
Abstract
Neuroplasticity refers to the changes in the molecular and cellular processes of neural circuits that occur in response to environmental experiences. Clinical and experimental studies have increasingly shown that estrogens participate in the neuroplasticity involved in cognition, behavior, and memory. It is generally accepted that estrogens exert their effects through genomic actions that occur over a period of hours to days. However, emerging evidence indicates that estrogens also rapidly influence the neural circuitry through nongenomic actions. In this review, we provide an overview of the genomic and nongenomic actions of estrogens and discuss how these actions may cooperate in synaptic plasticity. We then summarize the role of epigenetic modifications, synaptic protein synthesis, and posttranslational modifications, and the splice variants of estrogen receptors in the complicated network of estrogens. The combination of genomic and nongenomic mechanisms endows estrogens with considerable diversity in modulating neural functions including synaptic plasticity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neural circuit activity is essential for normal brain processes [1–3]. The fine regulation of this activity is driven by changes in synaptic structure and function, which is the cellular mechanism underlying cognition, behavior, and memory [4, 5]. Thereby, synaptic plasticity, which is the changes that occur in the number and/or morphology of neuronal synapses, is considered a fundamental feature of the nervous system, and it allows for adaptation to changing behavioral environments [6, 7]. Importantly, many of the cellular processes that contribute to synaptic plasticity are associated with neuronal disorders [8–10]. Understanding the abilities of synapses to modify their functional strength according to the environment and/or extracellular signals will help to delineate the underlying molecular mechanisms that allow these events to occur.
Estrogens are neuroactive steroids and/or neurosteroids that have the potential to influence the nervous system [11, 12]. Clinical and experimental studies have overwhelmingly demonstrated a modulatory role of estrogens in the brain and suggest their beneficial actions in neuronal plasticity [13–17]. For example, estrogen-primed animals exhibit decreased hippocampal seizure thresholds [18], and pretreatment with exogenous estrogens decrease rodent mortality and infarct size following middle cerebral artery occlusion [19–21]. Collectively, those studies reported an association between lower estrogen levels and increased risk of neuronal disorders which matched with human disease patterns that have long been known to differ between men and women and between premenopausal and postmenopausal women [22]. Consequently, interest in learning how estrogens affect the neuroplasticity of neural circuits and thus contribute to cognition, behavior, and memory is increasing. Thus, the underlying mechanisms of estrogenic action in neuroplasticity need to be clarified in order to better understand their functions in the normal activity of neural circuits and their potential therapeutic roles in brain disorders. In addition to the effects of estrogen on the regulation of gene transcription, which requires hours to days to manifest [5, 23], accumulating studies have shown that estrogens affect neuroplasticity within minutes [24–27]. Therefore, the relationships of estrogens with rapidly activated signaling cascades and transcriptional machinery and their potential crosstalk or convergence are evident [1, 28, 29].
Although genomic and nongenomic estrogenic effects are often viewed as distinct modes of estrogenic action, this may not necessarily hold true because the inhibition of one can limit the effectiveness of the other [30]. In this review, we focus on the relationship between the rapid nongenomic and genomic signaling of estrogens. First, we provide an overview of the advancements that have been made in the understanding of the rapid nongenomic and genomic signaling of estrogens in synaptic plasticity. Second, we discuss how the rapid nongenomic and genomic signaling actions of estrogens in the brain might rely on a cooperation or sequential modulatory system that ultimately results in changes in synaptic activity. Finally, we explore the molecular and cellular mechanisms that underlie the relationship between the rapid nongenomic and genomic signaling of estrogens.
The Genomic Actions of Estrogens in Synaptic Plasticity
Estrogens have a variety of effects on the central nervous system (CNS). These effects, which typically require hours or even days to take effect, are mediated through transcriptionally regulated changes in gene expression [31]. These estrogenic actions, which have been described as genomic effects, are characterized by the following features: a prolonged latency; long lasting; involving gene transcription and protein synthesis; initiation within the nucleus, usually through the nuclear receptor superfamily; ineffectiveness of steroid analogs that are unable to cross the plasma membrane [32, 33]; and functional at physiological levels [34]. Nuclear estrogen receptors (ERs) have two forms: ERα and ERβ [35–37]. Like other members of the nuclear receptor superfamily, ERα and ERβ contain a common structure of six functional domains (A/B, C, D, E, and F), as shown in Fig. 1 [36, 38]. In the inactive state, ERs usually exist as a monomer or complex with immunophilins and heat shock protein 90 (HSP-90) [39]. The estrogen-ER complex then binds to the estrogen response element (ERE) in the promoter region of the target genes and exerts its regulatory potential [40, 41].

Primary structure of the classic estrogen receptors (ER). ERα and ERβ share a common structure with six functional domains (A/B, C, D, E, and F). Domain A/B plays a role in protein-protein interactions and the transcriptional activation of target gene expression. Domain C is responsible for DNA binding and ER dimerization. Domain D, which is a hinge domain linking domains C and E, is involved in the nuclear localization of ERs. Domain E is the ligand-binding domain. Domain F contains cofactor recruitment regions. The similar structures of the receptors both contain a highly homologous DNA-binding region (95 %) and a hormone-binding region with weaker homology(69 %). However, the carboxy- and amino-terminal regions have the least homology (58 %)
Based upon the findings of previous studies on the genomic effects of estrogens on synaptic plasticity [39], novel research techniques, including pharmacological and gene manipulation, have recently been applied to better understand these genomic effects. HOXC10, which is one of the few neural gene targets of ERs, has been shown to play a critical role in spinal cord development and neuron formation [42, 43]. The authors of those studies found that the HOXC10 promoter contains multiple putative EREs, which indicates that HOXC10 might be transcriptionally regulated by estrogens. In addition, they showed that the ERE1 and ERE6 regions of the HOXC10 promoter are potentially involved in the transcriptional regulation of HOXC10 expression in the presence of estrogens [44]. Another neural gene target of ERs is Apo D, which encodes apolipoprotein D. Many studies have demonstrated a relationship between neuronal degeneration and Apo D expression [45, 46]. A recent study demonstrated that Apo D has three EREs in its promoter and that its expression can be modulated by these hormones [47]. Thus, these findings suggest that Apo D is partly responsible for the neuroprotective role of estrogens. In addition, the large-conductance voltage- and Ca(2+)-activated K(+) channel has been shown to play key roles in diverse body functions that are influenced by estrogens. The pore-forming alpha subunit (Slo, KCNMA1) promoters of the K(+) channel contain multiple EREs. A mutagenesis experiment further showed that the estrogen responsiveness of the mSlo gene involves a classical genomic mechanism that acts through ERE1 and ERE2 and that is facilitated by ERα, which therefore suggests that ERα exerts a genomic action on the mSlo gene promoter elements [48]. Luckily, the identification of estrogen target genes is greatly facilitated by transcriptomic methods, such as RNA sequencing, expression microarrays, and chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing (ChIP sequencing). Combining transcriptomic and ChIP sequencing data enables the discrimination of direct and indirect estrogen target genes [49]. Interestingly, Humphreys et al. analyzed the transcription of control and estradiol (E2)-treated animals with RNA sequencing and found significant alterations in the transcript levels of 88 genes in the treated animals [50]. Another gene assay and cell-based endogenous expression analysis revealed that estrogens significantly suppress the expression of brain size-related genes, including MCPH1, ASPM, CDK, RAP2, and WDR62 [51]. Intriguingly, when the EREs are deleted from the promoters, the suppressive effects are abolished, which suggests that EREs mediate the effects of estrogens on the brain size genes [51]. Ryokoet al. identified a classical ERE half-site on a TPH2 gene promoter that is functional because the deletion or mutation of this sequence blocks the E2-induced TPH2-luc activity. These results suggest that the ERE half-site plays an important role in the ER-mediated regulation of TPH2 transcriptional activity [52]. Hence, these studies indicate that advances in genomic technology allow for the identification of the neuronal target genes of estrogens and ERE-binding sites.
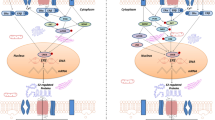
In addition to the mechanisms described above, estrogens serve as cofactors at non-ERE sites that interact with other DNA-binding elements, such as AP-1 or c-Jun [38]. However, ERs modulate chromatin states by associating with different classes of coregulators [53]. Steroid receptor coactivator-1 (SRC-1) is the predominant coactivator of p160 family members in the brain. Several studies have shown that the expression of SRC-1 in the hippocampus is highly correlated with several key synaptic proteins during development or after orchidectomies, but not after ovariectomies. These findings indicate that SRC-1 may be regulated by hippocampal-synthesized E2 and profoundly involved in the hippocampal E2 regulation of hippocampal synaptic plasticity [54]. While the roles of a large number of activator complexes and their associated enzymatic activities have been well established by the presence of the histone acetyltransferases (HATs) p300/CBP and the levels of H3K4me2 and H3K27Ac [55], the precise biochemical mechanisms by which so many of the coactivators that are required for the different functional activities are recruited at specific enhancer sites remain incompletely understood [56, 57]. Liu et al. reported a new signature of the functionally active estrogen-regulated enhancers that involve the selective trans-recruitment of an apparent complex of other DNA-binding transcription factors, including RARα/γ, GATA3, AP2γ, STAT1, AP1, and FoxA1 [58]. However, this phenomenon has only been described in MCF7 cells. Whether a similar conclusion can be made about the brain is still unclear (Fig. 2).

