
Abstract
Monoamine oxidases (MAO) are important components of the homeostatic machinery that maintains the levels of monoamine neurotransmitters, including dopamine, in balance. Given the imbalance in dopamine levels observed in Huntington disease (HD), the aim of this study was to examine MAO activity in a mouse striatal cell model of HD and in human neural cells differentiated from control and HD patient-derived induced pluripotent stem cell (hiPSC) lines. We show that mouse striatal neural cells expressing mutant huntingtin (HTT) exhibit increased MAO expression and activity. We demonstrate using luciferase promoter assays that the increased MAO expression reflects enhanced epigenetic activation in striatal neural cells expressing mutant HTT. Using cellular stress paradigms, we further demonstrate that the increase in MAO activity in mutant striatal neural cells is accompanied by enhanced susceptibility to oxidative stress and impaired viability. Treatment of mutant striatal neural cells with MAO inhibitors ameliorated oxidative stress and improved cellular viability. Finally, we demonstrate that human HD neural cells exhibit increased MAO-A and MAO-B expression and activity. Altogether, this study demonstrates abnormal MAO expression and activity and suggests a potential use for MAO inhibitors in HD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Huntington disease (HD) is an autosomal dominant neurodegenerative disorder that has no known cure. It is caused by an expansion of a CAG trinucleotide repeat within exon 1 of the huntingtin (HTT) gene, where the onset of motor symptoms and severity of neuropathology are dependent on the size of the trinucleotide expansion [1, 2]. The clinical profile of HD includes motor, cognitive, and psychiatric dysfunction. Pathologically, HD is characterized by preferential loss of neurons in the striatum and deep layers of the cortex.
Molecular assessments during disease progression show discrepancies in neurotransmitter levels [3, 4]. Dopamine, a monoamine, is one such neurotransmitter. Dopamine homeostasis and dopamine-regulated pathways are altered in HD mouse models [5, 6]. In the R6/1 mouse model of HD, the levels of striatal dopamine and its metabolite homovanillic acid are significantly reduced [7]. Dopamine turnover is also altered in R6/1 mice [7]. In R6/2 mice, significant deficits are observed in extracellular and tissue levels of striatal dopamine and in D1-class dopamine receptor signaling [5, 8–10]. Similarly, baseline and evoked extracellular levels of striatal dopamine are reduced in YAC128 HD mice [5, 10, 11]. In pre-symptomatic HD gene carriers, a progressive loss of dopamine D1 and D2 receptor (D1R and D2R) binding is observed by PET imaging [12–15]. Furthermore, in post mortem HD brains, there is a reduced number of striatal D1R- and D2R-positive cells as assessed by messenger RNA (mRNA) detection [1, 16], and the levels of striatal dopamine are also significantly reduced [3, 17].
One avenue of processing dopamine is through the activity of monoamine oxidase (MAO) enzymes. MAOs are flavin-containing enzymes located on the mitochondrial outer membrane which oxidatively deaminate monoamines, generating hydrogen peroxide as a by-product [5, 18]. Present as two isozymes, MAO-A and MAO-B are encoded by separate genes adjacent to each other on the short arm of the X chromosome. They also have identical exon-intron structural organization suggesting that they were derived as a result of ancestral gene duplication [5, 18]. The two isozymes share 70 % similarity in amino acid sequence. Dopamine is a common substrate for both isozymes but MAO-A has a higher affinity toward serotonin, norepinephrine, and epinephrine [5, 19, 20]. Although MAO proteins are found throughout the brain, MAO activity varies considerably from region to region, with the caudate showing the highest levels of activity [12, 21].
Abnormal MAO-A and MAO-B activity has been implicated in various mental and neurodegenerative disorders [16, 21], and pharmacological inhibitors of MAO have been shown to be of clinical benefit [17, 21]. Given the marked deficits in dopamine levels and signaling in HD, the aim of this study was to examine MAO activity in HD patient and rodent-derived cellular systems.
Materials and Methods
Cell Culture and Transfections
STHdh Q7/Q7 and STHdh Q111/Q111 cells were generated as described by [18, 22]. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) with 10 % fetal bovine serum (FBS; HyClone) and 5 mM glutamine (Gibco). To transfect the cells, cells were seeded and grown until 90 % confluency before FuGENE® 6 (Promega) was used according to the manufacturer’s instructions. In brief, a DNA/FuGENE ratio of 1:3 was used and the cells were incubated with the mix over 24 h before media was changed.
Human dermal fibroblasts and HD patient-derived induced pluripotent stem cell (hiPSC) lines from HD patients and controls were obtained from Coriell. The sample IDs are listed in Table 1. The dermal fibroblast cells were grown in α-MEM (HyClone) with 10 % FBS (HyClone) and 5 mM glutamine (Gibco). The hiPSC lines were grown as described in the “Derivation of Neural Progenitor Cells” section.
Serum Starvation and Drug Treatment
To starve STHdh Q7/Q7 and STHdh Q111/Q111 cells of serum, cells were seeded onto appropriate tissue culture plates and grown until confluent. The cells were washed once with PBS before medium without serum (DMEM with 5 mM glutamine) was added. If MAO inhibitors were included into the experiment, the drugs were added upon the start of 24-h serum starvation.
Derivation of Neural Progenitor Cells
CAG33 and CAG180 hiPSCs employed in this study were described by the HD iPSC Consortium, 2012 [23]. Neural progenitor cells (NPCs) were derived using the protocol described in Li et al. [24]. In brief, CAG33 and CAG180 hiPSCs at about 20 % confluence were treated with N2B27 media (DMEM/F12: neurobasal media (1:1), 1× N2, 1× B27, 1× pen/strep/glutamine, 10 ng/mL hLIF (Millipore), 5 μg/mL BSA) containing 3 μM CHIR99021 (Tocris), 2 μM SB431542 (Tocris), and 0.1 μM compound E for 7 days. The culture was then split 1:3 for the next six passages using Accutase without compound E on Matrigel-coated plates.
