
Abstract
Pseudomonas aeruginosa is an opportunistic pathogen of immunocompromised hosts. In cystic fibrosis (CF), P. aeruginosa causes acute and chronic lung infections that result in significant morbidity and mortality. P. aeruginosa possesses several traits that contribute to its ability to colonize and persist in acute and chronic infections. These include high resistance to antimicrobials, ability to form biofilms, plethora of virulence products, and metabolic versatility. In P. aeruginosa, a cell-to-cell communication process termed quorum sensing (QS) regulates many of these factors that contribute to its pathogenesis. Recent evidence suggests that the CF lung environment presents a specialized niche for P. aeruginosa. The relationship of P. aeruginosa QS, biofilm formation, and the CF lung environment is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pseudomonas aeruginosa is a ubiquitous Gram-negative microorganism found in many environments, such as soil and water. P. aeruginosa is also an opportunistic pathogen implicated in respiratory infections, urinary tract infections, gastrointestinal infections, keratitis, otitis media, and bacteremia. P. aeruginosa is found in an estimated 10–20% of all hospital-acquired infections [1]. Patients with compromised host defenses, such as those infected with human immunodeficiency virus, burn patients, or those with cystic fibrosis (CF), are susceptible to P. aeruginosa infections. These infections are often difficult to treat using conventional antibiotic therapies.
P. aeruginosa possesses a large genetic diversity that contributes to its ability to persist in the environment and to its pathogenesis. P. aeruginosa is able to metabolize various carbon and nitrogen sources and can grow in either an aerobic or anaerobic environment using several terminal electron acceptors. P. aeruginosa is intrinsically resistant to many antimicrobials, in part due to its multiple efflux systems. P. aeruginosa also produces numerous virulence factors, including secreted factors, such as elastase, proteases, phospholipase C, hydrogen cyanide, exotoxin A, and exoenzyme S, as well as cell-associated factors, such as lipopolysaccharide, flagella, and pili.
In P. aeruginosa, expression, production, and/or secretion of many virulence factors are controlled in a cell density-dependent manner known as quorum sensing (QS). Many bacteria, including P. aeruginosa, use QS to coordinate population behavior by producing, sensing, and responding to diffusible signaling molecules, termed autoinducers. Two complete QS systems, the las and rhl systems, have been described and well-studied in P. aeruginosa. These systems consist of the transcriptional regulatory proteins LasR (in the las system) and RhlR (in the rhl system), or R-proteins, and their cognate autoinducer signal molecules N-(3-oxododecanoyl) homoserine lactone (3O-C12-HSL) in the las system and N-butyryl homoserine lactone (C4-HSL) in the rhl system. Each R-protein possesses an autoinducer-binding domain as well as a deoxyribonucleic acid (DNA)-binding domain. The binding of the R-protein to its cognate autoinducer molecule produces a R-protein–autoinducer complex that modulates target gene expression, presumably by binding to conserved DNA elements termed las boxes located upstream of the translational start site of QS-regulated genes [2]. The las and rhl systems are not independent and are intertwined in a hierarchical manner, with the las system exerting control over the rhl system [3, 4]. P. aeruginosa also produces a third signal molecule, 2-heptyl-3-hydroxy-4-quinolone or the Pseudomonas quinolone signal (PQS), that links the las and rhl systems [5, 6]. Incredibly, transcriptome analyses have determined that between 6% and 10% of the P. aeruginosa genome is regulated by the las and/or rhl systems [7–9].
P. aeruginosa also readily forms communities of cells encased in an extracellular matrix, consisting of secreted proteins, polysaccharides, nucleic acids, and cellular debris, attached to abiotic or biotic surfaces [10]. These communities, known as biofilms, are of clinical relevance as the awareness of the prevalence of biofilm-centered infections increases. Classic examples include biofilm-mediated infections that occur on medical devices such as intravascular catheters, urinary catheters, orthopedic devices, and dialysis machines [11, 12]. More recently, there is evidence that chronic infections, such as recurrent ear infections (otitis media) and lung infections in CF patients, are caused by biofilm-dwelling bacteria [13–15]. The CF lung provides a particularly attractive environment for P. aeruginosa colonization and chronic lung infection and is the major causative agent of morbidity and mortality in CF patients [16]. Biofilms are now thought to be involved in 65–80% of all microbial infections [17, 18].
Biofilm growth is thought to offer a competitive advantage versus free-floating or planktonic growth. Bacteria in biofilms are more recalcitrant to disruption or killing by surfactants, biocides, grazing predators, and host defenses [12]. It is important to note that the biofilm mode of growth is often associated with antimicrobial resistance [12]. Studies indicate that bacteria growing in biofilms are up to 1,000 times more resistant to antimicrobial challenge versus planktonically grown cells [12]. As the knowledge of the importance of biofilms in infection increases, perception has altered research seeking to discover new therapies to treat these infections by necessitating an understanding of the mechanisms that govern biofilm development and its role in bacterial pathogenesis.
P. aeruginosa biofilm development and its relation to quorum sensing
P. aeruginosa biofilm formation has been divided into various stages, including bacterial attachment and irreversible adhesion to a substratum, microcolony development, maturation of the biofilm demonstrated by the appearance of large structures resembling mushrooms and stalks with water channels, and dispersion of bacterial cells from the biofilm (Fig. 1a, b) [19]. Heterogeneous microenvironments have been demonstrated to exist within biofilms due to oxygen and nutrient diffusional limitations [20], and recent evidence suggests that bacteria grow slowly within the biofilm [17]. The biofilm is also physiologically heterogeneous since each bacterium within the biofilm matrix experiences differing nutrient gradients. Bacteria within the biofilm are thought to possess a unique phenotype quite different from their planktonic counterparts [17].