Schematic of the genomic estrogenic actions in neurons. The estrogen-ER complex binds to the estrogen response element (ERE) in the promoter region of target genes or acts as a cofactor/coregulator at non-ERE sites that interact with other DNA-binding elements. E2, estradiol; SRC-1, steroid receptor coactivator-1
Notably, both ERα and ERβ are abundantly localized throughout the brain, but the relative local distributions of ERα and ERβ may differ [35, 59]. The differential local distribution of ERs most likely indicates the different effects of local E2 administration in the brain [60]. Evidence has revealed that the individual or simultaneous activation of ERα and ERβ has different effects [61]. These findings suggest the intriguing hypothesis that the interactions between the two ERs and the consequences of transcriptional modulation are complicated and delicately balanced. Several studies have reported a phenomenon of an opposite transcriptional response between ERα and ERβ that depended on the cellular context and associated cofactors [62, 63]. However, the exact relationship of ERα and ERβ and the precise actions of their interaction remain largely unknown due to a lack of systemic investigations and related methods.
The Rapid Nongenomic Actions of Estrogens in Synaptic Plasticity and Neuroprotection
Evidence that has been collected during the last decade has indicated that estrogens elicit cellular actions that occur as fast as seconds to minutes and mostly within 1 h [64, 65]. In vitro studies have shown that estrogens acutely modulate synaptic function in both sexes. Methods that are more sensitive have detected the subcellular location of ERs [66, 67], including nuclei, cytoplasm, plasma membranes, perimembrane spaces, endoplasmic reticuli, and mitochondria. Notably, the ERs located outside of the nucleus are suspected to be related to the rapid actions of estrogens [22]. Except for the established ERα and/or ERβ, there are at least three putative ERs that are involved in the rapid actions of estrogens in the brain: G protein-coupled receptor 30, G protein-coupled estrogen receptor 1, and the membrane-associated ER (mER) [68–70]. The putative ERs have the characteristics of initiating cell signaling from the membrane [71] with a companion G protein-coupled receptor [72]. Moreover, a high-affinity, saturable, and 3H-estradiol-binding site in the plasma membrane has been identified and designated as ER-X [69]. Using a novel putative mER agonist STX, it was reported that rapid membrane ER activation can initiate cellular signaling through the metabotropic glutamate receptor (mGluR) 1a [71]. Study also shows that the mER (not ERα or ERβ) mediates the estrogen-initiated inhibition of the expression of the ubiquitin-conjugating enzyme 9, which is the only known E2-conjugating enzyme and which is associated with neuroplasticity [73]. Therefore, the extranuclear receptors are thought to be essential for mediating the rapid nongenomic actions of estrogens. However, these putative ERs have not been structurally characterized [69].
A major debate on whether the circulating estrogens and/or local estrogens that are derived from the brain underlie the rapid effects of estrogens has existed for many years [74, 75]. Recent reports have demonstrated that the concentrations of estrogens are at nanomolar levels in some brain regions and at picomolar levels in the plasma [76]. Thus, the concentrations of circulating estrogens may be too low to initiate rapid actions [77]. These observations have suggested that the levels of the local estrogens that are synthesized within the brain, which fluctuate more rapidly than the levels of the circulating estrogens, play an essential role in the rapid actions of estrogens. This claim has been strengthened by the discovery of multiple enzymes that allow for the biosynthesis of brain-derived estrogens [78–81]. Estrogens undergo tissue and/or cell-specific enzymatic conversions into estrogen metabolites [82]. The neurons that express many estrogenic enzymes are considered important producers of brain estrogens.
Several key enzymes that are involved in the synthesis of testosterone and estrogens in the brain use dehydroepiandrosterone as a precursor [83]. For example, the rate-limiting step of steroidogenesis is mediated by the steroid acute regulatory protein and transporter protein, which are widely found in typical steroidogenic tissue (e.g., ovary and adrenal gland) as well as in neurons [84]. Increasingly, a number of other enzymes, including the estrogen-synthesizing enzyme, aromatase, have been widely found in different brain regions of male and female rats [85]. 17β-Hydroxysteroid dehydrogenase type 10 (17β-HSD10), which is encoded by the HSD17B10 gene that maps to Xp11.2, is a homotetrameric mitochondrial multifunctional enzyme that catalyzes the oxidation of neuroactive steroids. The brains of individuals with Alzheimer’s disease (AD) and animals in an AD mouse model exhibit abnormally increased levels of 17β-HSD10. The restoration of steroid homeostasis could be achieved through the supplementation of neuroactive steroids with a properly dosed treatment regimen or through the adjustment of 17β-HSD10 activity to protect neurons [86]. In addition, steroid sulfatase plays a key role in the intracrine conversion of dehydroepiandrosterone testosterone and estrogens [83]. Therefore, because neurons are equipped with all of the enzymes that are activated in steroidogenesis, they are therefore capable of synthesizing the so-called neurosteroids, which act directly and locally on neurons to exert neuroprotection. It is important to note that many of the enzymes involved in steroidogenesis are expressed in not only neurons in different brain regions but also astrocytes and even endothelial cells and oligodendrocytes [84]. It will be important to examine the astrocyte- and/or endothelial-specific enzymes in order to better clarify the contribution and/or crosstalk of these enzymes in non-neural cell types to the rapid actions of estrogens. Interestingly, a growing number of studies have found that the enzymatic activity that is responsible for the synthesis of estrogens can be modulated within minutes by mechanisms that cannot possibly involve changes in the concentrations of the enzymatic proteins [87, 88]. This mechanism underlying the rapid synthesis and metabolism of estrogens corresponds with the rapid action of estrogens in the brain. Collectively, these findings show that the presence and regulation of estrogenic enzymes in the brain and the extracellular localization of ERs are evidence of the rapid nongenomic actions of estrogens in synaptic plasticity.
To clarify the mechanisms through which estrogens exert their rapid effects on neuronal plasticity, two concepts need to be emphasized. The rapid nongenomic pathway is mediated by nonnuclear ERs and/or nuclear ERs [54]. The rapid signaling pathway whereby local estrogens modulate synaptic plasticity activate several cellular kinases, often through ion channels [89]. We use the term rapid nongenomic pathway to refer to processes that are coupled to the signaling cascade that requires ER participation, while rapid signaling pathway refers to the processes that are coupled to the signaling cascade in an ER-independent manner.
The Actions of the Rapid Nongenomic Pathway of Estrogens in Synaptic Plasticity
The rapid nongenomic pathway of estrogens in the nervous system usually involves the activation of multiple kinase pathways, including the mitogen-activated protein kinase (MAPK)/extracellular regulated kinase (ERK) pathway, phospholipase C pathway, phosphatidylinositol 3-kinase(PI3K)/Akt pathway, and protein kinase A (PKA) and protein kinase C (PKC) pathways [5, 21, 90]. Estrogens rapidly enhance ERK and Akt activation through phosphorylation in cortical neurons, and inhibitors of ERK and Akt activation significantly attenuate estrogen induction in excitatory glutamatergic synapses [91, 92]. Once Akt and ERK are activated, many essential cellular functions, such as survival, adhesion, metabolism, and proliferation, are initiated [93]. Interestingly, the ability of estrogens to phosphorylate ERK and Akt persists, even in ERα-knockout mice, thus implicating other ERs in these estrogen actions [94]. Another potential example of an estrogen-induced rapid nongenomic pathway involves the regulation of the JNK-c-jun signaling pathway which was well recognized as pro-apoptotic factors in ischemic brain [95–97]. The rapid nongenomic pathway of estrogens was also mimicked by the application of the PKA activator forskolin and the PKC activator phorbol-12,13-dibutyrate. In addition, the rapid nongenomic pathway mechanisms of estrogens include the interactions of estrogens with the signaling of neurotrophic factors, such as brain-derived neurotrophic factors, insulin-like growth factor-1 (IGF), and Wnt. A novel model of the interactions of estrogens and acute brain-derived neurotrophic factor signals suggests that they act in a cooperative manner, which results in dendritic spine formation and the subsequent stabilization of synaptic and circuit plasticity [98, 99]. Moreover, ERα regulates the IGF-type I receptor (IGF-IR) signaling pathways through the phosphorylation of ERK and Akt, and the interactions of the ER-IGF-IR pathway potentiates neural activities [100]. Taken together, these findings indicate that the interactions of ERs and IGF-IRs are one of the important mechanisms underlying the rapid nongenomic pathways of ERs [100]. Therefore, estrogens can be coupled with neurotrophin receptors, which results in the convergence or cross-coupling of specific signaling pathways, particularly at the level of the MAPK cascade [94].