RNA Isolation and Quantitative PCR
RNA from cells was extracted using RNeasy mini kit (QIAGEN) according to the manufacturer’s instructions. To generate complementary DNA (cDNA), Superscript® II Reverse Transcription Kit (Life Technologies) was used. A total of 20 μL of cDNA was generated per 1 μg of RNA. To perform quantitative PCR (qPCR), cDNA was diluted 10-fold and 2 μL was used per qPCR reaction. To complete the reaction volume, 0.67 mM primers and SYBR® Select Master Mix (Life Technologies) were added. The primers used were as follows:
-
mMAO-A-F: 5′ GCCCAGTATCACAGGCCAC 3′,
-
mMAO-A-R: 5′ GTCCCACATAAGCTCCACCA 3′
-
mMAO-B-F: 5′ ATGAGCAACAAAAGCGATGTGA 3′,
-
mMAO-B-R: 5′ TTCTAATTGTGTAAGTCCTGCCT 3′,
-
mActin-F: 5′ GGCTGTATTCCCCTCCATCG 3′,
-
mActin-F: 5′ CCAGTTGGTAACAATGCCATGT 3′,
-
hMAO-A-F: 5′ TTCAGGACTATCTGCTGCCAA 3′,
-
hMAO-A-R: 5′ GGTCCCACATAAGCTCCACC 3′,
-
hMAO-B-F: 5′ GCTCTCTGGTTCCTGTGGTATGTG 3′,
-
hMAO-B-R: 5′ TCCGCTCACTCACTTGACCAGATC 3′,
-
hActin-F: 5′ GTCTTCCCCTCCATCGTG 3′,
-
hActin-R: 5′ GATGGGGTACTTCAGGGTGA 3′
-
hOCT4-F: 5′ TCTTTCCACCAGGCCCCCGGCTC 3′
-
hOCT4-R: 3′ TGCGGGCGGACATGGGGAGATCC 3′
-
hNESTIN-F: 5′ GAAACAGCCATAGAGGGCAAA 3′
-
hNESTIN-R: 5′ TGGTTTTCCAGAGTCTTCAGTGA 3′
-
hPAX6-F: 5′ GTGTCTACCAACCAATTCCACAAC 3′
-
hPAX6-R: 5′ CCCAACATGGAGCCAGATG 3′
-
hSOX1-F: 5′ CAGTACAGCCCCATCTCCAAC 3′
-
hSOX1-R: 5′ GCGGGCAAGTACATGCTGA 3′
Immunoblotting
STHdh Q7/Q7 and STHdh Q111/Q111 cells were pelleted and lysed with homogenization buffer (50 mM Tris HCl pH 8.0, 150 mM NaCl, 1 % IGEPAL, 1 mM PMSF, 5 μM z-VAD and 1× complete protease inhibitor cocktail (Roche)). The cell pellets were resuspended in the homogenization buffer and incubated on ice for 20 min. The lysates were centrifuged at 4 °C at 13,300 rpm for 15 min and the supernatant was transferred to a new tube. The protein concentration was measured using the Bradford Assay (Bio-Rad). Five micrograms of protein was used for each immunoblotting analysis. Protein lysates were separated on a 7 % acrylamide gel and transferred onto a nitrocellulose membrane. Blots were incubated overnight at 4 °C. The antibodies used were anti-calnexin (Sigma C4731 1:2000 dilution), anti-MAO-A (Abcam ab126751 1:1000 dilution), anti-MAO-B (GeneTex GTX105790 1:1000 dilution). Alexa Fluor 680 goat anti-mouse and goat anti-rabbit (Molecular Probes) were used as secondary antibodies (1:3000 dilution). Li-Cor Odyssey Infrared Imaging system was used for detection and quantification.
Luciferase Assays
To perform the luciferase assays, manufacturer’s instructions were followed. The protocols used are briefly described below:
Dual-Glo Luciferase Assay System
In brief, the Dual-Glo Luciferase Assay System (Promega) was used to assay for MAO-A and MAO-B transcriptional activity. Cells were lysed using 5× passive lysis buffer, while shaking at room temperature for 15 min. Fifty microliters of the lysate was transferred into an opaque 96-well plate before equal volume of Luciferin Detection Reagent was added. After incubation at room temperature for 20 min, the signal was measured using FLUOstar Omega (BMG Labtech). To quench the firefly luciferase signal, Stop and Glo buffer was added and Renilla luciferase signal was measured.
CellTiter-Glo®
Cells were washed with PBS after drug treatment and 50 μL of fresh media was added to each well. Equal volume of CellTiter-Glo buffer (Promega) was added to each well and the plate was left to shake at room temperature for 10 min before readings were taken.
MTT Assay
After treatment, cells in a 96-well plate were washed once with 1× PBS and incubated with 100 μL of 1 mg/mL MTT (Sigma) in DMEM and incubated at 37 °C for 4 h. The cells were subsequently washed with 1× PBS before adding 100 μL of DMSO. MTT readings were taken at absorbance 570 nm.
GSH/GSSG-Glo™ Assay
Cells were washed after drug treatment and 50 μL of either total or oxidized glutathione reagent (Promega) was added to each well. After 5 min of shaking, equal volume of luciferin-generation reagent was added and incubated at room temperature for 30 min. Lastly, 100 μL of luciferin detection reagent was added and luminescence was measured.
MAO-Glo Assay System
Protein lysates were diluted to 0.5 mg/mL using lysis buffer. Twenty-five microliters of the diluted lysate was incubated with 25 μL of MAO substrate solution (1:250 dilution of provided MAO substrate to measure MAO-B activity and 1:25 of provided MAO substrate to measure MAO-A activity) (Promega) for 2 h at room temperature. Fifty microliters of luciferin detection reagent was then added and luminescence was measured.
ATP/ADP Measurements
To measure ATP/ADP ratios, ATP/ADP assay kit (Abcam) was used according to the manufacturer’s instructions. In brief, upon treatment in a 96-well plate, nucleotides were released using 100 μL of nucleotide-releasing buffer, before 50 μL of each well was taken to measure its ATP/ADP ratio.
Immunostaining
hiPSCs and NPCs were seeded on Matrigel-coated 13 mm coverslips in 24-well plates. Upon reaching the desired confluency, the wells were washed with 1× PBS and fixed with 4 % formaldehyde in PBS at room temperature for 30 min. The coverslips were washed three times for 5 min each using wash buffer (PBS with 0.3 % Triton X-100 and 2 % goat serum) and blocked with blocking buffer (PBS with 3 % Triton X-100 and 3 % goat serum) at room temperature for 1 h. For staining, the following antibodies and concentrations were used: anti-OCT3/4 (sc-9081; Santa Cruz) 1:100; anti-NESTIN (MAB5326; Millipore) 1:500. Antibodies were diluted in wash buffer and incubated at 4 °C overnight. The cells were then washed three times for 5 min each using wash buffer and stained with secondary antibodies (Alexa Fluor, Life Technologies) at a dilution of 1:200. After three times of 5-min washes, the cells were stained with DAPI and mounted onto microscope slides using ProLong® Gold Antifade Mountant (Life Technologies).
Statistical Analysis
Data are expressed as means ± SEM. Statistical significance was ascertained by one- or two-way ANOVA with appropriate post hoc testing or by Student’s t test. Where testing of normality was possible, the D’Agostino and Pearson omnibus normality test was used to assess distribution of values. Differences were considered statistically significant when p < 0.05.