a Model of P. aeruginosa biofilm development in a flow-through biofilm system. Bacteria are thought to attach to a surface reversibly (I), followed by irreversible attachment (II). Microcolony formation occurs (III), and complex three-dimensional structures know as stalks and towers form as the biofilm matures (IV). Dispersion or detachment of bacteria follows, which can occur by the separation of large pieces of encased bacteria or by a process in which bacteria become motile and tunnel out of the matrix (V). Bacteria are represented by gray ovals, with or without flagella, and the biofilm matrix in orange. b P. aeruginosa PAO1 expressing green fluorescent protein was grown for 4 days in a minimal media (modified FAB). A two-dimensional projection of the three-dimensional image is shown as a top-down view for 2 h (I), 1 day (II), 2 days (III), 3 days (IV), and 4 days (V) after inoculation. Maximal thickness of the biofilm is reported for each time period. Biofilm images were collected by a Leica confocal scanning light microscopy using a 488-nm wavelength. Images are at ×400 and represent a 250 × 250-μm field
The belief that biofilm development results from an organized set of molecular events has led to several studies to understand the mechanisms involved. The idea that biofilm development is a coordinated behavior was strengthened by evidence of a link between P. aeruginosa QS and biofilm development [21]. This study determined that lasI, which encodes for the synthetase responsible for 3O-C12-HSL production, is critical for mature biofilm formation in P. aeruginosa PAO1 [21]. Mutants lacking the lasI gene formed flat, thin biofilms with little of the dramatic three-dimensional structures which typify P. aeruginosa biofilms grown in a flow-through model system. It is exciting to note that the mutant was also more susceptible to removal by the surfactant sodium dodecyl sulfate (SDS). Further studies have demonstrated that QS mutant biofilms are also more susceptible to antimicrobial challenge and phagocytosis by polymorphonuclear leukocytes [7, 22]. This suggests that QS is not only important in the development of P. aeruginosa biofilms but also in its protective role. For example, the QS regulations of catalase and superoxide dismutase have been demonstrated to be important in P. aeruginosa PAO1 biofilm resistance to hydrogen peroxide [23]. Additionally, QS inhibitors, including a halogenated furanone synthesized by the red algae Delisea pulchra and several derivatives of this molecule, have been shown to not only interfere with P. aeruginosa PAO1 biofilm development but to enhance sensitivity to antimicrobial challenge with tobramycin or azithromycin, presumably by interfering with QS signaling [10].
Both flagellar and twitching motility have been demonstrated to be involved in P. aeruginosa attachment to surfaces and subsequent biofilm formation [24–26]. In P. aeruginosa, twitching motility is mediated by type IV pilin, and mutants in this pilin (ΔpilA) were unable to form the elaborate mushroom structures typical of P. aeruginosa biofilms. LecB, or the type II P. aeruginosa lectin, is also involved in pilus biogenesis [27] and has been reported to be involved in biofilm development [28]. Mutants in lecB formed weak biofilms as compared to the wild-type P. aeruginosa. LecB is an important virulence determinant in P. aeruginosa infections and has been shown in vitro to decrease the ciliary beat frequency of the airway epithelium [28]. Mutations in lecA, or the type I P. aeruginosa lectin, resulted in thin biofilms as compared to the wild-type P. aeruginosa [29]. It is interesting to note that a recent study reported that LecA binds to 3O-C12-HSL [30]. Genes involved in type IV biogenesis and LecA and LecB production are regulated by QS [9, 31].
Genetic analysis has identified several factors that are important for normal P. aeruginosa biofilm development. These include genes that encode structural components of the biofilm matrix. Overproduction of alginate, an exopolysaccharide, is the hallmark of the conversion of P. aeruginosa to a mucoid phenotype. The appearance of mucoid P. aeruginosa in CF infection is usually associated with chronic infection and poor clinical outcome [32, 33]. In mucoid P. aeruginosa, alginate is a matrix component in biofilms, and alginate-overproducing bacteria form complex-structured biofilms that are highly resistant to antimicrobials such as tobramycin [34]. Alginate also plays a protective role in P. aerugniosa infection and has been demonstrated to scavenge free radicals released by activated macrophages in vitro, to prevent phagocytic clearance, and to protect from defensins [35]. The regulation of alginate production has been well studied in P. aeruginosa [35]. However, in nonmucoid bacteria such as P. aeruginosa PAO1 and PA14, alginate is not the primary component of the biofilm matrix [36]. Instead, two loci, the psl (polysaccharide synthesis locus) in P. aeruginosa PAO1 and pel genes in P. aeruginosa PA14 and PAK [36], have been identified in in vitro assays and shown to be involved in the production of alternative polysaccharides. In PA14 and PAK, the pel genes produce a glucose-rich polysaccharide that is critical for pellicle formation and involved in biofilm development [36]. In P. aeruginosa PAO1, the psl genes have been shown to be important in the initiation and development of biofilms on both abiotic and biotic surfaces, such as mucin-coated surfaces and airway epithelial cells [36]. There is evidence that the psl and pel genes are regulated by the two-component system GacA/GacS and the small regulatory ribonucleic acid rsmZ [36]. The GacA/GacS system also positively regulates QS in P. aeruginosa PAO1 [37]. It is interesting to note that the gene pslB (PA2232) was demonstrated to be QS regulated in the P. aeruginosa PAO1 microarray analysis, suggesting there is a link between QS and the expression of the psl locus [9]. In P. aeruginosa PA14, the pel genes have also been shown to be QS regulated [38]. The role of these novel matrix components in nonmucoid biofilm development is of interest as nonmucoid P. aeruginosa initially colonizes the CF lung, most likely forming biofilms, followed by a conversion to mucoidy and chronic infection [35]. Although the PSL and PEL polysaccharides are now known to be an important part of the extracellular matrix, alginate may still play a role in the nonmucoid biofilm phenotype. For example, recent experiments have demonstrated that alginate is critical for the protection of P. aeruginosa PAO1, PA14, and FRD1 (a muciod P. aeruginosa isolate) biofilms to gamma-interferon-mediated macrophage killing in an in vitro assay [39]. There is also evidence that nonmucoid strains produce alginate in vitro under hypoxic conditions, such as those that are thought to exist in the CF lung and in a mouse model of acute pneumonia [35].