Along these lines, the rapid nongenomic pathway of estrogens can be presumed to be involved in the interactions of membrane-localized ERs with adaptor proteins, such as c-Src, and the downstream rapid signaling that occurs through the MAPK pathway, G proteins, PKA/PI3K pathway, or PKC pathway. These signaling pathways are rapidly activated, and they in turn trigger intracellular Ca2+ release, cAMP production, and/or c-Src activation with the subsequent activation of MAPK or calcium/calmodulin-dependent kinases. However, it is important to note that different receptors may be coupled with specific rapid signaling pathways in order to exert relative effects. The use of selective ER modulators and transgenesis (knockout and/or knockdown) mice in studies will help to clarify these issues.
Another question that should be addressed is how ERs couple with downstream cascade signaling. Intriguingly, outside the nervous system, ERα has been demonstrated to physically interact with CAV1, which is necessary for the trafficking of ERα to the membrane surface [101]. A subsequent study showed that membrane-localized ERs are localized within distinct caveolae and that CAV1 is necessary to couple ERα to the group I mGluRs and CAV3 is necessary for the association of ERα and ERβ with group II mGluRs [102]. However, recent studies on ER-interacting scaffold proteins have demonstrated that scaffold proteins, such as MNAR/PELP1, striatin, and p130Cas, might link ERs with kinases to potentially mediate estrogen-induced kinase signaling [103, 104].
The Actions of the Rapid Signaling Pathway of Estrogens in Synaptic Plasticity
Estrogens rapidly potentiate kinate-induced currents in hippocampal neurons from wild-type as well as ER knockout mice, thus suggesting that estrogens directly interact with ion channels in synapses then regulate the downstream signaling cascades [105–107]. L-type voltage-gated Ca2+channels (VGCCs) play important roles in dendritic development, neuronal survival, and synaptic plasticity [108]. In electrophysiological studies, estrogens acutely potentiate VGCCs in hippocampal neurons in an ER-independent manner by directly binding with a domain that overlaps with the dihydropyridine-binding site [17]. Calcium/calmodulin-dependent protein kinase-II (CaMKII) is a major neuronal protein that plays a significant role in the cellular processes of long-term potentiation (LTP) and the vesicular release of neurotransmitters [109, 110]. The loss of CaMKII in the forebrain has severe adverse effects on spatial learning in mice [110]. The activation of CaMKII by estrogen-modulated LTP induction stimulates the formation of new spines and enlarges existing spines [111, 112]. However, this effect of estrogens is not mediated by ER-dependent actions [113]. A number of reports have indicated that proline-rich tyrosine kinase 2(PYK2), which is a redox-sensitive kinase, is activated by estrogen [114]. The activation of PYK2 in cerebral ischemia is involved in the modulation of N-methyl-d-aspartate-type glutamate receptor activity and Ca2+ dynamics, which result in ischemic neuron death. However, the detailed mechanisms underlying the PYK2 activation by estrogens remain unclear. Several studies have shown that estrogens are able to interact with K+ channels in different types of cells, such as cardiac myocytes and neurons [115, 116]. Furthermore, tyrosine kinases seem to be involved in the activation of volume-sensitive K(+) channels, whereas tyrosine phosphatases appear to be involved in the inactivation of channels by estrogens [116–118]. Collectively, these findings suggest that estrogen regulation of PYK2 is mediated by direct interaction with potassium channels. Until recently, no evidence existed that estrogens modulate mGluRs directly without ERs [101, 119, 120]. Through these rapid pathways, estrogens play an important role in the modulation of synaptic plasticity (Fig. 3).

Schematic of the rapid nongenomic estrogenic actions in neurons. The expanded ERs are expressed throughout neurons, including the nuclei, cytoplasm, plasma membranes, perimembrane spaces, endoplasmic reticulum, and mitochondria. Local synthesis of estrogens, which is mediated by synaptic aromatase/steroid acute regulatory protein (StAR) or peripheral estrogens, results in the activation of ERs, ion channels, and/or other membrane receptors. Such activation couples with specific signaling cascades or second messenger systems through scaffold proteins (CAV/MNAR/PELP1/striatin/p130Cas) and ultimately leads to the remodeling of synaptic structure and function. ERGP, estrogen receptor G-protein; LTP, long-time potential; LGCC, L-type gate calcium channel
Cooperation of the Genomic and Rapid Nongenomic Actions of Estrogens in Synaptic Plasticity
In the classical mechanism, estrogens bind to the α or β isoform of the ERs, which then binds to EREs and alters gene transcription [31]. The nonclassical actions of ERα and ERβ possibly occur through alternative response elements in DNA or rapid changes in signaling cascades [121, 122]. All of these mechanisms operate in the CNS [123]. The nuclear accumulation of ERβ, which occurs 6–12 h after estrogen treatment, results in the increased expression of postsynaptic density (PSD)-95 and synaptophysin messenger RNA (mRNA), thus implicating the classical genomic estrogenic actions on synaptic plasticity. However, blocking PI3K signaling partially suppresses the estrogen-induced expression of PSD-95 and synaptophysin, which suggests a crosstalk between the genomic and nongenomic actions of estrogens [124]. As shown in Fig. 4, specific cellular and molecular mechanisms underlie the cooperation of genomic and rapid nongenomic actions of estrogens [125]. Previous studies have indicated that alternative rapid signaling cascades of estrogens act by interfering with activation of the ERK and PI3K signaling pathways that regulate on a transcriptional level [126, 127]. One of the most intensely studied mechanisms involves antiapoptotic genes, such as BCL-2. Several studies have shown that estrogens activate different pathways to modulate BCL-2 transcription. Wu et al. showed that estrogen induces rapid Ca2+ influx in hippocampal neurons, which results in the activation of the Scr/ERK signaling cascades and the upregulation of BCL-2 transcription [128]. Pugazhenthi et al. reported that estrogens regulate the activation of Akt/PKB pathways, which induce the expression of BCL-2 mRNA through the phosphorylation of the cAMP response element (CRE) binding (CREB) protein and its subsequent binding to the CRE in the BCL-2 promoter [129]. Indeed, estrogens are involved in the regulation of pCREB within 15 min of estrogen application, thus indicating the rapid involvement of multiple signaling pathways, such as Ca2+, CaMKII, PKA, and/or ERK [130]. Hence, we speculate that CREB is a critical transcription factor in the regulation of the transcription of many genes and a target for nonclassical estrogen signaling, which provides the most direct relationship between rapidly activated signaling cascades and transcriptional mechanisms [131]. Such systems underlie many regulatory mechanisms of synaptic plasticity in the brain, particularly the regulation of synaptic protein expression [132]. Estrogens regulate biphasic NPY gene expression at the level of the NPY promoter. However, the rapid nongenomic actions of estrogens, which are linked to the PI3K/Akt and ERK/MAPK pathways, are critical in this process. The corresponding pharmacological inhibitors of the PI3K/Akt or ERK/MAPK pathways block the effect of estrogens on NPY gene expression. These observations suggest that the rapid signaling events that are induced by estrogens potentiate the genomic actions of estrogens on NPY gene expression [30]. Although not all estrogenic mechanisms for modulating gene transcription depend on the rapid nongenomic effects and signaling pathways, a solid and classical mechanism underlies the cooperation of the rapid nongenomic and genomic signaling of estrogens in neuronal plasticity and neuroprotection. Notably, three sequences that appear to be EREs have been found in the promoter region of rat Nav1.7 (SCN9A). Similarly, two ERE-like sequences have been found in the promoter region of human Nav1.7 (SCN9A). Therefore, it is highly likely that estrogens regulate Nav1.7 mRNA expression [133]. It is also important to note that activation of estrogen receptor rapidly rescues the impairment of neuronal excitability through BK K+ channel-mediated mechanism in brain slices after oxygen-glucose deprivation [134], suggesting that this rapid signaling is also neuroprotective. Consistently, the increased BK K+ channel-mediated currents and related mRNA levels are found in neuronal cells treated with physiological concentrations of E2 [135]. Because of the important role of ion channels in the activation of signaling cascades, we hypothesize that the genomic effects of estrogens may also remodulate their rapid signaling cascades.

Schematic of the cooperation of the genomic and rapid nongenomic actions of estrogens in neurons. The cooperation of the genomic and rapid nongenomic actions of estrogens is involved in synaptic plasticity. Epigenetic modifications, synaptic protein synthesis, posttranslational modifications, and ER splice variants are thought to be the main molecular mechanisms that underlie the cooperation of the genomic and rapid nongenomic actions of estrogens
The Role of Epigenetic Modifications
The alterations in neuronal gene expression that result from the classical genomic actions of estrogens play an important role in neuroplasticity and neuroprotection, as described above. However, the current understanding of pretranscriptional regulatory processes is poor [136]. A growing number of lines of evidence indicate that epigenetic mechanisms regulate transcription without modifying gene sequences in memory formation, neuroplasticity, and neurodegeneration [137]. Indeed, extracellular cues, including synaptic activity, and neurotrophic factors might crosstalk with the neuronal transcriptional response through epigenetic modifications [138]. Histone phosphorylation or acetylation and DNA methylation are the main epigenetic mechanisms that control gene expression through modification of the chromatin structure, and these mechanisms are critically involved in the regulation of nuclear receptor-mediated transcription [139]. Indeed, the rapid signals that are initiated by estrogens impact the epigenetic modifications that are thought to be involved in the consolidation of memory and neuroplasticity [136, 140, 141].