Results
MAO-A/B Activity is Elevated in STHdh Q111/Q111 Cells
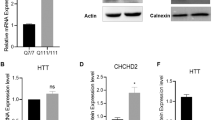
To determine MAO-A/B expression patterns in a HD model, we chose to study a set of well-characterized, immortalized mouse striatal cell lines: the STHdh Q7/Q7 and STHdh Q111/Q111 cells. STHdh Q7/Q7 and STHdh Q111/Q111 cells were derived from E14 striatal primordia of mice expressing 7 and 111 CAG repeats within exon 1 of the Hdh locus, respectively [18, 25–27]. Assessment of MAO-A/B mRNA levels in the two cell lines showed that MAO-A and MAO-B transcripts were expressed at levels approximately fivefold higher in STHdh Q111/Q111 than STHdh Q7/Q7 cells (unpaired two-tailed t test; p < 0.0001 for MAO-A, p = 0.0052 for MAO-B) (Fig. 1a). Consistent with the differences in transcript levels, quantification of protein levels showed that MAO-A and MAO-B protein levels were fourfold and twofold higher in STHdh Q111/Q111 than STHdh Q7/Q7 cells, respectively (unpaired two-tailed t test; p = 0.0003 for MAO-A, p = 0.0321 for MAO-B) (Fig. 1b).

STHdh Q111/Q111 cells exhibit increased MAO-A/B expression and activity when compared to STHdh Q7/Q7 cells. a Quantitative RT-PCR shows increased MAO-A and MAO-B expression in STHdh Q111/Q111 cells when compared to STHdh Q7/Q7 cells. Values shown were normalized to STHdh Q7/Q7 cells; n = 8; b A representative Western blot (left) showing increased MAO-A and MAO-B expression in STHdh Q111/Q111 cells when compared to STHdh Q7/Q7 cells. (right) Quantification and normalization of the band intensities demonstrate that MAO-A and MAO-B are expressed to a greater extent in STHdh Q111/Q111 cells than STHdh Q7/Q7 cells. Values shown were normalized to STHdh Q7/Q7 cells; n = 9. c Transcriptional activities at MAO-A and MAO-B promoter regions are elevated in STHdh Q111/Q111 cells compared to STHdh Q7/Q7 cells. STHdh Q111/Q111 cells exhibited increased luciferase activity across all promoter lengths, suggesting that transcriptional activity at MAO-A and MAO-B promoter regions are elevated in STHdh Q111/Q111 cells, compared to STHdh Q7/Q7 cells; n = 3. ***p < 0.001 when compared to all the respective controls. d MAO-A and MAO-B activities are elevated in STHdh Q111/Q111 cells. MAO-A and MAO-B activities were measured using MAO-Glo™ Assay System (Promega). STHdh Q111/Q111 cells displayed higher activity when compared to STHdh Q7/Q7 cells; n = 9. Error bars in the bar chart represent standard error of the mean, whereas the whiskers on the box plots represent minimum and maximum values. Q7 and Q111 refer to STHdh Q7/Q7 and STHdh Q111/Q111 cells, respectively. a–d *p < 0.05; **p < 0.01; ***p < 0.001 by unpaired two-tailed t test
Luciferase reporters that were previously established [17, 19, 20] were employed to examine transcriptional activities at MAO-A and MAO-B promoter loci (Fig. 1c). Regions spanning 0.24 to 2 kb upstream of the MAO-A/B transcriptional start site have been described as being critical to promote transcription [21, 28]. We show that MAO-A/B promoter fragments of up to 2 kb were transcriptionally active in both STHdh Q7/Q7 and STHdh Q111/Q111 cells and, importantly, that the transcriptional activity at the MAO-A/B promoter loci was significantly increased in STHdh Q111/Q111 cells compared to STHdh Q7/Q7 cells (unpaired two-tailed t test; for MAO-A p < 0.001 compared to respective STHdh Q7/Q7 controls; for MAO-B p < 0.001 for 0.24-, 0.86-, and 2-kb fragments, and p < 0.01 for 1.3-kb fragment compared to respective STHdh Q7/Q7 controls) (Fig. 1c). To determine the functional significance of the differences in MAO expression, MAO activity was measured in both cell lines. In line with the expression and transcriptional activity reporter data, MAO-A and MAO-B activity were elevated by approximately 1.5-fold and 2-fold in STHdh Q111/Q111 when compared to STHdh Q7/Q7 cells, respectively, (unpaired two-tailed t test; p < 0.05 for MAO-A, and p < 0.001 for MAO-B) (Fig. 1d).
Stress-induced Elevation in MAO Activity Contributes to Reduced Viability in STHdh Q111/Q111 Cells
To examine the effect of differential MAO activity in STHdh Q7/Q7 and STHdh Q111/Q111 cells, we employed a cellular stress paradigm to magnify effects induced by disparities in MAO activity. We chose the widely used serum starvation cellular stress paradigm which has previously been shown to increase MAO expression, and sought to determine the effect of serum starvation in STHdh Q7/Q7 and STHdh Q111/Q111 cells on MAO activity and cell viability [22, 29]. Serum starvation of STHdh Q7/Q7 and STHdh Q111/Q111 cells for 24 h led to increased MAO-A/B mRNA (Fig. 2a), and in particular, in STHdh Q111/Q111 cells compared to STHdh Q7/Q7 (two-way ANOVA; for MAO-A genotype: F(1,33) = 25.70, p < 0.0001; treatment: F(1,33) = 11.17, p = 0.0021; genotype × treatment: F(1,33) = 9.440, p = 0.0042; for MAO-B genotype: F(1,19) = 42.76, p < 0.0001; treatment: F(1,19) = 11.30, p = 0.0033; genotype × treatment: F(1,19) = 8.695, p = 0.0084; Fisher’s least significant difference (LSD) post hoc). Similarly, MAO-A/B protein levels were significantly higher in STHdh Q111/Q111 cells compared to STHdh Q7/Q7 following serum starvation (Fig. 2b) (two-way ANOVA; for MAO-A genotype: F(1,32) = 54.08, p < 0.0001; treatment: F(1,32) = 10.15, p = 0.0032; genotype × treatment: F(1,32) = 5.210, p = 0.0292; for MAO-B genotype: F(1,26) = 11.97, p < 0.0019; treatment: F(1,26) = 8.032, p = 0.0088; genotype × treatment: F(1,26) = 4.644, p = 0.0406; Fisher’s LSD post hoc). From an epigenetic perspective, MAO-A/B promoter regions (2 kb fragment) were more transcriptionally active upon serum deprivation, with significantly higher transcriptional activity in STHdh Q111/Q111 compared to STHdh Q7/Q7 cells (Fig. 2c) (two-way ANOVA; for MAO-A genotype: F(1,8) = 126.6, p < 0.0001; treatment: F(1,8) = 164.4, p < 0.0001; genotype × treatment: F(1,8) = 22.93, p = 0.0014; for MAO-B genotype: F(1,8) = 208.7, p < 0.0001; treatment: F(1,8) = 275.0, p < 0.0001; genotype × treatment: F(1,8) = 21.60, p = 0.0016; Fisher’s LSD post hoc). Similar results were obtained with the shorter MAO-A/B promoter fragments (data not shown). Consistent with these results, MAO-A and MAO-B activity were significantly elevated after 24 h of serum starvation in both STHdh Q7/Q7 and STHdh Q111/Q111 cells, but with a significantly greater increase in the STHdh Q111/Q111 cells (Fig. 2d) (two-way ANOVA; for MAO-A genotype: F(1,15) = 15.97, p = 0.0012; treatment: F(1,15) = 5.348, p = 0.0354; genotype × treatment: F(1,15) = 0.9548, p = 0.3440; for MAO-B genotype: F(1,46) = 12.90, p = 0.0008; treatment: F(1,46) = 14.42, p < 0.0004; genotype × treatment: F(1,46) = 0.1515, p = 0.6989; Fisher’s LSD post hoc).

