
Abstract
The microvasculature and immune cells are major components of the tumor microenvironment (TME). Hypoxia plays a pivotal role in the TME through hypoxia-inducible factor 1-alpha (HIF-1α) which upregulates vascular endothelial growth factor (VEGF). VEGF, an angiogenesis stimulator, suppresses tumor immunity by inhibiting the maturation of dendritic cells, and induces immunosuppressive cells such as regulatory T cells, tumor-associated macrophages, and myeloid-derived suppressor cells. HIF-1α directly induces immune checkpoint molecules. VEGF/VEGF receptor (VEGFR)-targeted therapy as a cancer treatment has not only anti-angiogenic effects, but also immune-supportive effects. Anti-angiogenic therapy has the potential to change the immunological “cold tumors” into the “hot tumors”. Glioblastoma (GB) is a hypervascular tumor with high VEGF expression which leads to development of an immuno suppressive TME. Therefore, in the last decade, several combination immunotherapies with anti-angiogenic agents have been developed for numerous tumors including GBs. In particular, combination therapy with an immune checkpoint inhibitor and VEGF/VEGFR-targeted therapy has been suggested as a synergic treatment strategy that may show favorable changes in the TME. In this article, we discuss the cross talk among immunosuppressive cells exposed to VEGF in the hypoxic TME of GBs. Current efficient combination strategies using VEGF/VEGFR-targeted therapy are reviewed and proposed as novel cancer treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The tumor microenvironment (TME) consists of immune cells, fibroblasts, endothelial cells, extracellular matrix, and some signaling molecules such as chemokines. The TME shows an immunosuppressive effect and plays critical roles in tumor growth, angiogenesis, and metastasis [1].
Glioblastomas (GBs) are the most aggressive and vascularized primary brain tumors [2]. Despite multimodal therapy including surgical removal, radiation, and chemotherapy, GBs are essentially incurable [3, 4]. The heterogeneity, infiltrative characteristics, presence of glioma stem cells, and function of blood-brain barrier have been appointed as the main causes of therapeutic resistance and malignant relapse [3, 4]. Furthermore, the lack of anti-tumor immune response due to an immunosuppressive TME also contributes to the treatment failure. Immune checkpoint molecules, exhaustion of cytotoxic T lymphocytes (CTLs), and immunosuppressive cells in hypoxic conditions induce the immunosuppressive TME. Tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs) are major components of the immunosuppressive cells in the TME of GBs [5]. Programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) are immune checkpoint molecules that are associated with immunosuppressive cells in the TME. PD-L1 expressed on tumor cells binds to PD-1 expressed on activated T cells and negatively regulates immune responses [6, 7].
Vascular endothelial growth factor (VEGF) is a key mediator of tumor angiogenesis and a major target for anti-angiogenic therapy for various malignant tumors including GBs [2, 8, 9]. VEGF is induced by hypoxia through a hypoxia-inducible factor 1 alpha (HIF-1α)-depending pathway, which contributes to immune suppression in the TME [10]. Therefore, anti-VEGF (bevacizumab; Bev) or VEGF receptor (VEGFR)-targeted (sunitinib, sorafenib) agents as a cancer treatment induce not only anti-angiogenic effects, but also immune-supportive effects [11,12,13,14]. Recently, the significance of the PD-1/PD-L1 immune checkpoint system has received attention in several types of tumors [6, 7]. Anti-PD-1 and PD-L1 antibodies exert a potent effect in inhibiting tumor growth in melanomas, non-small lung cancer, and kidney cancer [15]. However, immunologically “cold” tumors including GBs did not have advantages by immune checkpoint inhibitors [16]. Anti-angiogenic therapy may convert immunologically “cold” tumors to “hot” tumors. Therefore, combination usage of an anti-angiogenic agent and an immune checkpoint inhibitor may be a strategy to overcome the mechanism of resistance of immunotherapies for “cold” tumors [17]. Over the last decade, several anti-angiogenic therapies combined with chemotherapies and immunotherapies have been developed for elimination of the immunosuppressive TME. For further development of these strategies, we need to understand alterations in the TME following chemotherapies and immunotherapies with or without anti-VEGF/VEGFR therapy.
In this article, we discuss the cross talk among immunosuppressive cells exposed to VEGF in the hypoxic TME of GBs. The current efficient combination strategies using VEGF/VEGFR-targeted therapy are reviewed. Finally, alterations in the TME following these combination strategies are also summarized to understand the mechanisms of action and resistance, followed by a proposal for novel cancer therapies.
VEGF
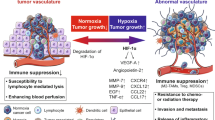
VEGF plays an important role in vascular development and in diseases involving abnormal growth of blood vessels. Since discovery of its dual roles in endothelial proliferation and vascular permeability [18, 19], VEGF has been considered a key mediator of neovascularization in tumors (Fig. 1). Elevated VEGF levels are associated with poor clinical outcomes in numerous tumors including GBs [2, 20]. In addition to angiogenic effects, VEGF suppresses the anti-tumor immune response [15]. VEGF inhibits maturation of dendritic cells (DCs), resulting in inactivation of CTLs [21]. VEGF also induces an immunosuppressive TME by strongly inducing Tregs, TAMs, and MDSCs [22]. Furthermore, VEGF enhances the expression of PD-1 on CD8+ CTLs and Tregs in a VEGFR2-dependent manner (Fig. 1) [23]. Tumor-derived VEGF, interleukin (IL)-10, and prostaglandin E3 cooperatively induced Fas ligand expression in endothelial cells, leading to exhaustion of CTL but not Tregs (Fig. 1) [24].

