
Abstract
Primary open angle glaucoma (POAG) is a major type of glaucoma characterized by progressive loss of retinal ganglion cells with associated visual field loss without an identifiable secondary cause. Genetic factors are considered to be major contributors to the pathogenesis of glaucoma. The aim of the study was to identify the causative gene in a large family with POAG by applying whole exome sequencing (WES). WES was performed on the DNA of four affected family members. Rare pathogenic variants shared among the affected individuals were filtered. Polymerase chain reaction and Sanger sequencing were used to analyze variants segregating with the disease in additional family members. WES analysis identified a variant in TP53BP2 (c.109G>A; p.Val37Met) that segregated heterozygously with the disease. In silico analysis of the substitution predicted it to be pathogenic. The variant was absent in public databases and in 180 population-matched controls. A novel genetic variant in the TP53BP2 gene was identified in a family with POAG. Interestingly, it has previously been demonstrated that the gene regulates apoptosis in retinal ganglion cells. This supports that the TP53BP2 variant may represent the cause of POAG in this family. Additional screening of the gene in patients with POAG from different populations is required to confirm its involvement in the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glaucoma is a leading cause of irreversible blindness worldwide, affecting more than 60 million people around the world [1]. Glaucoma comprises a group of heterogeneous optic neuropathies, characterized by progressive optic nerve degeneration. The diagnosis of glaucoma is usually late, since the loss of vision often starts in the periphery and progression to the loss of central vision is late. Due to this, glaucoma is also called a silent thief of sight, with devastating consequences to the patient’s quality of life. Glaucoma is classified into two main types: primary and secondary glaucoma. Among primary glaucoma subtypes, primary open angle glaucoma (POAG) represents the major type of glaucoma affecting about 35 million people worldwide and is characterized by a juvenile or adult onset. Patients with POAG have characteristic glaucomatous optic nerve damage with corresponding visual field defects and an open anterior chamber angle at gonioscopy, but no other (congenital) anomalies [2, 3]. One of the significant risk factors for POAG is elevation of intraocular pressure (IOP). However, POAG also occurs in patients without elevated IOP, and an elevated IOP does not necessarily lead to POAG [4]. The gradual loss of the retinal ganglion cells (RGCs) is a hallmark of the disease along with the increased IOP, but the exact pathophysiological mechanisms of the disease are not fully understood. Well-studied risk factors associated with POAG include age, family history, gender, ethnicity, central corneal thickness, and myopia. In addition, genetic factors play an important role in the disease etiology.
To date, more than 15 loci have been identified for glaucoma, and the causative gene has been identified for 5 of these loci: GLC1A (MYOC/TIGR) [5, 6], GLC1E (OPTN) [7, 8], GLC1F (ASB10) [9, 10], GLC1G (WDR36) [11], and GLC1H (EFEMP1) [12, 13]. In addition, mutations in the CYP1B1 [14] gene were identified in primary congenital, juvenile onset and adult onset POAG [15–17]. Finding the genes that cause glaucoma is the first step in improving early diagnosis and treatment of patients suffering from glaucoma. However, only less than 10% of POAG cases have pathogenic mutations in these disease-causing genes. It is therefore likely that the hereditary aspect of many of the remaining cases of POAG is either in the unidentified genes or due to the combined effects of several single nucleotide polymorphisms (SNPs). In recent years, several genome-wide association studies (GWAS) have identified several SNPs at different loci including CAV1/CAV2 [18], TMCO1 [19], CDKN2B-AS1 [20], CDC7-TGFBR3 [21], SIX1/SIX6 [22], GAS7, ATOH7, TXNRD2, ATXN2, and FOXC1 [23], to be associated with POAG, but they explain only a fraction of the disease heritability. In addition, the mechanisms how the associated loci influence the development of disease are often unclear. Therefore, additional genetic studies are required to explain the heritability, to gain a better understanding in the disease etiology, and to define new targets for treatment.
The goal of the current study was to identify the genetic cause of POAG in a large multiplex family using whole exome sequencing (WES).
Materials and Methods
Clinical Evaluation
A large family with eight individuals affected by POAG and one unaffected individual was ascertained. Affected and unaffected individuals were examined by an ophthalmologist at the Radboud University Medical Center in Nijmegen, the Netherlands. The study adhered to the principles of the Declaration of Helsinki and was approved by the Institutional Ethical Review Board of the Radboud University Medical Center in Nijmegen, the Netherlands. Blood samples were drawn from the family members, after obtaining written informed consent. DNA was extracted using standard methods.