It was reported that responses to estrogens involved highly specific changes in epigenetic modifications, dependent on cell group, gene, histone modification studied, promoter/enhancer site, and time following estrogen treatment [142]. A subsequent investigation showed that the memory-enhancing effects of estrogens are blocked by a potent HAT (whole name) inhibitor in vitro. In addition, the HAT inhibitor reverses the estrogen-induced increases in histone H3 acetylation, HAT activity, and levels of the de novo methyltransferase DNMT3B as well as the estrogen-induced decrease in the levels of the memory repressor protein histone deacetylase 2 [143]. Estrogens increase the histone H3 acetylation of target genes in the brain through its rapid signaling effects on the activation of ERK, which is essential for the consolidation of memory and neuroplasticity [144]. Wong et al. revealed that estrogens induce nongenomic ER signaling to activate PI3K/AKT, which results in AKT phosphorylation and inactivation of the histone methyltransferase EZH2, thus providing a direct link to disruption of the epigenome [145]. Although a variety of rapid nongenomic ER signals are involved in the epigenetic modifications of histone, estrogens significantly decrease the levels of expression of histone deacetylase. However, it is not clear if the mechanism is related to the rapid signaling actions of estrogens [146]. DNA methylation at CpG dinucleotides and two other alternative forms of methylation, non-CpG methylation and hydroxymethylation, has been reported in neurons [147, 148]. Altered DNA methylation in the brain has been implicated in epilepsy and AD [149, 150]. Thirty minutes after a single infusion of estrogens in the hippocampus, the infusion induced DNA methylation-dependent alterations in the transcription of immediate early genes and initiated a cascade of transcription factors, which contributed to long-term neuronal and circuit alterations [146]. Interestingly, the time course of the estrogen alterations of DNA methylation overlap with its rapid cellular actions, which occur as fast as seconds to minutes but mostly within 1 h. Therefore, it is likely that the activation of rapid signaling by estrogens promotes epigenetic reprogramming. Bredfeldtet al. demonstrated that the ER-mediated signaling that occurs through the PI3K/Akt pathway results in the phosphorylation of EZH2, which reduces the levels of methyltransferases [151, 152]. However, it is not clear if such mechanisms exist in the brain. Nonetheless, the results of these studies provide compelling evidence that communication between extracellular stimuli and chromatin occurs through the signal transduction pathways of estrogens. The mechanism for the rapid suppression of gene expression occurs through the epigenetic modification of methylation at the promoter regions [153]. In neurons, ERα expression rapidly increases after middle cerebral artery occlusion, which suggests a return to the developmental program of gene expression. The ERα gene is also methylated after neuronal injury, which suggests a role of DNA methylation in the regulation of ER expression in the brain. In addition, estrogens can induce differences in the DNA methylation of ERs in the brain [154]. Consequently, the altered expression of ERs in neurons may result in changes in its downstream effectors in both genomic and nongenomic pathways. However, these types of studies are still in their early stages, and advances are needed in the understanding of the hormonal regulation of the enzymes that control acetylation and methylation, transient versus stable DNA methylation patterns, and sex differences across the epigenome in order to fully understand the involvement of epigenetic modifications in brain function and behavior.
The Role of Synaptic Protein Synthesis and Posttranslational Modifications
Synapses are dynamic structures that are continually shaped and remodeled by a rich variety of highly sophisticated protein complexes called scaffold proteins [155]. Many investigations have shown that estrogens affect synaptic scaffold proteins [156, 157]. Increases in spine density involve the formation of new spines, which thus requires the synthesis of new proteins within the PSD [158, 159].The consolidation of long-term synaptic plasticity requires de novo protein synthesis, which involves a mechanism that modulates the translation of a subset of neuronal mRNAs [160, 161]. Estrogens induce the rapid protein synthesis of new PSD-95, but they do not cause a rapid and significant increase in PSD-95 mRNA levels [162]. This observation suggests that estrogens may modulate the de novo synthesis of a variety of scaffold proteins in synapses in accordance with neuronal plasticity. One of the signature actions of estrogens is to alter the morphology of neural processes [163]. Dendritic remodeling requires structural modifications of the cytoskeletal protein β-actin. The severing of filamentous actin is performed by the constitutively active enzyme cofilin, which is inactivated by phosphorylation. Membrane-initiated estrogen signaling that involves the mGluR 1 is responsible for the phosphorylation and subsequent deactivation of cofilin [164]. Thus, this phenomenon suggests that estrogens enhance the PTMs of cofilin. PTMs are ubiquitously involved in complex neuronal processing and are well-established general mechanisms required for learning and memory as well as the underlying cellular correlate, long-term synaptic plasticity [165]. Another study reported that rapid and transient ER signaling stimulation affected the PTMs of RGSz1 protein isoforms, which thereby attenuated 5-HT1AR signaling in the hypothalamus [166]. A wide variety of neurodegenerative diseases that involve impairments in the ubiquitin-proteasome system have been described as proteinopathies that are caused by aggregate-prone proteins that are not efficiently removed by proteasomes [167]. The treatment of cells with estrogens results in aggregate removal and increased cell survival due to activation of the autophagic pathway. Interestingly, previous observations have suggested that estrogens enhance the ubiquitination of calcium channels, which decreases Ca2+ influx. Such actions reversely activate different signaling cascades that mediate rapid estrogenic actions. Consequently, it is reasonable to presume that the regulation of PTM mechanisms by estrogens links the cooperation of the genomic and rapid nongenomic actions of estrogens in synaptic plasticity and neuroprotection.
Interestingly, the findings of recent studies have suggested a novel model whereby the concomitant translocation of proteins from the dendritic cytoplasm to synapses and the nucleus occurs in response to synaptic activity in order to control neuronal plasticity [168]. However, whether estrogens acutely regulate such a model at adult synapses and if such effects are responsible for its actions that link its genomic and rapid nongenomic effects are still unknown. Several synaptically synthesized proteins that are transported to the nucleus from synapses may link specific types of stimuli with the nucleus, which indicates that the synapse-to-nuclear transport of proteins dynamically informs the nucleus about synaptic activity [169, 170]. Signals generated at synapses trigger transcriptional changes that is essential for neuronal development, required for persistent forms of learning-related synaptic plasticity [171, 172]. However, the precise mechanisms underlying the information transfer from the cytoplasm to the nucleus of neurons are still poorly characterized [173]. Estrogens increase the concentration of filamentous actin in spines and strongly enhance its polymerization in association with LTP. A study of the origins of these effects showed that estrogens activate the small GTPase RhoA and phosphorylate (inactivate) the actin-severing protein cofilin, which is a downstream target of RhoA. Moreover, an antagonist of RhoA kinase (ROCK) blocks estrogens’ synaptic effects. Estrogens thus emerge as a positive modulator of the RhoA-cofilin pathway that regulates the subsynaptic cytoskeleton. Moreover, ovariectomies decrease RhoA activity, spine cytoskeletal plasticity, and LTP, whereas brief infusions of estrogens rescue plasticity, thus suggesting that the deficits in plasticity arise from acute as well as genomic consequences of hormone loss [174]. Therefore, estrogens affect synaptic physiology by partially activating the actin-signaling pathways that may participate in the translocation of synaptic proteins [175]. Such a mechanism acts in the ERs themselves, as in estrogen-treated cells, and the membrane and cytosolic ERβ levels gradually decrease, while those of nuclear ERβ progressively increase in a time-dependent manner, thus suggesting the estrogen-dependent nuclear translocation of ERβ [124]. Although the data strongly support a role of this pathway in the control of the translocation of proteins from the dendritic cytoplasm to synapses and the nucleus, the direct regulation has not been tested.
The Role of ER Splice Variants
The identification of multiple splice variants of ERs in rodents and humans has added a further layer of complexity to genomic regulation by estrogens [176]. In the hypothalamus, splice variants have been suggested to participate in membrane-initiated estrogenic signaling, which would connect the genomic and rapid nongenomic estrogenic effects [177]. The brain has the highest levels of exon skipping, which is the most common mechanism of alternative splicing [178, 179]. Canonical ER mRNA exons may generate a number of splice variants with single or multiple exon skipping, exon duplication, inserts, or partial exon deletions [180]. To date, the functional implications of ER splice variants in the CNS remain to be determined [3]. ERα splice variants have been investigated in the brain areas that regulate memory formation and that are affected in patients with AD [180]. The major ERα splice variants are the dominant negative del.7 isoform lacking exon 7, which encodes a substantial portion of the ERα ligand-binding domain. This alternative may protect tissues from excessive estrogenic effects [181]. However, the high concentrations of the dominant negative isoform del.7 that inhibits estrogenic signaling in the brains of the elderly and patients with AD suggest a possible reduction of the effects of estrogens on cognitive functions [180]. Importantly, the expression of multiple ER variants might be tissue-specific [182]. Indeed, Ishunina et al. found that the major ERα splice variant in the hippocampus was del.4 [183]. It is also worth noting that the ER splice variants were substantively different in structure. These structural differences suggest the intriguing possibility that these receptors may have specific functions not dependent on ligands and/or resistant to normal estrogenic effects, which could have detrimental consequences on therapeutic hormone treatment strategies during menopause or in disease states [184]. In addition to ERα splice variants, recent studies have identified those of ERβ in the CNS [60].