Serum deprivation increases MAO expression and activity in STHdh Q111/Q111 cells. a Quantitative RT-PCR shows increased MAO-A and MAO-B expression in STHdh Q111/Q111 cells upon serum starvation for 24 h. Values shown were normalized to untreated STHdh Q7/Q7 cells; n = 5; (two-way ANOVA; for MAO-A genotype: F(1,33) = 25.70, p < 0.0001; treatment: F(1,33) = 11.17, p = 0.0021; genotype × treatment: F(1,33) = 9.440, p = 0.0042; for MAO-B genotype: F(1,19) = 42.76, p < 0.0001; treatment: F(1,19) = 11.30, p = 0.0033; genotype × treatment: F(1,19) = 8.695, p = 0.0084; Fisher’s LSD post hoc); b MAO-A and MAO-B protein levels increase upon 24-h serum starvation in STHdh Q111/Q111 cells. Values shown were normalized to untreated STHdh Q7/Q7 cells; n = 9; (two-way ANOVA; for MAO-A genotype: F(1,32) = 54.08, p < 0.0001; treatment: F(1,32) = 10.15, p = 0.0032; genotype × treatment: F(1,32) = 5.210, p = 0.0292; for MAO-B genotype: F(1,26) = 11.97, p < 0.0019; treatment: F(1,26) = 8.032, p = 0.0088; genotype × treatment: F(1,26) = 4.644, p = 0.0406; Fisher’s LSD post hoc). c Transcriptional activity at MAO-A and MAO-B promoters increases upon serum starvation. Transcriptional activity at the MAO-A and MAO-B promoter regions (2-kb promoter fragment) increased upon serum starvation in STHdh Q7/Q7 and STHdh Q111/Q111 cells; n = 3; (two-way ANOVA; for MAO-A genotype: F(1,8) = 126.6, p < 0.0001; treatment: F(1,8) = 164.4, p < 0.0001; genotype × treatment: F(1,8) = 22.93, p = 0.0014; for MAO-B genotype: F(1,8) = 208.7, p < 0.0001; treatment: F(1,8) = 275.0, p < 0.0001; genotype × treatment: F(1,8) = 21.60, p = 0.0016; Fisher’s LSD post hoc) d MAO-A and MAO-B activities are enhanced upon serum starvation. MAO-A and MAO-B activities were measured using MAO-Glo™ Assay System (Promega). STHdh Q7/Q7 cells and STHdh Q111/Q111 cells displayed higher activity upon 24-h serum starvation, with the increase being significantly greater in STHdh Q111/Q111 cells; n = 11; (two-way ANOVA; for MAO-A genotype: F(1,15) = 15.97, p = 0.0012; treatment: F(1,15) = 5.348, p = 0.0354; genotype × treatment: F(1,15) = 0.9548, p = 0.3440; for MAO-B genotype: F(1,46) = 12.90, p = 0.0008; treatment: F(1,46) = 14.42, p < 0.0004; genotype × treatment: F(1,46) = 0.1515, p = 0.6989; Fisher’s LSD post hoc). e Serum withdrawal in STHdh Q7/Q7 cells and STHdh Q111/Q111 cells results in decreased ATP levels. ATP levels were measured using CellTiter-Glo® upon 24-h serum starvation; n = 4; (two-way ANOVA; genotype: F(1,12) = 137.4, p < 0.0001; treatment: F(1,12) = 44.80, p < 0.0001; genotype × treatment: F(1,12) = 9.469, p = 0.0096; Fisher’s LSD post hoc). f Serum withdrawal results in greater reduction in cell viability in STHdh Q111/Q111 cells as assessed by the MTT assay. STHdh Q7/Q7 and STHdh Q111/Q111 cells were serum starved for 24 h before incubation with 1 mg/mL MTT for 4 h; n = 4; (two-way ANOVA; genotype: F(1,12) = 148.5, p < 0.0001; treatment: F(1,12) = 44.85, p < 0.0001; genotype × treatment: F(1,12) = 6.167, p = 0.0288; Fisher’s LSD post hoc). Error bars in the bar chart represent standard error of the mean, whereas the whiskers on the box plots represent minimum and maximum values. Q7 and Q111 refer to STHdh Q7/Q7 and STHdh Q111/Q111 cells, respectively. *p < 0.05; **p < 0.01; ***p < 0.001
Serum starvation is a widely used method to induce cell death in many cellular systems, including the STHdh Q7/Q7 and STHdh Q111/Q111 cells [5, 25–27]. We deprived STHdh Q7/Q7 and STHdh Q111/Q111 cells of serum over 24 h and showed that ATP levels were reduced upon serum withdrawal (Fig. 2e), suggesting that serum starvation led to reduced cell viability. Interestingly, STHdh Q111/Q111 cells have inherently lower ATP levels compared to STHdh Q7/Q7 cells. In addition, serum starvation in STHdh Q7/Q7 cells led to a twofold reduction in cell viability, whereas serum starvation in STHdh Q111/Q111 cells resulted in a fourfold reduction in cell viability, suggesting that STHdh Q111/Q111 cells were more susceptible to serum withdrawal-induced reduction in cell viability (Fig. 2e) (two-way ANOVA; genotype: F(1,12) = 137.4, p < 0.0001; treatment: F(1,12) = 44.80, p < 0.0001; genotype × treatment: F(1,12) = 9.469, p = 0.0096; Fisher’s LSD post hoc). Similar results were obtained with the MTT cell viability assay (Fig. 2f) (two-way ANOVA; genotype: F(1,12) = 148.5, p < 0.0001; treatment: F(1,12) = 44.85, p < 0.0001; genotype × treatment: F(1,12) = 6.167, p = 0.0288; Fisher’s LSD post hoc).