Clinical characterization of the affected individuals included slit-lamp examination for iris diaphany, funduscopy, and IOP measurement with Goldmann applanation tonometry. Assessment of visual field defects was performed with a Humphrey Visual Field Analyzer (Carl Zeiss Humphrey Systems, Dublin, CA, USA). The decisions about glaucomatous damage on visual fields were based on the diagnostic criteria of the Hodapp et al. classification [24]. Evaluation of the anterior chamber angle was performed by gonioscopy, and corneal thickness was calculated by ultrasound pachymetry. In addition, a morphometric analysis of the optic disk was carried out by the Heidelberg Retina Tomograph II (HRT II; Heidelberg Engineering, Heidelberg, Germany), as described elsewhere [25]. An ophthalmic photographer masked to the results of the previous tests conducted the examination. The HRT Moorfields regression analysis (MRA) was used for classification of the optic disk [26]. The diagnosis of POAG was made when the following criteria were met: IOP higher than 22 mmHg (as measured by applanation tonometry in both eyes), glaucomatous optic neuropathy present in both eyes at funduscopy, visual field loss consistent with assessed optic neuropathy in at least one eye, and an open anterior chamber angle by gonioscopy.
Whole Exome Sequencing and Analysis
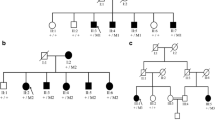
To identify the underlying genetic cause of the disease in this large family with POAG, WES was performed using genomic DNA of four affected individuals (III:1, III:5, III:6, and III:8) (Fig. 1). Enrichment of exonic sequences was achieved by using the SureSelectXT Human All Exon V.2 Kit (50 Mb) (Agilent Technologies, Inc., Santa Clara, CA, USA). Sequencing was performed on a SOLiD 4 sequencing platform (Life Technologies, Carlsbad, CA, USA). The hg19 reference genome was aligned with the reads obtained using SOLiD LifeScope software V.2.1 (Life Technologies).

Pedigree of a family with individuals affected by POAG. The (c.109G>A; p.Val37Met) variant in the TP53BP2 gene is indicated with M2, the variant (c.305G >A; p.Arg102His) in the MAPKAPK2 gene is indicated with M1, and the wild type allele is indicated with WT for both genes together with microsatellite markers haplotype. All affected individuals carry both variants heterozygously, while the unaffected individuals do not carry the variant
To identify the causative variant, only the variants shared by the four affected individuals were included for further analysis. All variants present within intergenic, intronic, and untranslated regions and synonymous substitutions were excluded. Variants present in the public genetic variant databases, including the Exome Variant Server (http://evs.gs.washington.edu/EVS/), dbSNP132 (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id=132), and 1000 Genomes (http://www.1000genomes.org/) with an allele frequency >0.5%, were excluded.
To evaluate the pathogenicity of the variants obtained from WES, bioinformatic analysis was performed using the PhyloP (nucleotide conservation in various species) and Grantham scores (difference in physicochemical nature of amino acid substitutions). Functional predictions were performed using publically available tools, i.e., SIFT (http://sift.bii.a-star.edu.sg/ Sorting Intolerant from Tolerant), MutationTaster (http://www.mutationtaster.org/), and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/ Polymorphism Phenotyping). Confirmation of variants and segregation analysis in all available family members was performed using PCR and Sanger sequencing. Sequencing was performed using the Big Dye Terminator Cycle Sequencing-Ready Reaction Kit (Applied Biosystems) on a 3730 DNA automated sequencer (Applied Biosystems, Foster City, CA, USA) using standard protocols. Segregating variants were analyzed in 180 population-matched controls by restriction fragment length analysis.
Linkage Analysis
Microsatellite markers with a genetic heterogeneity >60% were selected from the UCSC database. Microsatellite markers D1S2655, D1S2891, D1S2629, and D1S229 were amplified with M13 tailed primers, followed by a second PCR with fluorescently labeled M13 primers. Fluorescent amplification products were visualized on the ABI-310 genetic analyzer, and the size of the alleles was determined with 500LIZ (Applied Biosystems, Bleiswijk, the Netherlands) and analyzed with the GeneMapper software version 3.7 (Applied Biosystems, Bleiswijk, the Netherlands). Multipoint linkage analysis was performed for informative markers to determine the logarithm of the odds (LOD) score using the GeneHunter program (version 2.1).11 in the easyLINKAGE Software package (http://nephrologie.uniklinikum-leipzig.de/nephrologie.site,postext,easylinkage,a_id,797.html). For linkage analysis, autosomal dominant inheritance was assumed, with a disease-allele frequency of 0.0001 and 95% penetrance.