To date, three major splice variants of the classic ERβ receptor have been described in the rodent. These include a deletion of exon3, a deletion of exon 4, and an insert between exons 5 and 6 [185]. The human ERβ variants that have been identified to date contain variable length deletions and substitutions in exon 8 [176]. A novel finding demonstrated that human-specific ERβ splice variants exhibit marked constitutive activity in neuronal cells at both minimal and complex promoters. Furthermore, human-specific ERβ splice variants are largely unresponsive to ligands and induce modest increases of ERE-mediated promoter activity and robust decreases in AP-1-mediated promoter activity. Although the changes in the ERE-mediated promoter activity were modest, these fine-tuned changes could have important biological consequences [184]. Collectively, these findings suggest that the ER area constitutes part of the estrogenic rapid nongenomic pathway and that their splice variants, which are genomic changes, could undoubtedly affect the cooperation of the genomic and rapid nongenomic actions of estrogens.
In summary, although many questions remain to be resolved, there is substantial evidence that the biological effects of estrogenic actions constitute a complex interplay of genomic and nongenomic mechanisms and depend on the physiological and genetic context of the target cells. However, whether they rely on a cooperative or sequential modulatory system remains to be clarified. The combination of genomic and nongenomic mechanisms endows estrogens with considerable diversity, range, tissue, and power in modulating neural functions. Such cooperation may produce long-term changes in neuroplasticity. In this review, we highlighted the cellular and molecular mechanisms, including epigenetic modifications, synaptic protein synthesis, PTMs, and ER mutations to illustrate how estrogens induce changes in synaptic morphology and beneficial neuroprotection. Collectively, these literature investigations argue that the genomic and nongenomic estrogenic actions should probably be viewed as a unified mode.
References
Day JJ, Sweatt JD (2011) Epigenetic mechanisms in cognition. Neuron 70(5):813–829
Day JJ, Sweatt JD (2012) Epigenetic treatments for cognitive impairments. Neuropsychopharmacology 37(1):247–260
Srivastava DP, Woolfrey KM, Penzes P (2013) Insights into rapid modulation of neuroplasticity by brain estrogens. Pharmacol Rev 65(4):1318–1350
Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10(9):647–658
Srivastava DP, Waters EM, Mermelstein PG, Kramar EA, Shors TJ, Liu F (2011) Rapid estrogen signaling in the brain: implications for the fine-tuning of neuronal circuitry. J Neurosci Off J Soc Neurosci 31(45):16056–16063
Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61(3):340–350
Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R (2010) Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci 13(2):190–196
Kamat PK, Swarnkar S, Rai S, Kumar V, Tyagi N (2014) Astrocyte mediated MMP-9 activation in the synapse dysfunction: an implication in Alzheimer disease. Ther Targets Neurol Dis 1(1):e243
Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14(3):285–293
Popovych OV, Tass PA (2014) Control of abnormal synchronization in neurological disorders. Front Neurol 5:268
Paul SM, Purdy RH (1992) Neuroactive steroids. FASEB J 6(6):2311–2322
Ding D, Starke RM, Dumont AS et al (2014) Therapeutic implications of estrogen for cerebral vasospasm and delayed cerebral ischemia induced by aneurysmal subarachnoid hemorrhage. Biomed Res Int 2014:727428
DeVoogd T, Nottebohm F (1981) Gonadal hormones induce dendritic growth in the adult avian brain. Science 214(4517):202–204
Woolley CS, Gould E, Frankfurt M, McEwen BS (1990) Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci Off J Soc Neurosci 10(12):4035–4039
Toran-Allerand CD, Miranda RC, Bentham WD et al (1992) Estrogen receptors colocalize with low-affinity nerve growth factor receptors in cholinergic neurons of the basal forebrain. Proc Natl Acad Sci U S A 89(10):4668–4672
Bi R, Foy MR, Vouimba RM, Thompson RF, Baudry M (2001) Cyclic changes in estradiol regulate synaptic plasticity through the MAP kinase pathway. Proc Natl Acad Sci U S A 98(23):13391–13395
Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, Simpkins JW (2008) Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A 105(39):15148–15153
Terasawa E, Timiras PS (1968) Electrical activity during the estrous cycle of the rat: cyclic changes in limbic structures. Endocrinology 83(2):207–216
Jover T, Tanaka H, Calderone A et al (2002) Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci Off J Soc Neurosci 22(6):2115–2124
Yang LC, Zhang QG, Zhou CF et al (2010) Extranuclear estrogen receptors mediate the neuroprotective effects of estrogen in the rat hippocampus. PLoS One 5(5):e9851
Zhang QG, Han D, Wang RM et al (2011) C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection. Proc Natl Acad Sci U S A 108(35):E617–E624
Watson CS, Alyea RA, Jeng YJ, Kochukov MY (2007) Nongenomic actions of low concentration estrogens and xenoestrogens on multiple tissues. Mol Cell Endocrinol 274(1–2):1–7
Yi P, Driscoll MD, Huang J et al (2002) The effects of estrogen-responsive element- and ligand-induced structural changes on the recruitment of cofactors and transcriptional responses by ER alpha and ER beta. Mol Endocrinol 16(4):674–693
Prossnitz ER, Barton M (2011) The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol 7(12):715–726
Meyer MR, Amann K, Field AS et al (2012) Deletion of G protein-coupled estrogen receptor increases endothelial vasoconstriction. Hypertension 59(2):507–512
Meyer MR, Clegg DJ, Prossnitz ER, Barton M (2011) Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol (Oxf) 203(1):259–269
Meyer MR, Prossnitz ER, Barton M (2011) The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul Pharmacol 55(1–3):17–25
Hansen SF ALSL, Jensen TS, Leboeuf Yde C, Hestbaek L (2010) The Nordic maintenance care program: what are the indications for maintenance care in patients with low back pain? A survey of the members of the Danish Chiropractors’ Association. Chiropr Osteopat 18:25
Day JJ, Sweatt JD (2011) Cognitive neuroepigenetics: a role for epigenetic mechanisms in learning and memory. Neurobiol Learn Mem 96(1):2–12
Titolo D, Mayer CM, Dhillon SS, Cai F, Belsham DD (2008) Estrogen facilitates both phosphatidylinositol 3-kinase/Akt and ERK1/2 mitogen-activated protein kinase membrane signaling required for long-term neuropeptide Y transcriptional regulation in clonal, immortalized neurons. J Neurosci Off J Soci Neurosci 28(25):6473–6482
Phan A, Gabor CS, Favaro KJ et al (2012) Low doses of 17beta-estradiol rapidly improve learning and increase hippocampal dendritic spines. Neuropsychopharmacology 37(10):2299–2309
Guerriero G (2009) Vertebrate sex steroid receptors: evolution, ligands, and neurodistribution. Ann N Y Acad Sci 1163:154–168
Beato M (1989) Gene regulation by steroid hormones. Cell 56(3):335–344
Woolley CS (2007) Acute effects of estrogen on neuronal physiology. Annu Rev Pharmacol Toxicol 47:657–680
Mitterling KL, Spencer JL, Dziedzic N et al (2010) Cellular and subcellular localization of estrogen and progestin receptor immunoreactivities in the mouse hippocampus. J Comp Neurol 518(14):2729–2743
Smiley DA, Khalil RA (2009) Estrogenic compounds, estrogen receptors and vascular cell signaling in the aging blood vessels. Curr Med Chem 16(15):1863–1887
Morito K, Aomori T, Hirose T et al (2002) Interaction of phytoestrogens with estrogen receptors alpha and beta (II). Biol Pharm Bull 25(1):48–52
Varea O, Garrido JJ, Dopazo A, Mendez P, Garcia-Segura LM, Wandosell F (2009) Estradiol activates beta-catenin dependent transcription in neurons. PLoS One 4(4):e5153
Morissette M, Le Saux M, D’Astous M et al (2008) Contribution of estrogen receptors alpha and beta to the effects of estradiol in the brain. J Steroid Biochem Mol Biol 108(3–5):327–338
Barreto G, Santos-Galindo M, Diz-Chaves Y et al (2009) Selective estrogen receptor modulators decrease reactive astrogliosis in the injured brain: effects of aging and prolonged depletion of ovarian hormones. Endocrinology 150(11):5010–5015
Zhao L, O’Neill K, Diaz BR (2005) Selective estrogen receptor modulators (SERMs) for the brain: current status and remaining challenges for developing NeuroSERMs. Brain Res Brain Res Rev 49(3):472–493