Role of VEGF in the tumor microenvironment. VEGF has dual effects of vascular biological and tumor immunological regulation in the tumor microenvironment. VEGF plays a pivotal role in inducing vacular endothelial cells and immunosuppressive cells in hypoxic conditions. DCs dendritic cells, ECs endothelial cells, IFP interstial fluid pressure, MDSCs myeloid-derived suppressor cells, PD-1 programmed death-1, PD-L1 programmed death ligand 1, Tregs regulatory T cells, TAMs tumor-associated macrophages, TILs tumor-infiltrating lymphocytes, VEGFR vascular endothelial growth factor receptor, VVO vesiculo-vacuolar organelle
VEGF is induced by hypoxia via activation of the transcription factor, HIF-1, which could play an important role in triggering tumor angiogenesis [25, 26]. The TME is mainly altered by hypoxia and acidosis [27]. Hypoxia supports the escape of tumor cells from immune surveillance by recruiting TAMs, Tregs, and MDSCs into the TME directly or through upregulation of VEGF [28].
TAMs
Macrophages often behave as immunosuppressive cells and contribute to inflammatory diseases. Macrophages express different functional programs in response to microenvironmental signals, defined as M1/M2 polarization [29]. M2 macrophages produce growth factors and anti-inflammatory cytokines to suppress the host immune response, resulting in tumor progression [29].
TAMs typically behave as M2 macrophages [30], playing a pivotal role in the TME [28, 30]. The interaction between the tumor cells and TAMs is promoted via macrophage colony-stimulating factor and its receptor [29]. TAMs induce various growth factors such as basic fibroblast growth factor, epidermal growth factor, hepatocyte growth factor, and platelet-derived growth factor [29]. In addition, the lack of arginine by arginase I, and IL-10, transforming growth factor (TGF)-β, and prostaglandin F2 produced by TAMs suppress effector T cells [29]. TAMs produce VEGF and matrix metalloproteinase 9, which promote angiogenesis, invasion, and metastasis [29]. Although the existence of TAMs is still controversial as a prognostic biomarker for cancer patients, most reports have demonstrated that increased TAMs are related to poor prognosis in GBs; breast, esophagus, and liver cancers; and malignant lymphoma [31]. Interestingly, TAMs infiltration is associated with the resistance to anti-angiogenic therapy through downregulation of macrophage migration inhibitory factor in GBs [32]. VEGF in the hypoxic TME is a key factor for transitioning from the M1 to M2 macrophage phenotype [33]. Furthermore, HIF-1α promotes the migration and differentiation of TAMs from immature myeloid cells via VEGF exposure [17].
Tregs
Tregs (CD4 + CD25 + Foxp3+) play an active and significant role in the progression of tumors, and play an important role in suppressing tumor-specific immunity [34, 35]. Tregs suppress T cell-mediated immune responses via TGF-β, IL-10, and IL-35. Tregs induce the apoptosis of effector T cells through granzyme B. Consumption of IL-2 by Tregs inhibits effector T cells proliferation. Furthermore, Tregs directly suppress DCs [35]. Increased Tregs in tumor tissue or peripheral blood are significantly associated with shorter survival in most cancer patients including those with GBs [36]. Tregs may be critical for evaluation of the clinical significance of immunosuppressive effects after anti-angiogenic therapy. To detect Tregs more precisely, effector Tregs are defined as CD45RA-Foxp3highCD4+ which may reflect an immune suppressive function. Non Tregs (CD45RA-Foxp3lowCD4+), which produce inflammatory cytokines such as interferon-γ, are included in the CD4 + CD25 + Foxp3+ cell population [36].
The role of HIF-1α has also been implicated in direct regulation of the differentiation of Tregs and in promotion of the recruitment of Tregs to the TME via overexpression of CC chemokine ligand 22 and 28 [37]. HIF-1α induces VEGFR2-expressing Tregs through VEGF production [37].
MDSCs
MDSCs are a type of myeloid cells that can differentiate into macrophages, DCs and granulocytes, thus suppressing CTLs. In humans, MDSCs are CD11b + CD33 + Cd14 + HLA-DR- [38]. CD115, CD124, and VEGFR were also identified in MDSCs [39]. MDSCs are induced by IL-6, VEGF, and prostaglandin E2, which are produced by tumor cells. In addition, MDSCs are activated by interferon-γ, IL-4, IL-13, and TGF-β produced by CTLs and tumor stromal cells [38]. MDSCs also produce TGF-β, IL-10, and metalloproteinase 9 [40]. Interestingly, recent findings suggest that natural killer T cells activated by α-GalCer-loaded CD11b + Gr1 + MDSC can acquire the ability to convert immunosuppressive MDSCs into immunity-promoting antigen-presenting cells. Reprogramed MDSCs show upregulation of expression of CD11b, CD11c, and CD86, which support immunity by antigen-specific CTLs without increasing Tregs [22, 41]. GB patients with a more favorable prognosis exhibit decreased MDSCs and increased DCs compared to those with a worse prognosis [42]. Interactions between MDSCs and glioma stem cells via migration inhibitory factor enhances the function of MDSCs, which could be targeted to reduce the growth of GBs [42]. In addition, MDSCs directly promotes angiogenesis, which is associated with refractoriness to anti-angiogenic therapy [43]. VEGF is strongly associated with MDSC accumulation in GBs [44, 45]. HIF-1α has also been implicated in direct regulation of the function and differentiation of MDSCs in the hypoxic TME [46].