Reduced Viability Results from Excessive MAO A/B Activity in STHdh Q111/Q111 Cells and Is Associated with Increased Oxidative Stress
To determine if increased MAO activity plays a role in the reduction of cell viability, we introduced MAO inhibitors upon the start of serum deprivation. Three MAO inhibitors were used: (a) clorgyline, an irreversible MAO-A-specific inhibitor, (b) phenelzine sulfate, an irreversible non-selective MAO inhibitor, and (c) selegiline, an irreversible MAO-B specific inhibitor [17, 30]. The presence of MAO inhibitors at 1 μM led to improvements in cell viability as assessed by CellTiter-Glo assay following serum starvation in STHdh Q111/Q111 but not STHdh Q7/Q7 cells, suggesting that increased MAO activity in STHdh Q111/Q111 cells contributes to the serum starvation-induced reduction in cell viability (Fig. 3a) (one-way ANOVA, p = 0.0168; individual treatment versus “no serum” control comparisons by one-tailed t test). No improvements were observed with 0.1 or 0.01 μM of MAO inhibitors (data not shown). As MAO activity in STHdh Q7/Q7 cells was inherently lower, inhibition of MAO activity did not yield a significant rescue of the serum starvation-induced reduction in cell viability in these cells. Similar results were obtained using the MTT assay of cell viability (Fig. 3b) (one-way ANOVA for STHdh Q7/Q7 p = 0.6178; one-way ANOVA for STHdh Q111/Q111 p = 0.0027; individual treatment versus no serum control comparisons by one-tailed t test).

Elevated MAO-A/B activity compromises ATP metabolism and reduces cell viability in serum-deprived STHdh Q111/Q111 cells. a MAO inhibition rescues reduced ATP levels in serum-deprived STHdh Q111/Q111 cells. STHdh Q7/Q7 (left) and STHdh Q111/Q111 (right) cells were treated with 1 μM of MAO inhibitors upon the start of serum starvation over 24 h. ATP levels were measured using CellTiter-Glo®. Values shown were normalized to no serum controls. No significant improvements were observed in STHdh Q7/Q7 cells but inhibition of MAO in STHdh Q111/Q111 cells resulted in an increase of ATP levels; n = 12; (one-way ANOVA, p = 0.0168; individual treatment versus no serum control comparisons by one-tailed t test). b MAO inhibition rescues serum deprivation induced cell death in STHdh Q111/Q111 cells. STHdh Q7/Q7 (left) and STHdh Q111/Q111 (right) cells were treated with 1 μM of MAO inhibitors upon the start of serum starvation over 24 h before incubation with 1 mg/mL MTT for 4 h. Values shown was normalized to no serum controls. Inhibition of MAO in STHdh Q7/Q7 cells had no significant effect on cell death but inhibition of MAO in STHdh Q111/Q111 cells resulted in decreased cell death; n = 21; (one-way ANOVA for STHdh Q7/Q7 p = 0.6178; one-way ANOVA for STHdh Q111/Q111 p = 0.0027; individual treatment versus no serum control comparisons by one-tailed t test). c MAO inhibition rescues reduced ATP/ADP ratios in serum-deprived STHdh Q111/Q111 cells. STHdh Q7/Q7 (left) and STHdh Q111/Q111 (right) cells were treated with 1 μM of MAO inhibitors upon the start of serum starvation over 24 h. ATP/ADP ratios were measured using an ATP/ADP assay kit. Values shown were normalized to no serum controls. A significant improvement was only observed in STHdh Q111/Q111 cells where inhibition of MAO resulted in an increase in ATP/ADP ratio; n = 6; (p < 0.05 by one-tailed t test); d STHdh Q111/Q111 exhibit lower ATP/ADP ratios compared to STHdh Q7/Q7 cells. ATP/ADP ratios were measured in cells grown in normal culture conditions, in the presence of serum. Values shown were normalized to STHdh Q7/Q7 cells; n = 3; (one-way ANOVA; for STHdh Q7/Q7 p = 0.1677; for STHdh Q111/Q111 p = 0.0123, Fisher’s LSD post hoc); Error bars in the bar chart represent standard error of the mean, whereas the whiskers on the box plots represent minimum and maximum values. Q7 and Q111 refer to STHdh Q7/Q7 and STHdh Q111/Q111 cells, respectively. *p < 0.05; **p < 0.01; ***p < 0.001
The CAG tract expansion in HTT has been previously shown to result in compromised cellular metabolism and to lead to a reduced cellular ATP/ADP ratio [31, 32]. This phenomenon has been observed in a number of human and rodent HD cellular systems including HD patient lymphoblasts, hiPSC-derived neural cells, and the STHdh Q111/Q111 striatal cells [23, 31, 32]. Consistent with previous studies, we observed a significantly lower ATP/ADP ratio in STHdh Q111/Q111 compared to STHdh Q7/Q7 (Fig. 3c) (p < 0.05 by one-tailed t test). To examine the impact of MAO inhibition on this property, we measured the ATP/ADP ratio in serum-starved STHdh Q7/Q7 and STHdh Q111/Q111 cells at baseline and following treatment with MAO inhibitors. While we observed no effect of MAO inhibition on the ATP/ADP ratio in STHdh Q7/Q7 cells (Fig. 3d) (one-way ANOVA; p = 0.1677), treatment with MAO inhibitors resulted in a significant improvement of the ATP/ADP ratio in STHdh Q111/Q111 cells (Fig. 3d) (one-way ANOVA; p = 0.0123; Fisher’s LSD post hoc).
As the deamination activity of MAO isozymes triggers oxidative reactions, we quantified oxidized (GSSG) and reduced forms of glutathione (GSH) in both STHdh Q7/Q7 and STHdh Q111/Q111 cells upon serum starvation as a measure of oxidative stress. We demonstrated that oxidative stress increased upon the removal of serum (Fig. 4a) (two-way ANOVA; genotype: F(1,12) = 9.086, p = 0.0108; treatment: F(1,12) = 26.37, p = 0.0002; genotype × treatment: F(1,12) = 0.7452, p = 0.7452; Fisher’s LSD post hoc), reflecting a trend where both GSH/GSSG ratios and cell viability decrease upon serum withdrawal (Fig. 2e, f). This suggests that elevated oxidative stress may contribute to lower cell viability. STHdh Q111/Q111 cells experienced a higher oxidative stress burden upon serum deprivation, evident from a 3.2-fold decrease in GSH/GSSG ratio upon serum starvation, compared to a 1.8-fold decrease in STHdh Q7/Q7 cells. This was consistent with the reduction in cell viability observed in Fig. 2e, where serum deprivation resulted in fourfold reduction in viability in STHdh Q111/Q111 cells, compared to a twofold reduction in STHdh Q7/Q7 cells.