Extracellular DNA is also a major component of the biofilm matrix and has been shown to be important in the initial and early development of P. aeruginosa biofilms [40, 41]. Exposure of P. aeruginosa to DNaseI prevented biofilm development in a flow-through model system and dissolved young biofilms [41]. Further research with QS mutants in lasI and rhlI, the genes that direct the synthesis of the autoinducers 3O-C12-HSL and C4-HSL, respectively, determined that QS is important in regulating extracellular DNA release during both planktonic and biofilm growth [42]. Furthermore, PQS, the third QS autoinducer signal, was linked to extracellular DNA release during biofilm development [42]. The extracellular DNA has been localized within the biofilm structure and varies temporally as well as spatially. Early biofilms show large amounts of extracellular DNA on the substratum and coating the microcolonies, while in a mature biofilm, it is present primarily in the stalks of mushroom structures [42]. The source of extracellular DNA has been proposed to be due to either prophage-induced cell lysis or the release of membrane vesicles [42]. Previous work has demonstrated that Pf-1 phage is produced by P. aeruginosa PAO1 biofilms [40, 43], and the genes that encode for the Pf-1 phage in P. aeruginosa PAO1 have been demonstrated to be QS regulated during planktonic growth [9]. PQS has also been demonstrated to be involved in membrane vesicle formation in P. aeruginosa [44]. It is important to note that biofilms that are deficient in extracellular DNA have been shown to be more sensitive to SDS [42]. This suggests that DNase I treatment, such as the current CF treatment regime of inhaled, nebulized recombinant human DNase I, could be important in preventing the establishment of biofilms that are involved in chronic infection.
Several studies have reported that rhlA, one of the genes required for rhamnolipid biosynthesis, is important in initial P. aeruginosa biofilm development, maturation, and dispersion. Rhamnolipid is well known to be regulated by QS, where the RhlR–C4-HSL complex activates the transcription of rhlA [45]. A recent study by Pamp and Tolker-Nielsen [46] demonstrated that rhamnolipid is essential in the formation of the cap of the mushroom structure in mature biofilms. The study also suggested that rhlA is central in microcolony formation, as rhlA mutants formed thin, flat biofilms [46]. The deletion of rhlA results in thick biofilms with small or nonexistent water channels, suggesting that rhamnolipid is involved in the maintenance of these channels and/or dispersion of bacteria from the biofilm matrix [47–49]. Rhamnolipid has been demonstrated to be required for the dispersion of bacteria from the center of mushroom structures, where bacteria “tunnel” from the center of the macrocolony, and rhlA expression is localized to the center of macrocolonies about to undergo dispersion [47–49]. Bacteria that disperse from the biofilm often have altered phenotypes and are known as colony-morphotypic variants [50, 51]. Both P. aeruginosa PAO1 and clinical CF isolates have been shown to follow a pattern of biofilm development that includes dispersion, which is dependent upon QS and the presence of Pf-1 phage [50, 51]. Small-colony variants (SCVs) of P. aeruginosa PAO1 have been reported to display increased biofilm-forming capability, increased antimicrobial resistance, and enhanced dissemination [51], and SCVs have been isolated from CF patients [1]. It is interesting to note that more diverse morphotypes were reported from biofilm-dispersed CF isolates than PAO1. This may represent an in vivo selective adaptation process that maintains chronic infection.
Several genes encoding regulators have been identified that are important in P. aeruginosa biofilm development. These include gacA, the global carbon metabolism regulator crc, the stationary phase regulator rpoS, the three-component regulatory circuit sadARS, the response regulator for alginate production algR, and a hybrid sensor kinase/response regulator retS [52–57]. Mutations in gacA and crc caused defects in mircocolony formation, resulting in thin biofilms. As described previously, the GacA/GacS/rsmZ pathway is involved in the regulation of the extracellular matrix components psl and pel, which among its other influences may contribute to its regulation of normal biofilm development. Defects in type IV pilin production and twitching motility in crc mutants are thought to play a role in its defective biofilm phenotype [55].
AlgR has been previously shown to be required for full virulence in both acute septicemia and pneumonia infection models. In vitro algR mutant biofilms display normal early biofilm development but a defect in mature biofilm development resulting in thin, patchy biofilms [52]. AlgR is also involved in type IV-mediated twitching motility, which may contribute to its defective biofilm phenotype [53]. However, further study has demonstrated that AlgR specifically represses the rhl QS system and is responsible for the observed mutant phenotype [52]. The dependence of AlgR regulation of the rhl system was found to be surface contact or biofilm dependent. Excess production of rhamnolipid, a rhl-regulated product, was observed in algR deletion biofilms [52]. Excess rhamnolipid production has been previously correlated for dispersion in biofilm development, resulting in biofilms with a similar phenotype as the algR deletion mutants, suggesting that this is the reason for the defect in biofilm maturation [47–49].
In contrast, rpoS mutants form thick biofilms in vitro studies [56]. Transcriptome analysis of planktonic bacteria demonstrated that rpoS regulates expression of nearly 800 genes in stationary phase growth [58]. It is interesting to note that there is a circular link between rpoS and QS regulation, and microarray analysis of the rpoS regulon in P. aeruginosa PAO1 revealed that 40% of rpoS-regulated genes are also QS regulated [58]. However, it is currently unclear which of these genes may contribute to its aberrant biofilm phenotype.