PD-1/PD-L1
PD-1 is expressed on CD8+T cells and Tregs. PD-L1 is expressed on tumor cells in numerous malignant tumors including GBs and binds to PD-1 to negatively regulate the immune response of CD8+T cells [6, 7]. A recent study showed that PD-1 is expressed on TAMs and correlates negatively with phagocytic potency, demonstrating its relevance to tumor immunity [47]. PD-L1 protein expression was identified in 61 to 88% of patients with GBs [48]. PD-1/PD-L1 expression is associated with poor prognosis for patients with GBs [49, 50]. Garber et al. demonstrated that GB specimens have a higher frequency of PD-1+ tumor-infiltrating lymphocytes (TILs) compared with lower-grade gliomas, whereas PD-L1 expression does not significantly differ among malignant grades [51]. Isocitrate dehydrogenase (IDH)-wild-typed GBs are more “immunologically active” than IDH-mutated GBs. GBs with wild-type IDH display a higher number of TILs and elevated expression of PD-L1 compared with IDH-mutant GBs. Therefore, IDH-wild-type GBs may be more readily targeted by PD-1/PD-L1 checkpoint blockade [52]. In particular, PD-L1 is strongly expressed in the mesenchymal subgroup of GBs [53].
A previous study demonstrated that PD-1 expression on CD8+T cells and Tregs is induced by VEGF [23]. Similarly, PD-L1 is upregulated via VEGF exposure in some types of tumors including GBs [53]. Immunologically “cold” tumors including GBs are not good targets for immune checkpoint inhibitors [16]. Anti-angiogenic therapy has the possibility to change “cold” tumors into “hot” tumors with a favorable microenvironment. Therefore, combination therapy with an immune checkpoint inhibitor and VEGF-targeted therapy is expected to be an efficient treatment strategy [17]. In addition, hypoxia directly causes upregulation of PD-L1 and CTLA-4 on MDSCs, TAMs, DCs, and tumor cells through HIF-1α [54]. HIF-2α is also associated with PD-L1 expression on tumor cells in metastatic renal cell carcinoma [55]. Blocking HIF-1α by agonizing nitric oxide signaling decreased PD-L1 expression and promotes CTL-mediated lysis [56].
Cross talk in the TME
VEGF promotes the accumulation of TAMs, Tregs, and MDSCs in tumor tissue and secondary lymphoid organs [10]. In addition, HIF-1α activation is also a major component of the hypoxic TME. HIF-1α upregulation directly drives recruitment of immature myeloid cells and Tregs, and fosters their phenotypic conversion into highly suppressive MDSCs and TAMs [58]. Recent studies demonstrated functional cross talk among TAMs, Tregs, and MDSCs that was strongly associated with hypoxia-induced VEGF production [43, 57, 59]. Tregs modify the phenotype of TAMs to express inhibitory B7-H molecules. Tregs depletion significantly downregulates the expression of immune suppressive molecules such as B7-H1 on MDSCs and TAMs, and also reduces tumor growth. In addition, Tregs produce IL-10, IL-4, and IL-13, and induce monocyte differentiation toward TAMs [43, 57, 59]. MDSCs are reported to differentiate into TAMs in the hypoxic microenvironment, which is regulated by STAT3 activity [60]. Since these immunosuppressive cells and immune checkpoint molecules show cross talk in the hypoxic TME, anti-VEGF/VEGFR therapy can induce tumor oxygenation [61], resulting in an immune-supportive TME.
Alteration in the TME following chemotherapy with or without anti-VEGF/VEGFR therapy
Alteration of immunosuppressive cells and immune checkpoint molecules has been investigated utilizing clinical samples before and after chemotherapy [36, 49, 62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103], but results have been inconsistent (Tables 1 and 2) [36, 49, 62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. Metronomic cyclophosphamide decreases Tregs [62, 66,67,68, 83], and gemcitabine decreases both Tregs and MDSCs [35, 65, 103]. Tregs and MDSCs are not increased when chemotherapeutic agents are co-administered with immunomodulatory agents such as vaccination and IL-2 (Table 1) [49, 63, 75, 78]. However, most chemotherapeutic agents including temozolomide tend to increase Tregs and MDSCs [71, 72, 74, 79, 84]. In addition, chemotherapy also enhances PD-1/PD-L1 via TGF-β induced epithelial-mesenchymal transition [69].
In contrast, combinational chemotherapy with VEGF/VEGFR-targeted agents could make the TME immune supportive [23, 96]. VEGF/VEGFR-targeted therapy such as Bev, sorafenib, and sunitinib reduces the population of Tregs in the peripheral blood and tumor tissue in GBs, metastatic colorectal cancer, hepatocellular carcinoma, and renal cell carcinoma (Table 2) [86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. However, some studies using paired peripheral blood before and after VEGFR-targeted therapy demonstrated that Tregs and PD-1 were significantly increased [104]. The effect of anti-angiogenic therapy has been highly controversial regarding MDSCs [91, 105]. Serum VEGF-A levels are correlated with the population of MDSCs, and Bev may decrease MDSCs [90, 94, 95]. Sunitinib and sorafenib may also decrease the population of MDSCs and recover the Th1 reaction in the patients of hepatocellular carcinoma and metastatic kidney cancer [93, 105]. However, the population of MDSCs does not change following Bev and axitinib, despite decreasing in serum VEGF of patients with kidney cancer [106]. The status of MDSCs following anti-VEGF/VEGFR therapy is inconsistent [88, 90, 93,94,95, 97, 98, 104].
Furthermore, in the recurrent stage after anti-angiogenic therapy, the status of immunosuppressive cells and immune checkpoint molecules is also highly controversial (Table 2). VEGF-targeted therapy can lead to either tumor oxygenation or tumor hypoxia [14, 61, 107]. Therefore, both an immune-supportive TME in normoxic conditions and a hypoxia-induced immunosuppressive TME have been reported after VEGF-targeted therapy [14, 61, 107]. Tregs in the peripheral blood are increased in patients with recurrent GBs after development of resistance to VEGFR inhibitors [89]. An increased level of PD-1 expression on CD4 and CD8 T cells was reported in patients with GBs or metastatic renal cell carcinoma that is refractory to VEGFR-targeted therapy [89, 109]. Previous studies have demonstrated changes in the TME using tumor specimens resected under and after Bev therapy. Bev downregulates the expression of PD-1 and PD-L1 immune checkpoint molecules, suppresses the infiltration of TAMs and Tregs, and increases CTL infiltration. Importantly, the conditions are sustained during long-term Bev usage [14]. VEGF persistently contributes to tumor growth, even if secondary signaling pathways are upregulated. These findings support the concepts of continuous usage as Bev beyond progression [110]. The reason for the discrepancy in the status of immunosuppressive cells and molecules at the recurrent stage among relevant studies remains unclear. The reason may be the difference between peripheral blood and tumors, the difference in the target of inhibition, or the response rate of targeted therapies. Tregs, PD-L1, and TAMs are upregulated in patients showing partial response [101]. Tada et al. suggested that analyses of TILs and immune cells using tumor specimens are more important than analyses of peripheral blood for investigation of cancer immunology [36]. Further investigation using tumor samples as well as peripheral blood may be required for monitoring immunosuppressive cells and molecules and seeking for predictable and prognostic biomarkers.