Reduced viability resulting from excessive MAO-A/B activity is associated with increased oxidative stress. a Oxidative stress increases upon serum starvation in STHdh Q7/Q7 and STHdh Q111/Q111 cells. GSH/GSSG levels are a surrogate measure of oxidative stress. GSH/GSSG levels of STHdh Q7/Q7 and STHdh Q111/Q111 cells were measured in serum and in 24-h serum-starved conditions. Values shown were normalized to STHdh Q7/Q7 cells. Upon 24-h serum starvation, both STHdh Q7/Q7 and STHdh Q111/Q111 cells displayed a decrease in GSH/GSSG levels. Furthermore, GSH/GSSG levels were significantly lower in STHdh Q111/Q111 compared to STHdh Q7/Q7 cells following serum starvation; n = 4; (two-way ANOVA; genotype: F(1,12) = 9.086, p = 0.0108; treatment: F(1,12) = 26.37, p = 0.0002; genotype × treatment: F(1,12) = 0.7452, p = 0.7452; Fisher’s LSD post hoc) b MAO inhibition reduces oxidative stress levels in serum-deprived STHdh Q111/Q111 cells. GSH/GSSG levels were measured in 24-h serum-starved STHdh Q7/Q7 (left) and STHdh Q111/Q111 (right) cells in the presence of varying concentrations of MAO inhibitors. MAO inhibitors were added upon the start of serum starvation. Values shown were normalized to no serum controls. 1 μM of the inhibitors were capable of increasing GSH/GSSG levels, hence, reduce oxidative stress in STHdh Q111/Q111 cells but not STHdh Q7/Q7 cells; n = 4; (individual treatment versus no serum control comparisons by unpaired two-tailed t test). The whiskers on the box plots represent minimum and maximum values. Q7 and Q111 refer to STHdh Q7/Q7 and STHdh Q111/Q111 cells, respectively. *p < 0.05; **p < 0.01; ***p < 0.001
To understand if MAO-A/B isozymes play a role in oxidative stress, thereby influencing cell viability, we treated STHdh Q111/Q111 and STHdh Q7/Q7 cells with MAO inhibitors upon removal of serum and measured GSH/GSSG ratios (Fig. 4b). Similar to cell viability measurements, the presence of MAO inhibitors at 1 μM preferentially reduced oxidative stress in STHdh Q111/Q111, but not STHdh Q7/Q7 cells, reinforcing the notion that elevated expression and activity of MAO-A/B in STHdh Q111/Q111 cells contributes to increased oxidative stress, and thus impaired cell viability.
MAO-A/B Expression and Activity in HD Patient Fibroblasts and hiPSC-derived Neural Cells
Of the two MAO isozymes, only MAO-A is expressed in fibroblasts. To assess our findings from a clinical perspective, we measured MAO-A expression and activity in eight HD patient and eight control fibroblasts. Females and males were represented in both the control and HD donor groups; six control females (five HD females) and two control males (three HD males). Taking the longer CAG polynucleotide repeat length of the two alleles into consideration, the control group carried an average high CAG allele length of 19 ± 1.2 CAG repeats whereas the HD group carried an average high CAG allele length of 42 ± 6.1 CAG repeats. The mean ages of the control and HD donors were 59 ± 16.6 and 46 ± 23.6, respectively. Gender and age-related changes of platelet MAO have been reported where MAO activity increases with age [28]. Despite a lower average age of the HD donors, it is evident that a trend presents where HD patient fibroblasts exhibit higher levels of MAO-A expression and activity, although this difference did not reach statistical significance (Fig. 5a–b) (unpaired two-tailed t test; p = 0.08 for MAO-A mRNA and p = 0.06 for MAO-A activity).

MAO-A/B expression and activity are increased in human HD neural cells. a Eight HD and eight control fibroblasts were analyzed for MAO-A mRNA levels. Values shown were normalized to control fibroblasts. Without segregation of age and gender, HD patient fibroblasts display a trend toward increased MAO-A expression compared to their control counterparts; n = 8; (unpaired two-tailed t test; p = 0.08). b Using MAO-Glo™ Assay System, MAO-A activities in the fibroblast lines were tested. Compared against the control fibroblasts, HD patient fibroblasts exhibited a trend toward increased MAO-A activity; n = 8; (unpaired two-tailed t test; p = 0.06); c Quantitative RT-PCR shows increased MAO-A and MAO-B expression in CAG180 NPCs when compared to CAG33 NPCs. Values shown were normalized to CAG33 NPCs. CAG33 and CAG180 hiPSCs were described by the HD iPSC Consortium, 2012 [23] and were differentiated into NPCs according to the protocol described by Li et al., 2011 [24]; n = 3; (unpaired two-tailed t test; p < 0.001 for MAO-A mRNA and p = 0.007 for MAO-B mRNA). d MAO-A and MAO-B activities are elevated in CAG180 NPCs. MAO-A and MAO-B activities were measured using MAO-Glo™ Assay System (Promega). Values shown were normalized to CAG33 NPCs. CAG33 NPCs displayed higher activity when compared to CAG33 NPCs. Error bars in the bar chart represent standard error of the mean, whereas the whiskers on the box plots represent minimum and maximum values; n = 3-4; (unpaired two-tailed t test; p = 0.005 for MAO-A activity and p = 0.006 for MAO-B activity). **p < 0.01; ***p < 0.001
As many neuronal subtypes express both MAO-A and MAO-B, we sought to determine whether human HD neural cells exhibit elevated MAO levels and activity. To this end, we employed a well-established directed differentiation protocol [24] to obtain human neural progenitor cells (NPCs) from previously described HD hiPSC lines (Supplementary Figure 1) [23]. Both MAO-A and MAO-B mRNA levels were significantly elevated in the HD NPCs compared to control NPCs (Fig. 5c) (unpaired two-tailed t test; p < 0.001 for MAO-A mRNA and p = 0.007 for MAO-B mRNA). Similarly, MAO-A and MAO-B activity levels were elevated in HD NPCs compared to control NPCs (Fig. 5d) (unpaired two-tailed t test; p = 0.005 for MAO-A activity and p = 0.006 for MAO-B activity).
Discussion
This study demonstrates that MAO-A/B expression and activity are elevated in the presence of an expanded polyglutamine tract in HTT. Increased MAO-A/B activity plays a role in increasing oxidative stress and increasing susceptibility to stress-induced impairment in cell viability. Although oxidative stress is only one contributing factor to cell death, it is the most pertinent in the context of MAO isozymes, given their roles in oxidative metabolism. Elevation of MAO activity was not confined to a cellular model as a similar trend was observed in HD patient iPSC-derived neural cells. Overall, this is the first study that looks at the effect of enhanced MAO activity using biochemical assays in a well-established striatal and neural cell populations. In addition, this platform allowed us to evaluate the effects of pharmacological inhibitors of MAO activity and show that MAO inhibition alleviated stress-induced impairment in cell viability in HD mutant cells.
There have been previous reports on the analysis of MAO expression and activity in HD patients. With the first report in 1978, studies have shown that there is elevated MAO-B activity in HD patient platelet populations [33–35]. Increased post mortem MAO-B activity in patients suffering from HD has also been shown [36–38]. It is important to note, however, that most of these studies were performed before the causative mutation in HTT was identified. As a result, these studies did not exclude patients that were HD phenocopies, and thus were unable to ascertain that the effect is HD-specific.