Wu Y, Wang G, Scott SA, Capecchi MR (2008) Hoxc10 and Hoxd10 regulate mouse columnar, divisional and motor pool identity of lumbar motoneurons. Development 135(1):171–182
Hostikka SL, Gong J, Carpenter EM (2009) Axial and appendicular skeletal transformations, ligament alterations, and motor neuron loss in Hoxc10 mutants. Int J Biol Sci 5(5):397–410
Ansari KI, Hussain I, Kasiri S, Mandal SS (2012) HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol 48(1):61–75
Navarro A, del Valle E, Juarez A et al (2010) Apolipoprotein D synthesis progressively increases in frontal cortex during human lifespan. Age (Dordr) 32(1):85–96
Ordonez C, Navarro A, Perez C, Astudillo A, Martinez E, Tolivia J (2006) Apolipoprotein D expression in substantia nigra of Parkinson disease. Histol Histopathol 21(4):361–366
Ordonez C, Navarro A, Perez C, Martinez E, del Valle E, Tolivia J (2012) Gender differences in apolipoprotein D expression during aging and in Alzheimer disease. Neurobiol Aging 33(2):433 e11–20
Kundu P, Alioua A, Stefani E, Toro L (2007) Regulation of mouse Slo gene expression: multiple promoters, transcription start sites, and genomic action of estrogen. J Biol Chem 282(37):27478–27492
Handel AE (2016) Bioinformatics analysis of estrogen-responsive genes. Methods Mol Biol 1366:29–39
Humphreys GI, Ziegler YS, Nardulli AM (2014) 17beta-estradiol modulates gene expression in the female mouse cerebral cortex. PLoS One 9(11):e111975
Shi L, Lin Q, Su B (2015) Estrogen regulation of microcephaly genes and evolution of brain sexual dimorphism in primates. BMC Evol Biol 15:127
Hiroi R, Handa RJ (2013) Estrogen receptor-beta regulates human tryptophan hydroxylase-2 through an estrogen response element in the 5’ untranslated region. J Neurochem 127(4):487–495
Shao W, Halachmi S, Brown M (2002) ERAP140, a conserved tissue-specific nuclear receptor coactivator. Mol Cell Biol 22(10):3358–3372
Bian C, Zhu H, Zhao Y, Cai W, Zhang J (2014) Intriguing roles of hippocampus-synthesized 17beta-estradiol in the modulation of hippocampal synaptic plasticity. J Mole Neurosci MN 54(2):271–281
Rosenfeld MG, Lunyak VV, Glass CK (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20(11):1405–1428
Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K (2012) Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res 22(3):490–503
Li W, Notani D, Ma Q et al (2013) Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498(7455):516–520
Liu Z, Merkurjev D, Yang F et al (2014) Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 159(2):358–373
Milner TA, Ayoola K, Drake CT et al (2005) Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol 491(2):81–95
ter Horst GJ (2010) Estrogen in the limbic system. Vitam Horm 82:319–338
Sa SI, Fonseca BM, Teixeira N, Madeira MD (2015) Estrogen receptors alpha and beta have different roles in the induction and trafficking of progesterone receptors in hypothalamic ventromedial neurons. FEBS J 282(6):1126–1136
Koehler KF, Helguero LA, Haldosen LA, Warner M, Gustafsson JA (2005) Reflections on the discovery and significance of estrogen receptor beta. Endocr Rev 26(3):465–478
Pettersson K, Delaunay F, Gustafsson JA (2000) Estrogen receptor beta acts as a dominant regulator of estrogen signaling. Oncogene 19(43):4970–4978
Liu SB, Zhao MG (2013) Neuroprotective effect of estrogen: role of nonsynaptic NR2B-containing NMDA receptors. Brain Res Bull 93:27–31
Srivastava DP, Woolfrey KM, Liu F, Brandon NJ, Penzes P (2010) Estrogen receptor ss activity modulates synaptic signaling and structure. J Neurosci Off J Soc Neurosci 30(40):13454–13460
Watson CS, Alyea RA, Hawkins BE, Thomas ML, Cunningham KA, Jakubas AA (2006) Estradiol effects on the dopamine transporter—protein levels, subcellular location, and function. J Mol Signal 1:5
Zaitsu M, Narita S, Lambert KC et al (2007) Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol Immunol 44(8):1977–1985
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER (2005) A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307(5715):1625–1630
Toran-Allerand CD, Guan X, MacLusky NJ et al (2002) ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci Off J Soc Neurosci 22(19):8391–8401
Qiu J, Bosch MA, Tobias SC et al (2003) Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci Off J Soc Neurosci 23(29):9529–9540
Christensen A, Micevych P (2013) A novel membrane estrogen receptor activated by STX induces female sexual receptivity through an interaction with mGluR1a. Neuroendocrinology 97(4):363–368
Grove-Strawser D, Boulware MI, Mermelstein PG (2010) Membrane estrogen receptors activate the metabotropic glutamate receptors mGluR5 and mGluR3 to bidirectionally regulate CREB phosphorylation in female rat striatal neurons. Neuroscience 170(4):1045–1055
Martins WC, Tasca CI, Cimarosti H (2015) Battling Alzheimer’s disease: targeting SUMOylation-mediated pathways. Neurochem Res 41(3):568–578
Spiteri T, Musatov S, Ogawa S, Ribeiro A, Pfaff DW, Agmo A (2010) The role of the estrogen receptor alpha in the medial amygdala and ventromedial nucleus of the hypothalamus in social recognition, anxiety and aggression. Behav Brain Res 210(2):211–220
Pfaff DW, Ribeiro AC (2010) Theoretical consequences of fluctuating versus constant liganding of oestrogen receptor-alpha in neurones. J Neuroendocrinol 22(6):486–491
Konkle AT, McCarthy MM (2011) Developmental time course of estradiol, testosterone, and dihydrotestosterone levels in discrete regions of male and female rat brain. Endocrinology 152(1):223–235
Hojo Y, Higo S, Ishii H et al (2009) Comparison between hippocampus-synthesized and circulation-derived sex steroids in the hippocampus. Endocrinology 150(11):5106–5112
Pfrieger FW, Ungerer N (2011) Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 50(4):357–371
Rapp A, Gmeiner B, Huttinger M (2006) Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 88(5):473–483
Pelletier G (2010) Steroidogenic enzymes in the brain: morphological aspects. Prog Brain Res 181:193–207
Bruzzone F, Do Rego JL, Luu-The V, Pelletier G, Vallarino M, Vaudry H (2010) Immunohistochemical localization and biological activity of 3beta-hydroxysteroid dehydrogenase and 5alpha-reductase in the brain of the frog, Rana esculenta, during development. J Chem Neuroanat 39(1):35–50
Wicher G, Norlin M (2015) Estrogen-mediated regulation of steroid metabolism in rat glial cells; effects on neurosteroid levels via regulation of CYP7B1-mediated catalysis. J Steroid Biochem Mol Biol 145:21–27
Eghlidi DH, Urbanski HF (2015) Effects of age and estradiol on gene expression in the rhesus macaquehypothalamus. Neuroendocrinology 101(3):236–245
Chen C, Kuo J, Wong A, Micevych P (2014) Estradiol modulates translocator protein (TSPO) and steroid acute regulatory protein (StAR) via protein kinase A (PKA) signaling in hypothalamic astrocytes. Endocrinology 155(8):2976–2985
Tabatadze N, Sato SM, Woolley CS (2014) Quantitative analysis of long-form aromatase mRNA in the male and female rat brain. PLoS One 9(7):e100628
Yang SY, He XY, Isaacs C, Dobkin C, Miller D, Philipp M (2014) Roles of 17beta-hydroxysteroid dehydrogenase type 10 in neurodegenerative disorders. J Steroid Biochem Mol Biol 143:460–472
Cornil CA, Dalla C, Papadopoulou-Daifoti Z et al (2005) Rapid decreases in preoptic aromatase activity and brain monoamine concentrations after engaging in male sexual behavior. Endocrinology 146(9):3809–3820
Balthazart J, Baillien M, Ball GF (2006) Rapid control of brain aromatase activity by glutamatergic inputs. Endocrinology 147(1):359–366
Bernelot Moens SJ, Schnitzler GR, Nickerson M et al (2012) Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation 126(16):1993–2004
Scott E, Zhang QG, Wang R, Vadlamudi R, Brann D (2012) Estrogen neuroprotection and the critical period hypothesis. Front Neuroendocrinol 33(1):85–104
Dominguez R, Liu R, Baudry M (2007) 17-Beta-estradiol-mediated activation of extracellular-signal regulated kinase, phosphatidylinositol 3-kinase/protein kinase B-Akt and N-methyl-D-aspartate receptor phosphorylation in cortical synaptoneurosomes. J Neurochem 101(1):232–240
Li J, Siegel M, Yuan M et al (2011) Estrogen enhances neurogenesis and behavioral recovery after stroke. J Cereb Blood Flow Metab 31(2):413–425