Future direction
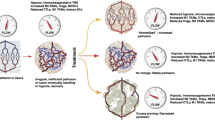
VEGF plays a key role in the development of the immunosuppressive TME by inhibition of DC maturation and enhancement of immunosuppressive cells and molecules [21, 23]. Immunologically “cold” tumors are unresponsive to immunotherapies including immune checkpoint inhibitors. Anti-angiogenic therapy may change the TME into an immunological favorable “hot” microenvironment. Bev suppresses immunosuppressive cells including TAMs, Tregs, and MDSCs, and improves the migratory capacity of CTLs [14]. Theoretically, Bev could enhance the effect of PD-1/PD-L1 inhibitors (Fig. 2). Therefore, many basic research studies have demonstrated that combination therapy of VEGF/VEGFR inhibitors and PD-1/PD-L1 inhibitors induces a synergistic effect on several types of tumors including GBs, melanoma, lung cancer, and hepatocellular carcinoma [92, 96, 111]. Patients with metastatic renal cell carcinoma show improvement in antigen-specific T cell migration after combination usage of anti-VEGF and anti-PD-L1 antibodies [111]. Currently, clinical trials testing theses combination therapies are being conducted for several types of tumors such as recurrent GBs, renal cell carcinoma, colorectal cancer, and ovarian cancer (NCT03024437, NCT02659384, NCT02873962, NCT02017717) [16]. This treatment strategy may lead to promising results for those malignant refractory tumors. However, high dose and long-term usage of anti-VEGF/VEGFR therapy is associated with hypoxia which is one mechanism of resistance to this combination therapy [108]. Further investigations are warranted to determine the adequate dosage and duration of anti-angiogenic agents for the combination usage with immune checkpoint inhibitors. Because immune checkpoint inhibitors and anti-VEGF/VEGFR treatment are immunological “break off” strategies, other immunotherapies such as DCs-based immunotherapy [112], tumor vaccine therapy [113], and chimeric antigen receptor T-cell therapy [114] should be added to them as “acceleration on” strategies to improve therapeutic efficacy. Furthermore, hypoxia-targeted therapy is another treatment strategy to overcome the mechanisms of resistance to immunotherapy, via suppressing Tregs, TAMs, and MDSCs [115].

Synergic treatment strategy of VEGF/VEGFR-targeted therapy and immune checkpoint inhibitor. Cytotoxic T cells (CTLs) are excluded by components of the tumor microenvironment (TME). Immune checkpoint molecules, and immunosuppressive cells including tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs) were induced into the TME via VEGF-A. Anti-angiogenic therapy, such as bevacizumab (Bev), changes the TME into an immunological favorable microenvironment. Bev improves the migratory capacity of CTLs. Although alteration of TME after Bev administration has been still inconsistent, Bev has the possibility to suppress some immunosuppressive molecules and cells. Therefore, Bev can enhance the effect of immune checkpoint inhibitors, leading to the synergic effect. Furthermore, immunological “acceleration on” therapies, such as dendritic cells (DCs)-based immunotherapy, tumor vaccine therapy, and chimeric antigen receptor T-cell therapy should be added to “break off” therapy including immune checkpoint inhibitors and anti-angiogenic therapy, to improve therapeutic efficacy. Bev bevacizumab, CAR chimeric antigen receptor, CTL cytotoxic T cell, DC dendritic cell, MDSC myeloid-derived suppressor cells, VEGF-A vascular endothelial growth factor, PD-1 programmed cell death 1, PD-L1 programmed cell death ligand-1, Treg regulatory T cell, TAM tumor-associated macrophage
As described above, reprogramming the TME to an immune-supportive microenvironment improves cancer immunotherapy [36, 49, 62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103]. To conquer therapeutic refractoriness to immunotherapy, the cross talk of immunosuppressive cells and molecules in the TME must be comprehensively regulated.
References
Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–8.
Chaudhry IH, O’Donovan DG, Brenchley PE, Reid H, Roberts IS. Vascular endothelial growth factor expression correlates with tumour grade and vascularity in gliomas. Histopathology. 2001;39:409–15.
Alphandéry E. Glioblastoma treatments: an account of recent industrial developments. Front Pharmacol. 2018;13(9):879.
Qazi MA, Vora P, Venugopal C, Sidhu SS, Moffat J, Swanton C, Singh SK. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol. 2017;28:1448–56.
Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest. 2017;97:498–518.
Iwai Y, Okazaki T, Nishimura H, Kawasaki A, Yagita H, Honjo T. Microanatomical localization of PD-1 in human tonsils. Immunol Lett. 2002;83:215–20.
Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med. 2003;198:39–50.
Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, Cloughesy T. Bevacizumab plus radiotherapy-temozolomide for new diagnosed glioblastoma. N Engl J Med. 2014;370:709–22.
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran WJ Jr, Mehta MP. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68.
Fricke I, Mirza N, Dupont J, Lockhart C, Jackson A, Lee JH, Sosman JA, Gabrilovich DI. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin Cancer Res. 2007;13:4840–8.
Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, Carbone DP. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–86.
Tamura R, Tanaka T, Miyake K, Yoshida K, Sasaki H. Bevacizumab for malignant gliomas: current indications, mechanisms of action and resistance, and markers of response. Brain Tumor Pathol. 2017;34:62–77.
Tamura R, Tanaka T, Ohara K, Miyake K, Morimoto Y, Yamamoto Y, Kanai R, Akasaki Y, Murayama Y, Tamiya T, Yoshida K, Sasaki H. Persistent restoration to the immunosupportive tumor microenvironment in glioblastoma by bevacizumab. Cancer Sci. 2019;110:499–508.
Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27:1492–504.
Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, Voloschin A, Ramkissoon SH, Ligon KL, Latek R, Zwirtes R, Strauss L, Paliwal P, Harbison CT, Reardon DA, Sampson JH. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20:674–86.
Ott PA, Hodi FS, Buchbinder E. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Front Oncol. 2015;5:202.
Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–39.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76.
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–27.
Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–103.
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138:105–15.
Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, Stockmann C, Combe P, Berger A, Zinzindohoue F, Yagita H, Tartour E, Taieb J, Terme M. VEGF-A modulates expression of inhibitory checkpoints on CD8+T cells in tumors. J Exp Med. 2015;212:139–48.
Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. 2014;20:607–15.
Blancher C, Moore JW, Talks KL, Houlbrook S, Harris AL. Relationship of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression to vascular endothelial growth factor induction and hypoxia survival in human breast cancer cell lines. Cancer Res. 2000;60:7106–13.
Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13.
Alfarouk KO, Verduzco D, Rauch C, Muddathir AK, Adil HH, Elhassan GO, Ibrahim ME, David Polo Orozco J, Cardone RA, Reshkin SJ, Harguindey S. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. 2014;1:777–802.
Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, Leblanc P, Munn LL, Huang P, Duda DG, Fukumura D, Jain RK, Poznansky MC. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA. 2012;109:17561–6.
Tamura R, Tanaka T, Yamamoto Y, Akasaki Y, Sasaki H. Dual role of macrophage in tumor immunity. Immunotherapy. 2018;10:899–909.
Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. 2005;23:953–64.
Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–12.
Castro BA, Flanigan P, Jahangiri A, Hoffman D, Chen W, Kuang R, De Lay M, Yagnik G, Wagner JR, Mascharak S, Sidorov M, Shrivastav S, Kohanbash G, Okada H, Aghi MK. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti-angiogenic therapy. Oncogene. 2017;36:3749–59.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63.
Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory Tcell lineage specification by the forkhead transcription factor Foxp3. Immunity. 2005;22:329–34.
Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32.
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, Hiraoka N, Fuller GN. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res. 2008;14:5166–72.
Wada J, Yamasaki A, Nagai S, Yanai K, Fuchino K, Kameda C, Tanaka H, Koga K, Nakashima H, Nakamura M, Tanaka M, Katano M, Morisaki T. Regulatory T-cells are possible effect prediction markers of immunotherapy for cancer patients. Anticancer Res. 2008;28:2401–8.
Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–506.
Dugast AS, Haudebourg T, Coulon F, Heslan M, Haspot F, Poirier N, de Silly RV, Usal C, Smit H, Martinet B, Thebault P, Renaudin K, Vanhove B. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. 2008;180:7898–906.
Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, Matrisian LM, Carbone DP, Lin PC. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21.
Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA, Kang CY. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol. 2009;182:1818–28.
Otvos B, Silver DJ, Mulkearns-Hubert EE, Alvarado AG, Turaga SM, Sorensen MD, Rayman P, Flavahan WA, Hale JS, Stoltz K, Sinyuk M, Wu Q, Jarrar A, Kim SH, Fox PL, Nakano I, Rich JN, Ransohoff RM, Finke J, Kristensen BW, Vogelbaum MA, Lathia JD. Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells. 2016;34:2026–39.
Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, Hockstein N, Guarino M, Masters G, Penman E, Denstman F, Xu X, Altieri DC, Du H, Yan C, Gabrilovich DI. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity. 2016;44:303–15.
Kumar R, de Mooij T, Peterson TE, Kaptzan T, Johnson AJ, Daniels DJ, Parney IF. Modulating glioma-mediated myeloid-derived suppressor cell development with sulforaphane. PLoS ONE. 2017;12:e0179012.
Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, Garcia J, Vogelbaum MA. Finke J Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol. 2011;13:591–9.
Kumar V, Gabrilovich DI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology. 2014;143:512–9.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, Ring AM, Connolly AJ, Weissman IL. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–9.
Vlahovic G, Fecci PE, Reardon D, Sampson JH. Programmed death ligand 1 (PD-L1) as an immunotherapy target in patients with glioblastoma. Neuro Oncol. 2015;17:1043–5.
Miyazaki T, Ishikawa E, Matsuda M, Akutsu H, Osuka S, Sakamoto N, Takano S, Yamamoto T, Tsuboi K, Matsumura A. Assessment of PD-1 positive cells on initial and secondary resected tumor specimens of newly diagnosed glioblastoma and its implications on patient outcome. J Neurooncol. 2017;133:277–85.
Xue S, Song G, Yu J. The prognostic significance of PD-L1 expression in patients with glioma: a meta-analysis. Sci Rep. 2017;7:4231.
Garber ST, Hashimoto Y, Weathers SP, Xiu J, Gatalica Z, Verhaak RG, Zhou S, Fuller GN, Khasraw M, de Groot J, Reddy SK, Spetzler D, Heimberger AB. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol. 2016;18:1357–66.
Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, Kurscheid S, Kresl P, Wöhrer A, Marosi C, Hegi ME, Preusser M. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 2017;19:1460–8.