In contrast, this study employs genetically defined mouse and human neural cell populations, highlighting that elevation of MAO activity is intrinsic to the presence of the mutant HTT allele. Although a number of studies reported increased MAO-B activity in HD patients and cells, one study did not observe differences in MAO-B activity in platelets among 59 HD patients, 20 at-risk individuals, 29 control individuals with an HD family history, and 64 healthy controls [29]. This discrepancy in reported MAO activity in HD patients may reflect lack of characterization of the study cohort. Indeed, lifestyle habits, such as smoking, have been shown to influence MAO-B activity (reviewed in [5]). As the study cohort comprised a mixture of HD patients with undefined smoking habits and receiving various drug interventions, these variations could have obscured differences in MAO activity. In general, the findings from previous studies are consistent with our data where we show that MAO-A/B activity and expression are increased in human neural cells originating from HD patients iPSCs.
MAO isozymes play a major role in metabolizing biogenic and dietary amines, generating hydrogen peroxide as a by-product. It is conceivable that increased MAO activity in the presence of mutant HTT results in an accumulation of reactive oxygen species (ROS). In support of this notion, recent studies have reported that STHdh Q111/Q111 cells exhibit increased mitochondrial ROS [30] and addition of a glutathione peroxidase (GPx) supplement results in the reduction of cell death [2, 39]. As heightened activity of MAO-A/B leads to an increase in ROS production, it is possible that MAO acts upstream of GPx to increase ROS production, thus contributing to cell death.
Analogous to our findings with MAO inhibitors, a recent study has demonstrated that knockdown of MAO-A expression leads to increased ATP levels [40]. MAO-A/B is located in the outer mitochondrial membrane and their activity leads to the production of metabolic by-products like H2O2. In HD mutant cells, increased MAO-A/B activity implicates an accumulation of ROS. Due to close proximity to the mitochondrial oxidative phosphorylation system, it is conceivable that electron flow through the respiratory chain may be impaired and ATP levels may be reduced by ROS, possibly contributing to the lower ATP/ADP ratios seen in HD mutant cells.
Several studies have reported deficits in dopamine levels and signaling in animal models and patients with HD (reviewed in [4, 6]). Although this study suggests that increased MAO expression and activity contribute to deficiencies in dopamine homeostasis in a HD mutant background, it is important to note that MAO isozymes metabolize other substrates. These include serotonin, epinephrine, norepinephrine, phenylethylamine, and benzylamine [6, 21]. As there is a dearth of studies measuring these neurotransmitter levels in HD, future studies should investigate whether other MAO-metabolized neurotransmitters are affected in a fashion similar to dopamine as well as the impact of MAO inhibitors on the levels of these neurotransmitters.
Despite the unclear link between the presence of mutant HTT and increased MAO transcriptional activity, there have been separate lines of evidence that may explain this phenomenon. HTT has been shown to regulate Sp1 transcription factor where mutant HTT forms an enhanced association with Sp1 [8–10, 41, 42]. As MAO-A/B are both transcriptionally regulated by Sp1 and Sp3 [10, 11, 21, 22], it is possible that close interaction between mutant HTT and Sp1 results in increased MAO expression. Sp1 also affects D2 receptor expression levels, perhaps perturbing the dopamine metabolic pathway, and indirectly resulting in increased MAO-A/B expression.
Although this study delineates the role of increased MAO activity in a HD mutant background, several questions remain. The present study was conducted mostly using a striatal cell line that carries 111 CAG repeats. The size of the CAG repeat length in HTT affects the age of onset of motor symptoms and severity of neuropathology [2, 13–15]; hence, it will be interesting to determine if there is a relationship between the CAG size and the magnitude of MAO activity. Although we demonstrate that human neural cells derived from HD patient iPSCs exhibit increased MAO activity, the next step would be to validate these findings in a larger cohort and to examine this phenomenon in other tissue types.
In summary, this study provides evidence that cellular HD models exhibit increased MAO expression and activity, highlighting the potential of MAO-A/B as pharmacological targets in HD. MAO-B inhibitors are currently employed in the clinical setting to treat Parkinsonian symptoms. In addition, MAO inhibitors have also been shown to possess anti-depressant properties. As the cardinal features of HD are cognitive, motor, and psychiatric deficits, MAO inhibitors represent potential candidates for therapeutic intervention in HD.
References
Augood SJ, Faull RL, Emson PC (1997) Dopamine D1 and D2 receptor gene expression in the striatum in Huntington’s disease. Ann Neurol 42:215–221. doi:10.1002/ana.410420213
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98. doi:10.1016/S1474-4422(10)70245-3
Kish SJ, Shannak K, Hornykiewicz O (1987) Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann Neurol 22:386–389. doi:10.1002/ana.410220318
Cha JH, Frey AS, Alsdorf SA et al (1999) Altered neurotransmitter receptor expression in transgenic mouse models of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci 354:981–989
Shih JC, Chen K, Ridd MJ (1999) Monoamine oxidase: from genes to behavior. Annu Rev Neurosci 22:197–217. doi:10.1146/annurev.neuro.22.1.197
Chen JY, Wang EA, Cepeda C, Levine MS (2013) Dopamine imbalance in Huntington’s disease: a mechanism for the lack of behavioral flexibility. Front Neurosci 7:114. doi:10.3389/fnins.2013.00114
Renoir T, Argyropoulos A, Chevarin C et al (2014) Sexually dimorphic dopaminergic dysfunction in a transgenic mouse model of Huntington’s disease. Pharmacol Biochem Behav. doi:10.1016/j.pbb.2014.10.004
Reynolds GP, Dalton CF, Tillery CL et al (1999) Brain neurotransmitter deficits in mice transgenic for the Huntington’s disease mutation. J Neurochem 72:1773–1776
Bibb JA, Yan Z, Svenningsson P et al (2000) Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc Natl Acad Sci U S A 97:6809–6814. doi:10.1073/pnas.120166397
Callahan JW, Abercrombie ED (2011) In vivo dopamine efflux is decreased in striatum of both fragment (R6/2) and full-length (YAC128) transgenic mouse models of Huntington’s disease. Front Syst Neurosci 5:61. doi:10.3389/fnsys.2011.00061
Madison JL, Wegrzynowicz M, Aschner M, Bowman AB (2012) Disease-toxicant interactions in manganese exposed Huntington disease mice: early changes in striatal neuron morphology and dopamine metabolism. PLoS One 7:e31024. doi:10.1371/journal.pone.0031024