Ramos JW (2008) The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol 40(12):2707–2719
Toran-Allerand CD, Singh M, Setalo G Jr (1999) Novel mechanisms of estrogen action in the brain: new players in an old story. Front Neuroendocrinol 20(2):97–121
Neubert M, Ridder DA, Bargiotas P, Akira S, Schwaninger M (2011) Acute inhibition of TAK1 protects against neuronal death in cerebral ischemia. Cell Death Differ 18(9):1521–1530
Nijboer CH, van der Kooij MA, van Bel F, Ohl F, Heijnen CJ, Kavelaars A (2010) Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav Immun 24(5):812–821
White BJ, Tarabishy S, Venna VR et al (2012) Protection from cerebral ischemia by inhibition of TGFbeta-activated kinase. Exp Neurol 237(1):238–245
Van Kempen TA, Gorecka J, Gonzalez AD, Soeda F, Milner TA, Waters EM (2014) Characterization of neural estrogen signaling and neurotrophic changes in the accelerated ovarian failure mouse model of menopause. Endocrinology 155(9):3610–3623
Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, Dreyfus CF (2014) Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci Off J Soc Neurosci 34(24):8186–8196
Yu Z, Gao W, Jiang E et al (2013) Interaction between IGF-IR and ER induced by E2 and IGF-I. PLoS One 8(5):e62642
Boulware MI, Heisler JD, Frick KM (2013) The memory-enhancing effects of hippocampal estrogen receptor activation involve metabotropic glutamate receptor signaling. J Neurosci Off J Soc Neurosci 33(38):15184–15194
Yang Q, Yang L, Zhang K et al (2015) Increased coupling of caveolin-1 and estrogen receptor alpha contributes to the fragile X syndrome. Ann Neurol 77(4):618–636
Raz L, Khan MM, Mahesh VB, Vadlamudi RK, Brann DW (2008) Rapid estrogen signaling in the brain. Neurosignals 16(2–3):140–153
Khan MM, Hadman M, Wakade C et al (2005) Cloning, expression, and localization of MNAR/PELP1 in rodent brain: colocalization in estrogen receptor-alpha- but not in gonadotropin-releasing hormone-positive neurons. Endocrinology 146(12):5215–5227
Gu Q, Moss RL (1998) Novel mechanism for non-genomic action of 17 beta-oestradiol on kainate-induced currents in isolated rat CA1 hippocampal neurones. J Physiol 506(Pt 3):745–754
Gu Q, Korach KS, Moss RL (1999) Rapid action of 17beta-estradiol on kainate-induced currents in hippocampal neurons lacking intracellular estrogen receptors. Endocrinology 140(2):660–666
MacLusky NJ, Luine VN, Hajszan T, Leranth C (2005) The 17alpha and 17beta isomers of estradiol both induce rapid spine synapse formation in the CA1 hippocampal subfield of ovariectomized female rats. Endocrinology 146(1):287–293
Feng Y, Wang B, Du F et al (2013) The involvement of PI3K-mediated and L-VGCC-gated transient Ca2+ influx in 17beta-estradiol-mediated protection of retinal cells from H2O2-induced apoptosis with Ca2+ overload. PLoS One 8(11):e77218
Hajimohammadreza I, Probert AW, Coughenour LL et al (1995) A specific inhibitor of calcium/calmodulin-dependent protein kinase-II provides neuroprotection against NMDA- and hypoxia/hypoglycemia-induced cell death. J Neurosci Off J Soc Neurosci 15(5 Pt 2):4093–4101
Achterberg KG, Buitendijk GH, Kool MJ et al (2014) Temporal and region-specific requirements of alphaCaMKII in spatial and contextual learning. J Neurosci OffJ Soc Neurosci 34(34):11180–11187
Lee SJ, Escobedo-Lozoya Y, Szatmari EM, Yasuda R (2009) Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458(7236):299–304
Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429(6993):761–766
Logan SM, Sarkar SN, Zhang Z, Simpkins JW (2011) Estrogen-induced signaling attenuates soluble Abeta peptide-mediated dysfunction of pathways in synaptic plasticity. Brain Res 1383:1–12
Yan XL, Liu DH, Zhang GL, Hu SQ, Chen YG, Xu T (2015) S-Nitrosylation of proline-rich tyrosine kinase 2 involves its activation induced by oxygen-glucose deprivation. Neurosci Lett 597:90–96
Druzin M, Malinina E, Grimsholm O, Johansson S (2011) Mechanism of estradiol-induced block of voltage-gated K+ currents in rat medial preoptic neurons. PLoS One 6(5):e20213
Hao X, Li X, Li X (2014) 17beta-estradiol downregulated the expression of TASK-1 channels in mouse neuroblastoma N2A cells. J Membr Biol 247(3):273–279
Kirkegaard SS, Lambert IH, Gammeltoft S, Hoffmann EK (2010) Activation of the TASK-2 channel after cell swelling is dependent on tyrosine phosphorylation. Am J Physiol Cell Physiol 299(4):C844–C853
Carrer HF, Araque A, Buno W (2003) Estradiol regulates the slow Ca2+−activated K+ current in hippocampal pyramidal neurons. J Neurosci Off J Soc Neurosci 23(15):6338–6344
Kuo J, Hamid N, Bondar G, Prossnitz ER, Micevych P (2010) Membrane estrogen receptors stimulate intracellular calcium release and progesterone synthesis in hypothalamic astrocytes. J Neurosci Off J Soc Neurosci 30(39):12950–12957
Huang GZ, Woolley CS (2012) Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR-dependent mechanism. Neuron 74(5):801–808
Lokuge S, Frey BN, Foster JA, Soares CN, Steiner M (2010) The rapid effects of estrogen: a mini-review. Behav Pharmacol 21(5–6):465–472
Srivastava DP, Penzes P (2011) Rapid estradiol modulation of neuronal connectivity and its implications for disease. Front Endocrinol 2:77
Chu Z, Andrade J, Shupnik MA, Moenter SM (2009) Differential regulation of gonadotropin-releasing hormone neuron activity and membrane properties by acutely applied estradiol: dependence on dose and estrogen receptor subtype. J Neurosci OffJ Soc Neurosci 29(17):5616–5627
Chamniansawat S, Chongthammakun S (2010) Genomic and non-genomic actions of estrogen on synaptic plasticity in SH-SY5Y cells. Neurosci Lett 470(1):49–54
Mittelman-Smith MA, Wong AM, Kathiresan AS, Micevych PE (2015) Classical and membrane-initiated estrogen signaling in an in vitro model of anterior hypothalamic kisspeptin neurons. Endocrinology 156(6):2162–2173
Galluzzo P, Ascenzi P, Bulzomi P, Marino M (2008) The nutritional flavanone naringenin triggers antiestrogenic effects by regulating estrogen receptor alpha-palmitoylation. Endocrinology 149(5):2567–2575
Li M, Guo J, Gao W et al (2014) Bisphenol AF-induced endogenous transcription is mediated by ERalpha and ERK1/2 activation in human breast cancer cells. PLoS One 9(4):e94725
Wu TW, Wang JM, Chen S, Brinton RD (2005) 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience 135(1):59–72
Pugazhenthi S, Nesterova A, Sable C et al (2000) Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem 275(15):10761–10766
Cheong RY, Kwakowsky A, Barad Z, Porteous R, Herbison AE, Abraham IM (2012) Estradiol acts directly and indirectly on multiple signaling pathways to phosphorylate cAMP-response element binding protein in GnRH neurons. Endocrinology 153(8):3792–3803
Kwakowsky A, Cheong RY, Herbison AE, Abraham IM (2014) Non-classical effects of estradiol on cAMP responsive element binding protein phosphorylation in gonadotropin-releasing hormone neurons: mechanisms and role. Front Neuroendocrinol 35(1):31–41
Tremere LA, Kovaleski RF, Burrows K, Jeong JK, Pinaud R (2012) Mechanistic basis and functional roles of long-term plasticity in auditory neurons induced by a brain-generated estrogen. J Neurosci OffJ Soc Neurosci 32(46):16478–16495
Bi RY, Ding Y, Gan YH (2015) A new hypothesis of sex-differences in temporomandibular disorders: estrogen enhances hyperalgesia of inflamed TMJ through modulating voltage-gated sodium channel 1.7 in trigeminal ganglion? Med Hypotheses 84(2):100–103
Zhang H, Xie M, Schools GP et al (2009) Tamoxifen mediated estrogen receptor activation protects against early impairment of hippocampal neuron excitability in an oxygen/glucose deprivation brain slice ischemia model. Brain Res 1247:196–211
Nishimura I, Ui-Tei K, Saigo K, Ishii H, Sakuma Y, Kato M (2008) 17beta-estradiol at physiological concentrations augments Ca(2+) -activated K+ currents via estrogen receptor beta in the gonadotropin-releasing hormone neuronal cell line GT1-7. Endocrinology 149(2):774–782
Sultan FA, Wang J, Tront J, Liebermann DA, Sweatt JD (2012) Genetic deletion of Gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity. J Neurosci Off J Soc Neurosci 32(48):17059–17066
Vukojevic V, Kolassa IT, Fastenrath M et al (2014) Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors. J Neurosci Off J Soci Neurosci 34(31):10274–10284
Riccio A (2010) Dynamic epigenetic regulation in neurons: enzymes, stimuli and signaling pathways. Nat Neurosci 13(11):1330–1337