Results
Clinical Evaluation
Table 1 provides detailed clinical information of the eight affected family members diagnosed with POAG. All individuals had open drainage angles on gonioscopy (at least Shaffer grade III) and normal results of corneal thickness evaluation. The mean age at diagnosis was 54.8 years (range 49–60 years), with a mean IOP of 13.2 ± 2.2 mmHg (after use of IOP lowering medications). All individuals had bilateral glaucoma: they had glaucomatous optic neuropathy on funduscopy with reproducible compatible glaucomatous visual field loss, and all individuals showed abnormal results on Heidelberg Retina Tomograph II testing. A representative color photo of the optic disk of individual III-9 with POAG, with the corresponding superior arcuate scotoma on Humphrey visual field testing is shown in Fig. 2. From the medical chart, we distilled that the mean highest IOP recorded on diurnal testing was 23.6 ± 4.7 mmHg.

Photograph of the optic disk (a) and Humphrey visual field testing (b) of the left eye in a 76-years-old patient (III-9) with POAG and a corresponding visual acuity of 20/32. a Photograph shows a pallor, glaucomatous excavated optic disk. b Visual field testing shows a superior arcuate scotoma as well as inferior defects that are congruent with the excavation of the optic disk
Mutation Detection
In Table 2, the number of variants that passed the various filtering steps per individual for the four affected individuals, as well as the variants shared by all four affected individuals is shown. The mean coverage of the WES data was 100X. Filtering for variants shared between all four affected individuals (III:1, III:5, III:6, and III:8) resulted in nine variants for further analysis (Table 3). Segregation analysis was performed for nine variants with a phyloP >2.7 or a Grantham score >80 (Table 3). Two novel heterozygous variants in the TP53BP2 and MAPKAPK2 genes were found to be segregating with the disease in the family (Fig. 1). The variant in the TP53BP2 gene (c.109G>A; p.Val37Met) was predicted to be deleterious by SIFT, probably damaging by PolyPhen-2 and disease-causing by Mutation Taster (Table 3). The wild type nucleotide was highly conserved (phyloP score 4.08), and the amino acid residue p.Val37 was completely conserved among vertebrates (Fig. 3a). The second variant that segregated with the disease in the family was identified in the MAPKAPK2 gene (c.305G>A; p.Arg102His) (Fig. 1). This variant was also predicted to be deleterious by SIFT, probably damaging by PolyPhen and disease-causing by Mutation Taster (Table 3). The wild type nucleotide was highly conserved (phyloP score 5.69), and the amino acid residue p.Arg102 is completely conserved among vertebrates (Fig. 3b). Both variants were not identified in 180 population-matched controls and were not present in the Exome Variant Server, dbSNP132 and 1000 genomes.

a Evolutionary conservation of valine at position 37 is represented by alignment of the human TP53BP2 (ASPP2) protein sequence to orthologous protein sequences of various vertebrate species. b Evolutionary conservation of arginine at position 102 is represented by alignment of the human MAPKAPK2 protein sequence to orthologous protein sequences of various vertebrate species
Linkage Analysis
Twelve microsatellite markers were used for linkage analysis of the genomic region encompassing the MAPKAPK2 and TP53BP2 genes, but only four markers were informative. A multipoint LOD score of 2.48 was obtained for markers D1S2655 and D1S2891, which is suggestive of linkage and is in accordance with the maximum LOD score that can be achieved considering the structure of the pedigree. The disease-associated haplotype encompasses markers D1S2655, the MAPKAPK2 variant, D1S2891, D1S2629, D1S229, and the TP53BP2 variant. All affected individuals carry the disease haplotype that includes both genetic variants, which indicates that both variants are in the same linkage interval and are in cis configuration.
Discussion
In the current study, we used WES to identify the genetic defect in a large family with POAG. We identified potentially pathogenic variants in the TP53BP2 (c.109G>A; p.Val37Met) and MAPKAPK2 (c.305G>A; p.Arg102His) genes. A disease haplotype carrying both variants segregates with the disease, with a maximum LOD score of 2.4. Variants in both TP53BP2 and MAPKAPK2 segregate with the disease, since both of them are on the same haplotype. Therefore, segregation of the MAPKAPK2 variant with the disease is a coincidental finding. Since the inheritance pattern was not obvious from the pedigree, we considered both recessive and dominant inheritance patterns during the variant prioritization process. However, no homozygous or compound heterozygous variants were identified among the putative pathogenic variants that were shared between affected individuals, and this thus does not support recessive inheritance. Since phenotype data and DNA were not available from the deceased parents, we can only speculate that the inheritance pattern may be autosomal dominant.