Xue S, Hu M, Li P, Ma J, Xie L, Teng F, Zhu Y, Fan B, Mu D, Yu J. Relationship between expression of PD-L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget. 2017;8:49702–12.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1α and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90.
Dogan HT, Kiran M, Bilgin B, Kiliçarslan A, Sendur MAN, Yalçin B, Ardiçoglu A, Atmaca AF, Gumuskaya B. Prognostic significance of the programmed death ligand 1 expression in clear cell renal cell carcinoma and correlation with the tumor microenvironment and hypoxia inducible factor expression. Diag Pathol. 2018;13:60.
Barscum IB, Smallwod CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adoptive immunity in cancer cells. Cancer Res. 2014;74:665–74.
Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH. Regulatory T cells stimulate B7-H1 expression in myeloid derived suppressor cells in rat melanoma. J Invest Dermatol. 2012;132:1239–46.
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. HIF-1α regulates function and differentiation of myeloid-suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–53.
Fujimura T, Kambayashi Y, Aiba S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology. 2012;1:1433–4.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–20.
Tamura R, Tanaka T, Miyake K, Tabei Y, Ohara K, Sampetrean O, Kono M, Mizutani K, Yamamoto Y, Murayama Y, Tamiya T, Yoshida K, Sasaki H. Histopathological investigation of glioblastomas resected under bevacizumab treatment. Oncotarget. 2016;7:52423–35.
Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, Valle J, Smith D, Wadsley J, Cunningham D, Pandha H, Neoptolemos JP, Middleton G. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived supperssor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother. 2014;63:175–83.
Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, Norberg P, Xie W, Herndon JE 2nd, Healy P, McLendon RE, Friedman AH, Friedman HS, Bigner D, Vlahovic G, Mitchell DA, Sampson JH. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. 2017;23:1898–909.
Ellebaek E, Engell-Noerregaard L, Iversen TZ, Froesig TM, Munir S, Hadrup SR, Andersen MH, Svane IM. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: results from a phase II trial. Cancer Immunol Immunother. 2012;61:1791–804.
Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 2016;14:282.
Ge Y, Domschke C, Stoiber N, Schott S, Heil J, Rom J, Blumenstein M, Thum J, Sohn C, Schneeweiss A, Beckhove P, Schuetz F. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother. 2012;61:353–62.
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–8.
Greten TF, Ormandy LA, Fikuart A, Höchst B, Henschen S, Hörning M, Manns MP, Korangy F. Low-dose cyclophosphamide treatment impairs regulatory T cells and unmasks AFP-specific CD4+ T-cell responses in patients with advanced HCC. J Immunother. 2010;33:211–8.
Haratake N, Toyokawa G, Tagawa T, Kozuma Y, Matsubara T, Takamori S, Akamine T, Yamada Y, Oda Y, Maehara Y. Positive conversion of PD-L1 expression after treatments with chemotherapy and nivolmab. Anticancer Res. 2017;37:5713–7.
Homma Y, Taniguchi K, Nakazawa M, Matsuyama R, Mori R, Takeda K, Ichikawa Y, Tanaka K, Endo I. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin Transl Oncol. 2014;16:330–5.
Kim HS, Kim JY, Lee YJ, Kim SH, Lee JY, Nam EJ, Kim S, Kim SW, Kim YT. Expression of programmed cell death ligand 1 and immune checkpoint markers in residual tumors after neoadjuvant chemotherapy for advanced high-grade serous ovarian cancer. Gynecol Oncol. 2018;151:414–21.
Koumarianou A, Christodoulou MI, Patapis P, Papadopoulos I, Liakata E, Giagini A, Stavropoulou A, Poulakaki N, Tountas N, Xiros N, Economopoulos T, Pectasides D, Tsitsilonis OE, Pappa V. The effect of metronomic versus standard chemotherapy on the regulatory to effector T-cell equilibrium in cancer patients. Exp Hematol Oncol. 2014;3:3.
Liang Y, Lü W, Zhang X, Lü B. Tumor-infiltrating CD8+ and FOXP3+ lymphocytes before and after neoadjuvant chemotherapy in cervical cancer. Diagn Pathol. 2018;24:93.
Mesnage SJL, Auguste A, Genestie C, Dunant A, Pain E, Drusch F, Gouy S, Morice P, Bentivegna E, Lhomme C, Pautier P, Michels J, Le Formal A, Cheaib B, Adam J, Leary AF. Neoadjuvant chemotherapy (NACT) increases immune infiltration and programmed death-ligand 1 (PD-L1) expression in epithelial ovarian cancer (EOC). Ann Oncol. 2017;28:651–7.
Mitchell DA, Cui X, Schmittling RJ, Sanchez-Perez L, Snyder DJ, Congdon KL, Archer GE, Desjardins A, Friedman AH, Friedman HS, Herndon JE 2nd, McLendon RE, Reardon DA, Vredenburgh JJ, Bigner DD, Sampson JH. Monoclonal antibody blockade of IL-2 receptor during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood. 2011;118:3003–12.
Pelekanou V, Carvajal-Hausdorf DE, Altan M, Wasserman B, Carvajal-Hausdorf C, Wimberly H, Brown J, Lannin D, Pusztai L, Rimm DL. Effect of neoadjuvant chemotherapy on tumor-infiltrating lymphocytes and PD-L1 expression in breast cancer and its clinical significance. Breast Cancer Res. 2017;19:91.
Rojko L, Reiniger L, Téglási V, Fábián K, Pipek O, Vágvölgyi A, Agócs L, Fillinger J, Kajdácsi Z, Tímár J, Döme B, Szállási Z, Moldvay J. Chemotherapy treatment is associated with altered PD-L1 expression in lung cancer patients. J Cancer Res Clin Oncol. 2018;144:1219–26.