A retS mutant also formed more robust biofilms in contrast to wild-type P. aeruginosa [57]. Microarray analysis of a retS mutant also indicated that it is involved in the regulation of both the psl and pel loci, which likely is responsible for its enhanced biofilm formation in vitro [57]. The regulator retS appears to be involved in QS-mediated phenotypes including autoinducer production via the GacA/GacS/rsmZ pathway [57]. This regulator appears to activate genes implicated in acute phase infection, such as the type III secretion system, type IV pilin, and secreted virulence molecules including exotoxin A and lipase [57]. The study authors suggest that this gene is involved in mediating the transition of the P. aeruginosa phenotype from an acute to chronic phase infection [57].
Similarly, the sadARS system appears to regulate virulence factors, such as the type III secretion system, in addition to being crucial for normal P. aeruginosa biofilm formation [59]. Mutation in this loci results in biofilms with abnormal maturation, where the maintenance of the complex three-dimensional water channels is defective. The gene sadA has been previously demonstrated to be QS regulated [9]. It is interesting to note that SadR possesses an glutamate–alanine–leucine domain, which has been linked with a phospodiesterase activity that degrades bis-(3′,5′)-cyclic-dimeric-guanosine monophosphate (cyclic di-GMP), producing guanosine monophosphate [59]. Increasing cyclic di-GMP levels have been reported to promote biofilm development [60, 61], and several recent studies have revealed a role for cyclic di-GMP in P. aeruginosa biofilm formation [62–66]. In P. aeruginosa PAO1, decreased cyclic di-GMP levels inhibited biofilm formation, while increased cyclic di-GMP levels promoted biofilm formation in a flow-through system [63]. Protein receptors for cyclic di-GMP have been identified that regulate the production of biofilm extracellular matrix components [65, 66]. For example, cyclic-di-GMP has been demonstrated to bind to one of the proteins in the pel operon, PelD, and modulate production of the extrapolysaccharide matrix protein PEL in P. aeruginosa PA14 [65]. Cyclic di-GMP has also been demonstrated to be required for alginate biosynthesis in mucoid P. aeruginosa, where the protein Alg44 binds cyclic di-GMP and then activates polymerization or transport of the alginate polysaccharide [66]. A role for cyclic di-GMP has also been proposed in biofilm dispersion. Recently, a novel regulator, bdlA, has been reported to be involved in the nutrient-induced dispersion of P. aeruginosa PAO1 biofilms [67]. BdlA has been proposed to transduce environmental signals that modulate the intracellular levels of cyclic di-GMP, thereby promoting biofilm dispersion [67].
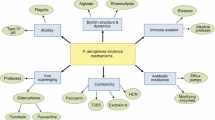
Although there is clear evidence for the role of QS in biofilm development (Fig. 2), other studies have been performed with varying conclusions as to the relative importance of QS in biofilms [10]. These inconsistencies may be attributed to the different conditions, including hydrodynamics and media, and/or P. aeruginosa strains utilized in these studies. Nevertheless, several genes that have been identified as QS regulated, including rhlA, rpoS, sadA, and those in the denitrification pathway, have been shown to be important in P. aeruginosa biofilm development, maintenance, and dispersion [10, 59, 68, 69]. A recent study of P. aeruginosa biofilms in the mouse thermal injury model suggested that QS was not important in the formation of biofilms in acute infection [70]; however, several previous studies have clearly demonstrated a role for QS in this model [71–74]. Further investigation is clearly needed to precisely define the role of QS in both in vitro and in vivo biofilm development.

Genes and factors implicated during P. aeruginosa biofilm development that are QS regulated. Genes and factors are listed below the developmental stage of biofilm formation in which they have been reported to be important
The CF lung and P. aeruginosa biofilms
There are several lines of evidence that support the notion that P. aeruginosa exists in a biofilm in the CF lung. These include microscopic analysis of CF sputum, increased antimicrobial resistance of P. aeruginosa in the CF lung, and the ratio of the P. aeruginosa autoinducer molecules, 3O-C12-HSL and C4-HSL, in sputum from CF patients mirrors those of biofilm-grown P. aeruginosa [75, 76]. The environment of the CF lung is thought to promote P. aeruginosa biofilm formation. Mutations in the CF transmembrane regulator (CFTR) cause a defective function of CFTR in airway epithelium and submucosal glands, leading to decreased mucociliary clearance and altered airway surface liquid (ASL) concentrations [77]. In addition, the nitric oxide (NO) system, involved in bacterial killing, is impaired in CF patients as reflected by a reduction in expression of the inducible NO synthase (iNOS) responsible for NO production in neutrophils, macrophages, and epithelial cells [16].
The ability of P. aeruginosa to grow anaerobically is believed to be important in biofilm development in the CF lung [76, 78]. Worlitzsch et al. [79] reported that oxygen gradients exist in thick mucus plugs present in the lower airway of CF patients. As neutrophils infiltrate and mucus secretion increases, metabolically active bacteria and epithelial cells consume the available oxygen, and the surroundings quickly become oxygen limited [79]. The conversion of P. aerugiosa to a mucoid phenotype during the progression of chronic CF lung disease may further restrict the diffusion of oxygen and contribute to the microaerobic or anaerobic environments in CF infections. Recent data suggest that anaerobic environments promote the conversion of nonmucoid P. aeruginosa to mucoidy [79]. CF airway mucus has been demonstrated to support anaerobic growth of P. aeruginosa [79]. P. aeruginosa is able to grow anaerobically using the alternative electron acceptors nitrate, nitrite, and nitrous oxide through a respiratory process termed denitrification [80]. Nitrate and nitrite concentrations in CF sputum are able to support anaerobic respiration of P. aeruginosa [79, 81–83]. In the absence of nitrate or nitrite, arginine can be catabolized to ornithine using substrate-level phosphorylation and used as an energy source for anaerobic growth [84, 85]. P. aeruginosa can also use pyruvate to sustain long-term survival, but this fermentation process can not support growth of the bacterium, under hypoxic conditions [86]. Anaerobic conditions promote biofilm development in both P. aeruginosa PAO1 and in CF isolates, suggesting that P. aeruginosa readily adapts and perhaps favors anaerobic biofilm growth in vivo [87].