O’Carroll AM, Fowler CJ, Phillips JP et al (1983) The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedebergs Arch Pharmacol 322:198–202
Antonini A, Leenders KL, Spiegel R et al (1996) Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain 119(6):2085–2095
Andrews TC, Weeks RA, Turjanski N et al (1999) Huntington’s disease progression. PET and clinical observations. Brain 122(12):2353–2363
van Oostrom JCH, Dekker M, Willemsen ATM et al (2009) Changes in striatal dopamine D2 receptor binding in pre-clinical Huntington’s disease. Eur J Neurol 16:226–231. doi:10.1111/j.1468-1331.2008.02390.x
Bortolato M, Shih JC (2011) Behavioral outcomes of monoamine oxidase deficiency: preclinical and clinical evidence. Int Rev Neurobiol 100:13–42. doi:10.1016/B978-0-12-386467-3.00002-9
Youdim MBH, Edmondson D, Tipton KF (2006) The therapeutic potential of monoamine oxidase inhibitors. Nat Rev Neurosci 7:295–309. doi:10.1038/nrn1883
Trettel F, Rigamonti D, Hilditch-Maguire P et al (2000) Dominant phenotypes produced by the HD mutation in STHdh (Q111) striatal cells. Hum Mol Genet 9:2799–2809
Chen K, Ou X-M, Wu JB, Shih JC (2011) Transcription factor E2F-associated phosphoprotein (EAPP), RAM2/CDCA7L/JPO2 (R1), and simian virus 40 promoter factor 1 (Sp1) cooperatively regulate glucocorticoid activation of monoamine oxidase B. Mol Pharmacol 79:308–317. doi:10.1124/mol.110.067439
Wu JB, Chen K, Ou X-M, Shih JC (2009) Retinoic acid activates monoamine oxidase B promoter in human neuronal cells. J Biol Chem 284:16723–16735. doi:10.1074/jbc.M901779200
Wong WK, Chen K, Shih JC (2001) Regulation of human monoamine oxidase B gene by Sp1 and Sp3. Mol Pharmacol 59:852–859
Ou X-M, Chen K, Shih JC (2006) Monoamine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc Natl Acad Sci U S A 103:10923–10928. doi:10.1073/pnas.0601515103
HD iPSC Consortium (2012) Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11:264–278. doi:10.1016/j.stem.2012.04.027
Li W, Sun W, Zhang Y et al (2011) Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. PNAS 108:8299–8304. doi:10.1073/pnas.1014041108
Magnani E, Bettini E (2000) Resazurin detection of energy metabolism changes in serum-starved PC12 cells and of neuroprotective agent effect. Brain Res Brain Res Protoc 5:266–272
Gerber HP, Dixit V, Ferrara N (1998) Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem 273:13313–13316
Sugiyama H, Savill JS, Kitamura M et al (1999) Selective sensitization to tumor necrosis factor-alpha-induced apoptosis by blockade of NF-kappaB in primary glomerular mesangial cells. J Biol Chem 274:19532–19537
Veral A, Alper G, Menteş G, Ersöz B (1997) Age and sex related alterations in serum and platelet monoamine oxidase. Eur J Clin Chem Clin Biochem 35:265–268
Markianos M, Panas M, Kalfakis N, Vassilopoulos D (2004) Platelet monoamine oxidase activity in subjects tested for Huntington’s disease gene mutation. J Neural Trans (Vienna, Austria : 1996) 111:475–483. doi:10.1007/s00702-003-0103-x
Ribeiro M, Silva AC, Rodrigues J et al (2013) Oxidizing effects of exogenous stressors in Huntington’s disease knock-in striatal cells—protective effect of cystamine and creatine. Toxicol Sci 136:487–499. doi:10.1093/toxsci/kft199
Gines S, Seong I-S, Fossale E et al (2003) Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet 12:497–508
Seong I-S, Ivanova E, Lee J-M et al (2005) HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet 14:2871–2880. doi:10.1093/hmg/ddi319
Mann J, Chiu E (1978) Platelet monoamine oxidase activity in Huntington’s chorea. J Neurol Neurosurg Psychiatr 41:809–812
Belendiuk K, Belendiuk GW, Freedman DX (1980) Blood monoamine metabolism in Huntington’s disease. Arch Gen Psychiatry 37:325–332
Norman TR, Chiu E, French MA (1987) Platelet monoamine oxidase activity in patients with Huntington’s disease. Clin Exp Pharm Phys 14:547–550
Mann JJ, Stanley M, Gershon S, Rossor M (1980) Mental symptoms in Huntington’s disease and a possible primary aminergic neuron lesion. Science 210:1369–1371
Mann JJ, Kaplan RD, Bird ED (1986) Elevated postmortem monoamine oxidase B activity in the caudate nucleus in Huntington’s disease compared to schizophrenics and controls. J Neural Trans (Vienna, Austria : 1996) 65:277–283
Richards G, Messer J, Waldvogel HJ et al (2011) Up-regulation of the isoenzymes MAO-A and MAO-B in the human basal ganglia and pons in Huntington’s disease revealed by quantitative enzyme radioautography. Brain Res 1370:204–214. doi:10.1016/j.brainres.2010.11.020
Mason RP, Casu M, Butler N et al (2013) Glutathione peroxidase activity is neuroprotective in models of Huntington’s disease. Nat Genet 45:1249–1254. doi:10.1038/ng.2732
Fitzgerald JC, Ugun-Klusek A, Allen G et al (2014) Monoamine oxidase-A knockdown in human neuroblastoma cells reveals protection against mitochondrial toxins. FASEB J 28:218–229. doi:10.1096/fj.13-235481
Li S-H, Cheng AL, Zhou H et al (2002) Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 22:1277–1287
Dunah AW, Jeong H, Griffin A et al (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 296:2238–2243. doi:10.1126/science.1072613
Acknowledgments
We are grateful to Dr. Marcy MacDonald for sharing STHdh Q7/Q7 and STHdh Q111/Q111 cells with us and the HD iPSC Consortium for the CAG33 and CAG180 human-induced pluripotent stem cells. We are also thankful for Dr. Jean C. Shih’s kind contribution of MAO-A and MAO-B luciferase reporter constructs.
Conflict of Interest
The authors declare no conflict of interest.
Funding
The work was funded by the Agency for Science Technology and Research and the National University of Singapore, Singapore.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
Characterization of CAG33 and CAG180 NPCs. a Immunostaining of CAG33 and CAG180 NPCs shows an induction of early neuronal marker, Nestin. CAG33 and CAG180 hiPSCs were differentiation over 7 days, and passaged once before immunostaining. Expression of Nestin (green) indicates the acquisition of neuronal identity, whereas the loss of Oct3/4 (red) indicates the loss of pluripotency. The images were taken at ×20 magnification. Scale bar = 100 μM. b Quantitative RT-PCR shows increased neuronal markers Nestin, Pax6 and Sox1, and reduced pluripotency-associated markers Oct3/4. NPCs passaged once after 7 days of differentiation were analyzed for their gene expression profile. Successful neuronal differentiation was shown by upregulation of neuronal markers and down-regulation of pluripotency-associated markers. (Comparisons to respective undifferentiated hiPSC line by unpaired two-tailed t test);**p < 0.01; ***p < 0.001. (PDF 3416 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Ooi, J., Hayden, M.R. & Pouladi, M.A. Inhibition of Excessive Monoamine Oxidase A/B Activity Protects Against Stress-induced Neuronal Death in Huntington Disease. Mol Neurobiol 52, 1850–1861 (2015). https://doi.org/10.1007/s12035-014-8974-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8974-4