Sampson JH, Schmittling RJ, Archer GE, Congdon KL, Nair SK, Reap EA, Desjardins A, Friedman AH, Friedman HS, Herndon JE 2nd, Coan A, McLendon RE, Reardon DA, Vredenburgh JJ, Bigner DD, Mitchell DA. A pilot study of IL-2Rα blockade during lymphopenia depletes regulatory T-cells and correlates with enhanced immunity in patients with glioblastoma. PLoS ONE. 2012;e7:e31046.
Shinto E, Hase K, Hashiguchi Y, Sekizawa A, Ueno H, Shikina A, Kajiwara Y, Kobayashi H, Ishiguro M, Yamamoto J. CD8+ and FOXP3+ tumor-infiltrating T cells before and after chemoradiotherapy for rectal cancer. Ann Surg Oncol. 2014;21:S414–21.
Takahashi H, Sakakura K, Mito I, Ida S, Chikamatsu K. Dynamic changes in immune cell profile in head and neck squamous cell carcinoma: immunomodulatory effects of chemotherapy. Cancer Sci. 2016;107:1065–71.
Tarhini AA, Edington H, Butterfield LH, Lin Y, Shuai Y, Tawbi H, Sander C, Yin Y, Holtzman M, Johnson J, Rao UN, Kirkwood JM. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE. 2014;9:e87705.
Teng F, Meng X, Kong L, Mu D, Zhu H, Liu S, Zhang J, Yu J. Tumor-infiltrating lymphocytes, forkhead box P3, programmed death ligand-1, and cytotoxic T lymphocyte-associated antigen-4 expressions before and after neoadjuvant chemoradiation in rectal cancer. Transl Res. 2015;166:721–32.
Verma C, Eremin JM, Robins A, Bennett AJ, Cowley GP, El-Sheemy MA, Jibril JA, Eremin O. Abnormal T regulatory cells (Tregs: FOXP3+, CTLA-4+), myeloid-derived suppressor cells (MDSCs: monocytic, granulocytic) and polarised T helper cell profiles (Th1, Th2, Th17) in women with large and locally advanced breast cancers undergoing neoadjuvant chemotherapy (NAC) and surgery: failure of abolition of abnormal treg profile with treatment and correlation of treg levels with pathological response to NAC. J Transl Med. 2013;11:16.
Wesolowski R, Duggan MC, Stiff A, Markowitz J, Trikha P, Levine KM, Schoenfield L, Abdel-Rasoul M, Layman R, Ramaswamy B, Macrae ER, Lustberg MB, Reinbolt RE, Mrozek E, Byrd JC, Caligiuri MA, Mace TA, Carson WE 3rd. Circulating myeloid-derived suppressor cells increase in patients undergoing neo-adjuvant chemotherapy for breast cancer. Cancer Immunol Immunother. 2017;66:1437–47.
Fadul CE, Fisher JL, Gui J, Hampton TH, Côté AL, Ernstoff MS. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 2011;13:393–400.
Adotevi O, Pere H, Ravel P, Haicheur N, Badoual C, Merillon N, Medioni J, Peyrard S, Roncelin S, Verkarre V, Mejean A, Fridman WH, Oudard S, Tartour E. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J Immunother. 2010;33:991–8.
Desar IME, Jacobs JH, Hulsbergen-vandeKaa CA, Oyen WJ, Mulders PF, van der Graaf WT, Adema GJ, van Herpen CM, de Vries IJ. Sorafenib reduces the percentage of tumour infiltrating regulatory T cells in renal cell carcinoma patients. Int J Cancer. 2011;129:507–12.
Donini M, Buti S, Lazzarelli S, Bozzetti R, Rivoltini L, Camisaschi C, Castelli C, Bearz A, Simonelli C, Lo Re G, Mattioli R, Caminiti C, Passalacqua R. Dose-finding/phase II trial: bevacizumab, immuneotherapy, and chemotherapy (BIC) in metastatic renal cell cancer (mRCC). Antituor effects and variation of circulating T regulatory cells (Treg). Targ Oncol. 2015;10:277–86.
Du Four S, Maenhout SK, Benteyn D, De Keersmaecker B, Duerinck J, Thielemans K, Neyns B, Aerts JL. Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol Immunother. 2016;65:727–40.
Feng PH, Chen KY, Huang YC, Luo CS, Wu SM, Chen TT, Lee CN, Yeh CT, Chuang HC, Han CL, Lin CF, Lee WH, Kuo CH, Lee KY. Bevacizumab reduces S100A9-positive MDSCs linked to intracranial control in patients with EGFR-mutant lung adenocarcinoma. J Thorac Oncol. 2018;13:958–67.
Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol. 2011;11:856–61.
Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, Zeng W, Giobbie-Hurder A, Atkins MB, Ibrahim N, Friedlander P, Flaherty KT, Murphy GF, Rodig S, Velazquez EF, Mihm MC Jr, Russell S, DiPiro PJ, Yap JT, Ramaiya N, Van den Abbeele AD, Gargano M, McDermott D. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol Res. 2014;2:632–42.
Kalathil SG, Lugade AA, Miller A, Iyer R, Thanavala Y. PD-1+ and Foxp3+ T cell reduction correlates with survival of HCC patients after sorafenib therapy. JCI Insight. 2016;1:e86182.
Koinis F, Vetsika EK, Aggouraki D, Skalidaki E, Koutoulaki A, Gkioulmpasani M, Georgoulias V, Kotsakis A. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulatins in the peripheral blood of patients with non-small cell lung cancer. J Thorac Oncol. 2016;11:1263–72.
Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, Boidot R, Végran F, Bonnefoy N, Vincent J, Bengrine-Lefevre L, Ladoire S, Delmas D, Apetoh L, Ghiringhelli F. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016;76:5241–52.
Liu XD, Hoang A, Zhou L, Kalra S, Yetil A, Sun M, Ding Z, Zhang X, Bai S, German P, Tamboli P, Rao P, Karam JA, Wood C, Matin S, Zurita A, Bex A, Griffioen AW, Gao J, Sharma P, Tannir N, Sircar K, Jonasch E. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol Res. 2015;3:1017–29.