Matsuda KI, Mori H, Nugent BM, Pfaff DW, McCarthy MM, Kawata M (2011) Histone deacetylation during brain development is essential for permanent masculinization of sexual behavior. Endocrinology 152(7):2760–2767
Mahan AL, Mou L, Shah N, Hu JH, Worley PF, Ressler KJ (2012) Epigenetic modulation of Homer1a transcription regulation in amygdala and hippocampus with pavlovian fear conditioning. J Neurosc Off J Soc Neurosci 32(13):4651–4659
Koshibu K, Graff J, Beullens M et al (2009) Protein phosphatase 1 regulates the histone code for long-term memory. J Neurosci Off J Soc Neurosci 29(41):13079–13089
Gagnidze K, Weil ZM, Faustino LC, Schaafsma SM, Pfaff DW (2013) Early histone modifications in the ventromedial hypothalamus and preoptic area following oestradiol administration. J Neuroendocrinol 25(10):939–955
Zhao Z, Fan L, Fortress AM, Boulware MI, Frick KM (2012) Hippocampal histone acetylation regulates object recognition and the estradiol-induced enhancement of object recognition. J Neurosci OffJ Soc Neurosci 32(7):2344–2351
Frick KM, Zhao Z, Fan L (2011) The epigenetics of estrogen: epigenetic regulation of hormone-induced memory enhancement. Epigenetics 6(6):675–680
Wong RL, Walker CL (2013) Molecular pathways: environmental estrogens activate nongenomic signaling to developmentally reprogram the epigenome. Clin Cancer Res 19(14):3732–3737
Zhao Z, Fan L, Frick KM (2010) Epigenetic alterations regulate estradiol-induced enhancement of memory consolidation. Proc Natl Acad Sci U S A 107(12):5605–5610
Kinde B, Gabel HW, Gilbert CS, Griffith EC, Greenberg ME (2015) Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A 112(22):6800–6808
Ma DK, Jang MH, Guo JU et al (2009) Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323(5917):1074–1077
Feng J, Zhou Y, Campbell SL et al (2010) Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13(4):423–430
Lord J, Cruchaga C (2014) The epigenetic landscape of Alzheimer’s disease. Nat Neurosci 17(9):1138–1140
Bannister AJ, Zegerman P, Partridge JF et al (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410(6824):120–124
Bredfeldt TG, Greathouse KL, Safe SH, Hung MC, Bedford MT, Walker CL (2010) Xenoestrogen-induced regulation of EZH2 and histone methylation via estrogen receptor signaling to PI3K/AKT. Mol Endocrinol 24(5):993–1006
Wilson ME, Westberry JM (2009) Regulation of oestrogen receptor gene expression: new insights and novel mechanisms. J Neuroendocrinol 21(4):238–242
Nugent BM, Schwarz JM, McCarthy MM (2011) Hormonally mediated epigenetic changes to steroid receptors in the developing brain: implications for sexual differentiation. Horm Behav 59(3):338–344
Beales PA, Ciani B, Cleasby AJ (2015) Nature’s lessons in design: nanomachines to scaffold, remodel and shape membrane compartments. Phys Chem Chem Phys 17(24):15489–15507
Li C, Brake WG, Romeo RD et al (2004) Estrogen alters hippocampal dendritic spine shape and enhances synaptic protein immunoreactivity and spatial memory in female mice. Proc Natl Acad Sci U S A 101(7):2185–2190
Waters EM, Thompson LI, Patel P et al (2015) G-protein-coupled estrogen receptor 1 is anatomically positioned to modulate synaptic plasticity in the mouse hippocampus. J Neurosci OffJ Soc Neurosci 35(6):2384–2397
Garner CC, Nash J, Huganir RL (2000) PDZ domains in synapse assembly and signalling. Trends Cell Biol 10(7):274–280
Sheng M (2001) Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci U S A 98(13):7058–7061
Kelleher RJ 3rd, Govindarajan A, Tonegawa S (2004) Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44(1):59–73
O’Donnell C, Sejnowski TJ (2014) Selective memory generalization by spatial patterning of protein synthesis. Neuron 82(2):398–412
Akama KT, McEwen BS (2003) Estrogen stimulates postsynaptic density-95 rapid protein synthesis via the Akt/protein kinase B pathway. J Neurosci Off J Soc Neurosci 23(6):2333–2339
Calizo LH, Flanagan-Cato LM (2000) Estrogen selectively regulates spine density within the dendritic arbor of rat ventromedial hypothalamic neurons. J Neurosci Off J Soc Neurosci 20(4):1589–1596
Christensen A, Dewing P, Micevych P (2011) Membrane-initiated estradiol signaling induces spinogenesis required for female sexual receptivity. J Neurosci Off J Soc Neurosci 31(48):17583–17589
Lee L, Dale E, Staniszewski A et al (2014) Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer’s disease. Sci Reports 4:7190
McAllister CE, Mi Z, Mure M, Li Q, Muma NA (2014) GPER1 stimulation alters posttranslational modification of RGSz1 and induces desensitization of 5-HT1A receptor signaling in the rat hypothalamus. Neuroendocrinology 100(2–3):228–239
D’Alessandro A, D’Aguanno S, Cencioni MT et al (2012) Protein repertoire impact of Ubiquitin-Proteasome System impairment: insight into the protective role of beta-estradiol. J Proteomics 75(4):1440–1453
VanLeeuwen JE, Rafalovich I, Sellers K et al (2014) Coordinated nuclear and synaptic shuttling of afadin promotes spine plasticity and histone modifications. J Biol Chem 289(15):10831–10842
Ch’ng TH, Uzgil B, Lin P, Avliyakulov NK, O’Dell TJ, Martin KC (2012) Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150(1):207–221
Abe K, Takeichi M (2007) NMDA-receptor activation induces calpain-mediated beta-catenin cleavages for triggering gene expression. Neuron 53(3):387–397
Yoshida T, Uchigashima M, Yamasaki M et al (2011) Unique inhibitory synapse with particularly rich endocannabinoid signaling machinery on pyramidal neurons in basal amygdaloid nucleus. Proc Natl Acad Sci U S A 108(7):3059–3064
Brunet I, Weinl C, Piper M et al (2005) The transcription factor Engrailed-2 guides retinal axons. Nature 438(7064):94–98
Stipanovich A, Valjent E, Matamales M et al (2008) A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature 453(7197):879–884
Kramar EA, Chen LY, Brandon NJ et al (2009) Cytoskeletal changes underlie estrogen’s acute effects on synaptic transmission and plasticity. J Neurosci Off J Soc Neurosci 29(41):12982–12993
Chen LY, Rex CS, Casale MS, Gall CM, Lynch G (2007) Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci Off J Soc Neurosci 27(20):5363–5372
Handa RJ, Ogawa S, Wang JM, Herbison AE (2012) Roles for oestrogen receptor beta in adult brain function. J Neuroendocrinol 24(1):160–173
Dominguez R, Micevych P (2010) Estradiol rapidly regulates membrane estrogen receptor alpha levels in hypothalamic neurons. J Neurosci Off J Soc Neurosci 30(38):12589–12596
Ishunina TA, Swaab DF (2008) Estrogen receptor-alpha splice variants in the human brain. Gynecol Endocrinol 24(2):93–98
Wang ET, Sandberg R, Luo S et al (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456(7221):470–476
Ishunina TA, Swaab DF (2012) Decreased alternative splicing of estrogen receptor-alpha mRNA in the Alzheimer’s disease brain. Neurobiol Aging 33(2):286–296, e3
Wong J, Weickert CS (2009) Transcriptional interaction of an estrogen receptor splice variant and ErbB4 suggests convergence in gene susceptibility pathways in schizophrenia. J Biol Chem 284(28):18824–18832
Perlman WR, Matsumoto M, Beltaifa S et al (2005) Expression of estrogen receptor alpha exon-deleted mRNA variants in the human and non-human primate frontal cortex. Neuroscience 134(1):81–95
Ishunina TA, Fischer DF, Swaab DF (2007) Estrogen receptor alpha and its splice variants in the hippocampus in aging and Alzheimer’s disease. Neurobiol Aging 28(11):1670–1681
Mott NN, Pak TR (2012) Characterisation of human oestrogen receptor beta (ERbeta) splice variants in neuronal cells. J Neuroendocrinol 24(10):1311–1321
Chung WC, Pak TR, Suzuki S, Pouliot WA, Andersen ME, Handa RJ (2007) Detection and localization of an estrogen receptor beta splice variant protein (ERbeta2) in the adult female rat forebrain and midbrain regions. J Comp Neurol 505(3):249–267
Funding Information
This work was supported by NSFC grants (numbers 81171197 and 81220108010) and a Bureau of Health of Chongqing Medical Research Grant (number 2011-1-018) to G-J. C.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lai, YJ., Yu, D., Zhang, J.H. et al. Cooperation of Genomic and Rapid Nongenomic Actions of Estrogens in Synaptic Plasticity. Mol Neurobiol 54, 4113–4126 (2017). https://doi.org/10.1007/s12035-016-9979-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9979-y