiers in Cardiovascular Medicine, 8, 644279.
Mauvais-Jarvis, F. (2011). Estrogen and androgen receptors: Regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends in Endocrinology and Metabolism, 22(1), 24–33.
Herring, M. J., Oskui, P. M., Hale, S. L., & Kloner, R. A. (2013). Testosterone and the cardiovascular system: A comprehensive review of the basic science literature. Journal of the American Heart Association, 2(4), e000271.
Yang, J., Samal, E., Burgos Angulo, M., Bertalovitz, A., & McDonald, T. V. (2021). Establishment of an arrhythmogenic right ventricular cardiomyopathy derived iPSC cell line (USFi004-A) carrying a heterozygous mutation in PKP2 (c.1799delA). Stem Cell Research, 54, 102398.
Xia, S., Wang, X., Yue, P., Li, Y., & Zhang, D. (2020). Establishment of induced pluripotent stem cell lines from a family of an ARVC patient receiving heart transplantation in infant age carrying compound heterozygous mutations in DSP gene. Stem Cell Research, 48, 101977.
Buljubasic, F., El-Battrawy, I., Lan, H., et al. (2020). Nucleoside diphosphate kinase B contributes to arrhythmogenesis in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with arrhythmogenic right ventricular cardiomyopathy. Journal of Clinical Medicine, 9(2), 486.
Parrotta, E. I., Procopio, A., Scalise, S., et al. (2021). Deciphering the role of wnt and rho signaling pathway in iPSC-derived ARVC cardiomyocytes by in silico mathematical modeling. International Journal of Molecular Sciences, 22(4), 2004.
Bondue, A., Arbustini, E., Bianco, A., et al. (2018). Complex roads from genotype to phenotype in dilated cardiomyopathy: Scientific update from the Working Group of Myocardial Function of the European Society of Cardiology. Cardiovascular Research, 114(10), 1287–1303.
Karbassi, E., Fenix, A., Marchiano, S., et al. (2020). Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nature Reviews Cardiology, 17(6), 341–359.
Sun, X., & Nunes, S. S. (2016). Biowire platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Methods, 101, 21–26.
Correia, C., Koshkin, A., Duarte, P., et al. (2017). Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Science and Reports, 7(1), 8590.
Dai, D. F., Danoviz, M. E., Wiczer, B., Laflamme, M. A., & Tian, R. (2017). Mitochondrial maturation in human pluripotent stem cell derived cardiomyocytes. Stem Cells International, 2017, 5153625.
Mills, R. J., Titmarsh, D. M., Koenig, X., et al. (2017). Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proceedings of the National Academy of Sciences of the United States of America, 114(40), E8372–E8381.
Bhute, V. J., Bao, X., Dunn, K. K., et al. (2017). Metabolomics identifies metabolic markers of maturation in human pluripotent stem cell-derived cardiomyocytes. Theranostics, 7(7), 2078–2091.
Liu, Y., Bai, H., Guo, F., et al. (2020). PGC-1alpha activator ZLN005 promotes maturation of cardiomyocytes derived from human embryonic stem cells. Aging (Albany NY), 12(8), 7411–7430.
Miki, K., Deguchi, K., Nakanishi-Koakutsu, M., et al. (2021). ERRgamma enhances cardiac maturation with T-tubule formation in human iPSC-derived cardiomyocytes. Nature Communications, 12(1), 3596.
Giacomelli, E., Meraviglia, V., Campostrini, G., et al. (2020). Human-iPSC-derived cardiac stromal cells enhance maturation in 3d cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell, 26(6), 862-879 e811.
Guo, F., Sun, Y., Wang, X., et al. (2019). Patient-specific and gene-corrected induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of short QT syndrome. Circulation Research, 124(1), 66–78.
Joung, J. K., & Sander, J. D. (2013). TALENs: A widely applicable technology for targeted genome editing. Nature Reviews Molecular Cell Biology, 14(1), 49–55.
Nemudryi, A. A., Valetdinova, K. R., Medvedev, S. P., & Zakian, S. M. (2014). TALEN and CRISPR/Cas genome editing systems: Tools of discovery. Acta Naturae, 6(3), 19–40.
Shah, S. Z., Rehman, A., Nasir, H., et al. (2019). Advances In research on genome editing Crispr-Cas9 technology. Journal of Ayub Medical College, Abbottabad, 31(1), 108–122.
Zhang, F., Wen, Y., & Guo, X. (2014). CRISPR/Cas9 for genome editing: Progress, implications and challenges. Human Molecular Genetics, 23(R1), R40-46.
Broughton, K. M., & Sussman, M. A. (2016). Empowering adult stem cells for myocardial regeneration V2.0: Success in small steps. Circulation Research, 118(5), 867–880.
Acknowledgements
We thank the Chinese Scholarship Council (CSC) for the financial support for Xuehui Fan.
Funding
Open Access funding enabled and organized by Projekt DEAL. We would like to acknowledge funding support from the National Nature Science Foundation of China (82200361), Natural Science Foundation of Sichuan Province (No. 22NSFSC3053), Foundation of the Education Department of Sichuan Province (No.17ZB0476), the Foundation of Office of Science & Technology of Luzhou City (No. 2021-JYJ-70) and Southwest Medical University Scientific Research Fund (No. 2021ZKMS035).
Author information
Authors and Affiliations
Contributions
Conceptualization, I.A., X.Z., A.M., I.B.; Writing-Original Draft Preparation, X.F., G.Y.; Writing-review and editing, F.D.,M.G., X.Z.; Visualization, X.F., G.Y.; Funding Acquisition, X.F.; All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable
Consent to Participate
Not applicable
Consent for Publication
Not applicable
Competing Interests
All authors disclosed no relevant relationships.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, X., Yang, G., Duru, F. et al. Arrhythmogenic Cardiomyopathy: from Preclinical Models to Genotype–phenotype Correlation and Pathophysiology. Stem Cell Rev and Rep 19, 2683–2708 (2023). https://doi.org/10.1007/s12015-023-10615-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-023-10615-0