Quillien V, Carpentier AF, Gey A, Avril T, Tartour E, Sejalon F, Campillo-Gimenez B, Vauleon E. Absolute numbers of regulatory T cells and neutrophils in corticosteroid-free patients are predictive for response to bevacizumab in recurrent glioblastoma patients. Cancer Immunol Immunother. 2019;68:871–82.
Tamma R, Rutigliano M, Lucarelli G, Annese T, Ruggieri S, Cascardi E, Napoli A, Battaglia M, Ribatti D. Microvascualr density, macrophages, and mast cells in human clear cell renal carcinoma with and without bevacizumab treatment. Urol Oncol. 2019;6:S1078–439.
Terme M, Colussi O, Marcheteau E, Tanchot C, Tartour E, Taieb J. Modulation of immunity by antiangiogenic molecules in cancer. Clin Dev Immunol. 2012. https://doi.org/10.1155/2012/492920.
Thomas AA, Fisher JL, Hampton TH, Christensen BC, Tsongalis GJ, Rahme GJ, Whipple CA, Steel SE, Davis MC, Gaur AB, Lewis LD, Ernstoff MS, Fadul CE. Immune modulation associated with vascular endothelial growth factor (VEGF) blockade in patients with glioblastoma. Cancer Immunol Immunother. 2017;66:379–89.
Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, Denker M, Leveque V, Cañamero M, Babitski G, Koeppen H, Ziai J, Sharma N, Gaire F, Chen DS, Waterkamp D, Hegde PS, McDermott DF. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624.
Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, Riedemann L, Taylor J, Ivy P, Duda DG, Ancukiewicz M, Plotkin SR, Chi AS, Gerstner ER, Eichler AF, Dietrich J, Stemmer-Rachamimov AO, Batchelor TT, Jain RK. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013;15:1079–87.
Vizio B, Novarino A, Giacobino A, Cristiano C, Prati A, Ciuffreda L, Montrucchio G, Bellone G. Potential plasticity of T regulatory cells in pancreatic carcinoma in relation to disease progression and outcome. Exp Ther Med. 2012;4:70–8.
Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, Doi T, Wada H, Nishikawa H, Shitara K. Targeting VEGFR2 with Ramucirumab strongly impacts effector/activated regulatory T cells and CD8+ T cells in the tumor microenvironment. J Immunother Cancer. 2018;6:106.
Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, Michielin O, Romano E, Speiser DE. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63:247–57.
Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, Finke JH. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–57.
Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, Ochoa AC. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. 2009;69:1553–60.
Yamamoto Y, Tamura R, Tanaka T, Ohara K, Tokuda Y, Miyake K, Takei J, Akasaki Y, Yoshida K, Murayama Y, Sasaki H. “Paradoxical” findings of tumor vascularity and oxygenation in recurrent glioblastomas refractory to bevacizumab. Oncotarget. 2017;8:103890–9.
Shin SJ, Jeon YK, Cho YM, Lee JL, Chung DH, Park JY, Go H. The association between PD-L1 expression and the clinical outcomes to vascular endothelial growth factor-targeted therapy in patients with metastatic clear renal cell carcinoma. Oncologist. 2015;20:1253–60.
Grothey A, Sugrue MM, Purdie DM, Dong W, Sargent D, Hedrick E, Kozloff M. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol. 2008;26:5326–34.
Rexer H, Doehn C. First-line treatment for advanced renal cell carcinoma: a phase 3, open-label, randomized study of Atezolizumab (Anti-PD-L1-Antibody) in combination with Bevacizumab versus Sunitinib in patients with untreated advanced renal cell carcinoma (“IMmotion”)—AN 37/15 der AUO. Urologe A. 2016;55:1242–3.
Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, Tasaki T, Hayashi K, Komita H, Watanabe N, Suzuki Y, Yamamoto Y, Mori R, Arai T, Tanaka T, Joki T, Yanagisawa T, Murayama Y. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol Immunother. 2016;65:1499–509.
Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, Heth JA, Salacz M, Taylor S, D’Andre SD, Iwamoto FM, Dropcho EJ, Moshel YA, Walter KA, Pillainayagam CP, Aiken R, Chaudhary R, Goldlust SA, Bota DA, Duic P, Grewal J, Elinzano H, Toms SA, Lillehei KO, Mikkelsen T, Walbert T, Abram SR, Brenner AJ, Brem S, Ewend MG, Khagi S, Portnow J, Kim LJ, Loudon WG, Thompson RC, Avigan DE, Fink KL, Geoffroy FJ, Lindhorst S, Lutzky J, Sloan AE, Schackert G, Krex D, Meisel HJ, Wu J, Davis RP, Duma C, Etame AB, Mathieu D, Kesari S, Piccioni D, Westphal M, Baskin DS, New PZ, Lacroix M, May SA, Pluard TJ, Tse V, Green RM, Villano JL, Pearlman M, Petrecca K, Schulder M, Taylor LP, Maida AE, Prins RM, Cloughesy TF, Mulholland P, Bosch ML. First results on survival from a large phase 3 trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16:142.
O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot JM, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9:0984.
Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, Ager C, Nicholas C, Jaiswal AR, Sun Y, Shah K, Balasubramanyam S, Li N, Wang G, Ning J, Zal A, Zal T, Curran MA. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest. 2018;128:5137–49.
Funding
R Tamura has been supported by a Grant-in-Aid for Scientific Research (KAKENHI) by the Ministry of Education, Culture, Sports, Science and Technology and the Japan Society for the Promotion of Science (Grant Numbers 18J21382).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
No object.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tamura, R., Tanaka, T., Akasaki, Y. et al. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol 37, 2 (2020). https://doi.org/10.1007/s12032-019-1329-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-019-1329-2