Anaerobiosis is not only important in P. aeruginosa survival in the CF lung environment but also within the biofilm itself. Within an aerobically grown biofilm, oxygen depletion has been reported to occur within 30 μm of the surface of the biofilm [20]. As P. aeruginosa forms biofilms in the CF mucus, the bacteria undergo a switch from an aerobic to an anaerobic metabolism [78]. P. aeruginosa existing within the biofilms experience oxygen gradients, in which the base and the center of the biofilm are anaerobic [20, 79, 88]. Genes involved in denitrification have been identified as QS regulated, suggesting a link between anaerobic metabolism and QS [9]. A further study reported that during anaerobic biofilm growth of P. aeruginosa, the rhl QS system was required for proper balance of the denitrification pathway [89]. Dysregulation of the pathway resulted in the accumulation of toxic intermediates, such as nitrite and nitrous oxide, which caused cell death to occur within the biofilm.
It is interesting to note that nitrous oxide has been demonstrated to be a signal involved in P. aeruginosa biofilm dispersal [90]. Exposure of P. aeruginosa biofilms to NO in combination with antimicrobials or SDS enhanced the activity of these agents. In addition, recent data suggest that mucoid P. aeruginosa CF isolates are exquisitely sensitive to nitrite due to an inability to remove the toxic accumulation of NO [91]. The fact that iNOS activity decreases as the chronic CF infection progresses may contribute to the ability of P. aeruginosa to persist as a biofilm.
The thick mucus present in the airways of CF lungs provides a diffusion-restricted environment. A recent study demonstrated that in ASL, P. aeruginosa PAO1 formed macrocolonies resembling biofilms dependent upon the concentration of solids [92]. In normal ASL, PAO1 failed to form macrocolonies. However, in concentrated ASL mimicking ASL found in CF patients, PAO1 readily formed biofilms under either aerobic or anaerobic conditions. Furthermore, the formation of these macrocolonies was dependent upon a functional las QS system. The mutation in lasI abrogated macrocolony formation. Diffusion studies demonstrated that concentrated ASL greatly restricted the movement of 3O-C12-HSL, the product of the lasI gene. The constrained microenvironment in the CF ASL is thought to promote P. aeruginosa biofilm formation by concentrating 3O-C12-HSL and activating QS-regulated factors involved in biofilm development.
Quorum sensing in P. aeruginosa pathogenesis and implications in vivo
Both in vitro and in vivo models have clearly demonstrated the importance of QS in P. aeruginosa pathogenesis. Numerous models of infection, including plant, invertebrate, and animal models, have supported the premise that QS contributes to P. aeruginosa pathogenesis [71–74, 93–101]. In animal models of lung pathogenesis, including the mouse model of acute pneumonia, the mouse model of chronic lung infection, and the rat chronic lung infection model, lasR, lasI, and rhlI have been demonstrated to be required for virulence [73, 93, 102–105]. Other QS-regulated factors, such as type IV pilin biosynthesis and type III secretion pathways, have been clearly linked to P. aeruginosa pathogenesis [9, 98].
Evidence exists that P. aeruginosa QS is active in CF patients. Several studies have reported the presence of transcripts for known QS regulators and QS-regulated genes in CF patient samples, suggesting a role for QS in vivo. Transcripts for lasR, lasI, lasA, lasB, and toxA have been detected in sputa samples from chronically infected CF patients [106, 107]. There appeared to be a correlation of lasR to the expression lasI, lasR, lasA, lasB, and toxA, and as lasR is known to regulate expression of these genes in vitro, this relationship suggested that lasR also regulates these genes in vivo [106]. A further study reported a statistically significant correlation between lasR and algD transcription [106]. This suggested that lasR might to some extent regulate algD or, alternatively, be activated by a common environmental trigger in vivo. AlgD catalyzes the first step in alginate biosynthesis, which is responsible for the mucoid phenotype often observed in clinical isolates from P. aeruginosa chronically infected CF patients [108]. The autoinducer molecules 3O-C12-HSL, C4-HSL, and PQS are also present in CF sputa samples, and PQS has been detected in bronchoalveolar lavage fluid and mucopurulent fluid from distal airways of end-stage lungs removed at transplant in CF patients [106, 109, 110].
Curiously, the isolation of P. aeruginosa from acute and chronic infections, including the infected CF lung, with defects in QS suggests that the role of QS is diminished or perhaps not important in vivo [111, 112]. The dichotomy between the isolation of QS-deficient P. aeruginosa from infections and the importance of QS in the infectious process has been a conundrum. This is important as QS is thought to be an attractive target for the development of novel therapeutics [78, 113]. However, a recent study reported that in instances of high QS activity, spontaneous QS mutants, usually in the main regulator lasR, occur [112]. Mutants in lasR are more resistant to cell lysis and death at high cell densities and at alkaline pH. These “social cheaters” appear to have a competitive advantage over P. aeruginosa with intact QS systems. The appearance of QS mutants suggests, in contrast to previous belief, high QS activity due to the metabolic burden QS places on the bacteria.
Summary
P. aeruginosa possesses many traits, including intrinsic antimicrobial resistance, ability to form biofilms, arsenal of virulence products, and metabolic versatility that contribute to its ability to colonize and persist in acute and chronic infections such as those of CF patients. In P. aeruginosa, QS regulates many of these phenotypes (Fig. 3). The CF lung presents an ideal niche for P. aeruginosa. Dysregulation in mucocilliary clearance and NO-mediated killing are thought to contribute to colonization of P. aeruginosa in the CF airways. A restricted microenvironment due to thick airway liquid results in decreased oxygen concentrations and increased localized autoinducer concentrations that have been demonstrated to promote biofilm development. The biofilm mode of growth subsequently provides protection against numerous host factors and antimicrobial challenge. Due to its role in pathogenesis, biofilm development, and antimicrobial resistance, QS represents an attractive target for the development of novel therapeutics. Further understanding of the QS mechanisms involved in these phenotypes will substantially aid in this goal.

Quorum sensing regulates multiple phenotypes in P. aeruginosa. Several genes that have been identified to contribute to each phenotype are listed
References
Ikeno T et al (2007) Small and rough colony Pseudomonas aeruginosa with elevated biofilm formation ability isolated in hospitalized patients. Microbiol Immunol 51(10):929–938
Whiteley M, Greenberg EP (2001) Promoter specificity elements in Pseudomonas aeruginosa quorum-sensing-controlled genes. J Bacteriol 183(19):5529–5534
Pesci EC, Iglewski BH (1997) The chain of command in Pseudomonas quorum sensing. Trends Microbiol 5(4):132–134 discussion 134–135
de Kievit TR et al (2002) Role of the Pseudomonas aeruginosa las and rhl quorum-sensing systems in rhlI regulation. FEMS Microbiol Lett 212(1):101–106
Pesci E, Iglewski B (1999) Quorum sensing in Pseudomonas aeruginosa, in cell–cell signaling in bacteria. American Society for Microbiology, Washington DC, pp 147–155
McKnight SL, Iglewski BH, Pesci EC (2000) The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol 182(10):2702–2708
Hentzer M et al (2003) Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. Embo J 22(15):3803–3815
Schuster M et al (2003) Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol 185(7):2066–2079
Wagner VE et al (2003) Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol 185(7):2080–2095
Parsek MR, Greenberg EP (2005) Sociomicrobiology: the connections between quorum sensing and biofilms. Trends Microbiol 13(1):27–33
Costerton JW, Stewart PS (2001) Battling biofilms. Sci Am 285(1):74–81
Costerton J, Stewart P, Greenberg E (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322
Govan JR, Deretic V (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60(3):539–574
Donlan RM, Costerton JW (2002) Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 15(2):167–193
Costerton JW (2002) Anaerobic biofilm infections in cystic fibrosis. Mol Cell 10(4):699–700
Gomez MI, Prince A (2007) Opportunistic infections in lung disease: Pseudomonas infections in cystic fibrosis. Curr Opin Pharmacol 7(3):244–251
Hentzer M, Eberl L, Givskov M (2005) Transcriptome analysis of Pseudomonas aeruginosa biofilm development: anaerobic respiration and iron limitation. Biofilms 2:37–61
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284(5418):1318–1322
Sauer K et al (2002) Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J Bacteriol 184(4):1140–1154
Xu KD et al (1998) Spatial physiological heterogeneity in Pseudomonas aeruginosa biofilm is determined by oxygen availability. Appl Environ Microbiol 64(10):4035–4039
Davies DG et al (1998) The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280(5361):295–298
Bjarnsholt T et al (2005) Pseudomonas aeruginosa tolerance to tobramycin, hydrogen peroxide and polymorphonuclear leukocytes is quorum-sensing dependent. Microbiology 151(Pt 2):373–383
Hassett DJ et al (1999) Quorum sensing in Pseudomonas aeruginosa controls expression of catalase and superoxide dismutase genes and mediates biofilm susceptibility to hydrogen peroxide. Mol Microbiol 34(5):1082–1093
Klausen M et al (2003) Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol Microbiol 48(6):1511–1524
Klausen M et al (2003) Involvement of bacterial migration in the development of complex multicellular structures in Pseudomonas aeruginosa biofilms. Mol Microbiol 50(1):61–68
O’Toole G, Kolter R (1998) Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol 30(2):295–304
Sonawane A, Jyot J, Ramphal R (2006) Pseudomonas aeruginosa LecB is involved in pilus biogenesis and protease IV activity but not in adhesion to respiratory mucins. Infect Immun 74(12):7035–7039
Tielker D et al (2005) Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology 151(Pt 5):1313–1323
Diggle SP et al (2006) The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ Microbiol 8(6):1095–1104
Boteva RN, Bogoeva VP, Stoitsova SR (2005) PA-I lectin from Pseudomonas aeruginosa binds acyl homoserine lactones. Biochim Biophys Acta 1747(2):143–149
Winzer K et al (2000) The Pseudomonas aeruginosa lectins PA-IL and PA-IIL are controlled by quorum sensing and by RpoS. J Bacteriol 182(22):6401–6411
Pedersen SS (1992) Lung infection with alginate-producing, mucoid Pseudomonas aeruginosa in cystic fibrosis. Acta Pathol Microbiol Immunol Scand Suppl 28:1–79
Pedersen SS et al (1992) Role of alginate in infection with mucoid Pseudomonas aeruginosa in cystic fibrosis. Thorax 47(1):6–13
Hentzer M et al (2001) Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J Bacteriol 183(18):5395–5401
Ramsey DM, Wozniak DJ (2005) Understanding the control of Pseudomonas aeruginosa alginate synthesis and the prospects for management of chronic infections in cystic fibrosis. Mol Microbiol 56(2):309–322
Ryder C, Byrd M, Wozniak DJ (2007) Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr Opin Microbiol 10(6):644–648
Reimmann C et al (1997) The global activator GacA of Pseudomonas aeruginosa PAO positively controls the production of the autoinducer N-butyryl-homoserine lactone and the formation of the virulence factors pyocyanin, cyanide, and lipase. Mol Microbiol 24(2):309–319
Sakuragi Y, Kolter R (2007) Quorum-sensing regulation of the biofilm matrix genes (pel) of Pseudomonas aeruginosa. J Bacteriol 189(14):5383–5386
Leid JG et al (2005) The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol 175(11):7512–7518
Allesen-Holm M et al (2006) A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol 59(4):1114–1128
Whitchurch CB et al (2002) Extracellular DNA required for bacterial biofilm formation. Science 295(5559):1487
Yang L et al (2007) Effects of iron on DNA release and biofilm development by Pseudomonas aeruginosa. Microbiology 153(Pt 5):1318–1328
Whiteley M et al (2001) Gene expression in Pseudomonas aeruginosa biofilms. Nature 413(6858):860–864
Mashburn LM, Whiteley M (2005) Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature 437(7057):422–425
Pearson JP, Pesci EC, Iglewski BH (1997) Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J Bacteriol 179(18):5756–5767
Pamp SJ, Tolker-Nielsen T (2007) Multiple roles of biosurfactants in structural biofilm development by Pseudomonas aeruginosa. J Bacteriol 189(6):2531–2539
Boles BR, Thoendel M, Singh PK (2005) Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol Microbiol 57(5):1210–1223
Davey ME, Caiazza NC, O’Toole GA (2003) Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J Bacteriol 185(3):1027–1036
Lequette Y, Greenberg EP (2005) Timing and localization of rhamnolipid synthesis gene expression in Pseudomonas aeruginosa biofilms. J Bacteriol 187(1):37–44
Kirov SM, Webb JS, Kjelleberg S (2005) Clinical significance of seeding dispersal in biofilms. Microbiology 151(Pt 11):3452–3453 discussion 3453
Kirov SM et al (2007) Biofilm differentiation and dispersal in mucoid Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 153(Pt 10):3264–3274
Morici LA et al (2007) Pseudomonas aeruginosa AlgR represses the Rhl quorum-sensing system in a biofilm-specific manner. J Bacteriol 189(21):7752–7764
Whitchurch CB et al (2002) Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J Bacteriol 184(16):4544–4554
Parkins MD, Ceri H, Storey DG (2001) Pseudomonas aeruginosa GacA, a factor in multihost virulence, is also essential for biofilm formation. Mol Microbiol 40(5):1215–1226
O’Toole GA et al (2000) The global carbon metabolism regulator Crc is a component of a signal transduction pathway required for biofilm development by Pseudomonas aeruginosa. J Bacteriol 182(2):425–431
Heydorn A et al (2002) Statistical analysis of Pseudomonas aeruginosa biofilm development: impact of mutations in genes involved in twitching motility, cell-to-cell signaling, and stationary-phase sigma factor expression. Appl Environ Microbiol 68(4):2008–2017
Goodman AL et al (2004) A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell 7(5):745–754
Schuster M et al (2004) The Pseudomonas aeruginosa RpoS regulon and its relationship to quorum sensing. Mol Microbiol 51(4):973–985
Kuchma SL, Connolly JP, O’Toole GA (2005) A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J Bacteriol 187(4):1441–1454
Romling U, Gomelsky M, Galperin MY (2005) C-di-GMP: the dawning of a novel bacterial signalling system. Mol Microbiol 57(3):629–639
Romling U, Amikam D (2006) Cyclic di-GMP as a second messenger. Curr Opin Microbiol 9(2):218–228
Guvener ZT, Harwood CS (2007) Subcellular location characteristics of the Pseudomonas aeruginosa GGDEF protein, WspR, indicate that it produces cyclic-di-GMP in response to growth on surfaces. Mol Microbiol 66(6):1459–1473
Hickman JW, Tifrea DF, Harwood CS (2005) A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci USA 102(40):14422–14427
Kulasakara H et al (2006) Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3¢-5¢)-cyclic-GMP in virulence. Proc Natl Acad Sci USA 103(8):2839–2844
Lee VT et al (2007) A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol 65(6):1474–1484
Merighi M et al (2007) The second messenger bis-(3¢-5¢)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol Microbiol 65(4):876–895
Morgan R et al (2006) BdlA, a chemotaxis regulator essential for biofilm dispersion in Pseudomonas aeruginosa. J Bacteriol 188(21):7335–7343
Branda SS et al (2005) Biofilms: the matrix revisited. Trends Microbiol 13(1):20–26
Kulasekara HD et al (2005) A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol 55(2):368–380
Schaber JA et al (2007) Pseudomonas aeruginosa forms biofilms in acute infection independent of cell-to-cell signaling. Infect Immun 75(8):3715–3721
Rahme LG et al (1995) Common virulence factors for bacterial pathogenicity in plants and animals. Science 268(5219):1899–1902
Rumbaugh KP, Griswold JA, Hamood AN (1999) Contribution of the regulatory gene lasR to the pathogenesis of Pseudomonas aeruginosa infection of burned mice. J Burn Care Rehabil 20(1):42–49
Mahajan-Miklos S, Rahme LG, Ausubel FM (2000) Elucidating the molecular mechanisms of bacterial virulence using non-mammalian hosts. Mol Microbiol 37(5):981–988
Hendrickson EL et al (2001) Differential roles of the Pseudomonas aeruginosa PA14 rpoN gene in pathogenicity in plants, nematodes, insects, and mice. J Bacteriol 183(24):7126–7134
Singh PK et al (2000) Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407(6805):762–764
Costerton JW (2002) Anaerobic biofilm infections in cystic fibrosis. Mol Cell 10(4):699–700
Thelin WR, Boucher RC (2007) The epithelium as a target for therapy in cystic fibrosis. Curr Opin Pharmacol 7(3):290–295
Hassett DJ et al (2002) Anaerobic metabolism and quorum sensing by Pseudomonas aeruginosa biofilms in chronically infected cystic fibrosis airways: rethinking antibiotic treatment strategies and drug targets. Adv Drug Deliv Rev 54(11):1425–1443
Worlitzsch D et al (2002) Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109(3):317–325
Hass D, Gamper M, Zimmermann A (1992) Anaerobic control in Pseudomons aeruginosa. In: Galli E, Silver S, Witholt B (eds) Pseudomonas: molecular biology and biotechnology. American Society for Microbiology, Washington, DC, pp 177–187
Hassett DJ (1996) Anaerobic production of alginate by Pseudomonas aeruginosa: alginate restricts diffusion of oxygen. J Bacteriol 178(24):7322–7325
Linnane SJ et al (1998) Total sputum nitrate plus nitrite is raised during acute pulmonary infection in cystic fibrosis. Am J Respir Crit Care Med 158(1):207–212
Grasemann H (1999) Total sputum nitrate plus nitrite is raised during acute pulmonary infection in cystic fibrosis. Am J Respir Crit Care Med 159(2):684–685
Mercenier A et al (1980) Regulation of enzyme synthesis in the arginine deiminase pathway of Pseudomonas aeruginosa. J Bacteriol 144(1):159–163
Vander Wauven C et al (1984) Pseudomonas aeruginosa mutants affected in anaerobic growth on arginine: evidence for a four-gene cluster encoding the arginine deiminase pathway. J Bacteriol 160(3):928–934
Eschbach M et al (2004) Long-term anaerobic survival of the opportunistic pathogen Pseudomonas aeruginosa via pyruvate fermentation. J Bacteriol 186(14):4596–4604
O’May CY, Reid DW, Kirov SM (2006) Anaerobic culture conditions favor biofilm-like phenotypes in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. FEMS Immunol Med Microbiol 48(3):373–380
Govan J, Deretic V (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60(3):539–574
Yoon SS et al (2002) Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev Cell 3(4):593–603
Barraud N et al (2006) Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J Bacteriol 188(21):7344–7353
Yoon SS et al (2006) Anaerobic killing of mucoid Pseudomonas aeruginosa by acidified nitrite derivatives under cystic fibrosis airway conditions. J Clin Invest 116(2):436–446
Matsui H et al (2006) A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA 103(48):18131–18136
Tang HB et al (1996) Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect Immun 64(1):37–43
Rahme LG et al (1997) Use of model plant hosts to identify Pseudomonas aeruginosa virulence factors. Proc Natl Acad Sci USA 94(24):13245–13250
Preston MJ et al (1997) Contribution of proteases and LasR to the virulence of Pseudomonas aeruginosa during corneal infections. Infect Immun 65(8):3086–3090
Tan MW, Mahajan-Miklos S, Ausubel FM (1999) Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci USA 96(2):715–720
Mahajan-Miklos S et al (1999) Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa–Caenorhabditis elegans pathogenesis model. Cell 96(1):47–56
de Kievit TR, Iglewski BH (2000) Bacterial quorum sensing in pathogenic relationships. Infect Immun 68(9):4839–4849
Plotnikova JM, Rahme LG, Ausubel FM (2000) Pathogenesis of the human opportunistic pathogen Pseudomonas aeruginosa PA14 in Arabidopsis. Plant Physiol 124(4):1766–1774
Rahme LG et al (2000) Plants and animals share functionally common bacterial virulence factors. Proc Natl Acad Sci USA 97(16):8815–8821
Jander G, Rahme LG, Ausubel FM (2000) Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol 182(13):3843–3845
Tang H, Kays M, Prince A (1995) Role of Pseudomonas aeruginosa pili in acute pulmonary infection. Infect Immun 63(4):1278–1285
Pearson JP et al (2000) Pseudomonas aeruginosa cell-to-cell signaling is required for virulence in a model of acute pulmonary infection. Infect Immun 68(7):4331–4334
Potvin E et al (2003) In vivo functional genomics of Pseudomonas aeruginosa for high-throughput screening of new virulence factors and antibacterial targets. Environ Microbiol 5(12):1294–1308
Hoffmann N et al (2005) Novel mouse model of chronic Pseudomonas aeruginosa lung infection mimicking cystic fibrosis. Infect Immun 73(4):2504–2514
Erickson DL et al (2002) Pseudomonas aeruginosa quorum-sensing systems may control virulence factor expression in the lungs of patients with cystic fibrosis. Infect Immun 70(4):1783–1790
Storey D et al (1998) Pseudomonas aeruginosa lasR transcription correlates with the transcription of lasA, lasB, and toxA in chronic lung infections associated with cystic fibrosis. Infect Immun 66(6):2521–2528
Deretic V, Gill JF, Chakrabarty AM (1987) Gene algD coding for GDPmannose dehydrogenase is transcriptionally activated in mucoid Pseudomonas aeruginosa. J Bacteriol 169(1):351–358
Collier DN et al (2002) A bacterial cell to cell signal in the lungs of cystic fibrosis patients. FEMS Microbiol Lett 215(1):41–46
Smith KM, Bu Y, Suga H (2003) Library screening for synthetic agonists and antagonists of a Pseudomonas aeruginosa autoinducer. Chem Biol 10(6):563–571
Schaber JA et al (2004) Analysis of quorum sensing-deficient clinical isolates of Pseudomonas aeruginosa. J Med Microbiol 53(Pt):841–853
Sandoz KM, Mitzimberg SM, Schuster M (2007) Social cheating in Pseudomonas aeruginosa quorum sensing. Proc Natl Acad Sci USA 104(40):15876–15881
Smith RS, Iglewski BH (2003) Pseudomonas aeruginosa quorum sensing as a potential antimicrobial target. J Clin Invest 112(10):1460–1465
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wagner, V.E., Iglewski, B.H. P. aeruginosa Biofilms in CF Infection. Clinic Rev Allerg Immunol 35, 124–134 (2008). https://doi.org/10.1007/s12016-008-8079-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-008-8079-9