TP53BP2 encodes a member of the ASPP (apoptosis-stimulating protein of p53) family of p53-interacting proteins comprised of three members: ASPP1, ASPP2, and iASPP. Both ASPP1 and ASPP2 are proapoptotic proteins involved in the regulation of the apoptosis and are encoded by the TP53BP2, and iASPP is encoded by PPP1R13B genes, respectively [27]. ASPP2 is well known for its binding and activation of the apoptotic function of p53, p63, and p73 by selectively enhancing their DNA-binding and transactivation activities on proapoptotic genes such as BAX and PIG3 [27, 28]. Apoptosis is tightly regulated during normal development. In the case of abnormal regulation, it mediates cell death of neuronal cells in neurodegenerative diseases such as Alzheimer’s disease or Parkinson’s disease or death of RGCs in glaucoma due to overexpression of p53 [29–31]. Recently, the role of ASPP1 and ASPP2 proteins in neuronal apoptosis and their involvement in the regulation of adult RGCs after injury have been investigated. The results indicated that both ASPP1 and ASPP2 are highly expressed in RGCs and contribute to p53-dependent death of RGCs.
In glaucoma cell death of the post-mitotic neurons, i.e., RGCs, occurs due to an increased rate of apoptosis. The ASPP proteins are involved in the regulation of apoptosis by activating p53. The expression of ASPP2 affects the DNA binding activity of p53 on the Bax promoter or downstream targets involved in apoptosis [32]. In the current study, we speculate that binding between ASPP2 and p53 may be affected by an amino acid variant (c.109G>A; p.Val37Met) in the ASPP2 protein, which leads to the increased accumulation of p53, followed by an increase in cell death of the RGCs, subsequently leading to glaucoma. Previously, it has been reported that normal ASSP2 protein is required for the activation of apoptosis in a controlled manner. It was observed that the blockade of the ASPP-p53 pathway is important for the survival of neurons after axonal injury [33]. The results of Wilson et al. are further supported by a recent study in an in vivo model of acute optic nerve damage, in which it was shown that iASPP is expressed by injured RGCs and short interference RNA (siRNA)-induced iASPP knockdown exacerbates RGC death, while RGC survival was enhanced by adeno-associated virus (AAV)-mediated iASPP expression. Increased expression of iASPP in RGCs downregulates p53 activity and blocks the expression of proapoptotic targets PUMA and Fas/CD95 [34]. Since iASPP is an inhibitor of p53-mediated apoptosis, it is possible that the mutation in the ASPP2 protein influences the expression of iASPP. Subsequently, it would not be able to perform actively in the survival of retinal ganglion cells due to apoptosis.
In a recent study, it has been observed that siRNA interfering the expression of ASPP2 is involved in the development of the proliferative vitreoretinopathy (PVR). Using epiretinal membranes of PVR patients, they examined the expression of ASPP2 using immunohistochemistry and observed reduced expression of ASPP2 in PVR membranes. In addition, knockdown of ASPP2 is involved in increased expression of cytokines such as TGF-β, CTGF, VEGF, TNF-α, and interleukins [35]. In glaucoma, the role of inflammatory cytokines is well known, and it is possible that the amino acid variant identified in the ASPP2 protein affects the expression of inflammatory cytokines and interleukins, which mediate apoptosis of retinal ganglion cells in glaucoma. In another recent study, the neuroprotective effect of minocycline in rats with glaucoma was evaluated, and downregulation of TP53BP2 was observed upon treatment [36]. Minocycline is a tetracycline with anti-inflammatory and anti-apoptotic properties. In previous studies, it has been shown that minocycline significantly delays RGC death in models of experimental glaucoma and optic nerve transaction [37].
Taken together, these studies support the involvement of the TP53BP2 gene in glaucoma and suggest that the genetic variant identified by WES in the large POAG family may be relevant to the disease.
The second variant that segregates with the disease in the family was identified in MAPK-activated protein kinase 2 (MAPKAPK2, also known as MK2), which is one of the downstream targets of p38 MAPK. The Ocular Tissue Database (OTDB, http://genome.uiowa.edu/otdb/) demonstrates a minimal expression in the eye for MAPKAPK2 in contrast to TP53BP2. Therefore, TP53BP2 gene seems to be the strongest candidate to be associated with the disease in this particular family.
In conclusion, through WES in a large POAG family, we identified a novel genetic variant in the TP53BP2 gene, which is predicted to be pathogenic and affects a highly conserved amino acid residue. Since it has been demonstrated that the gene regulates apoptosis in RGCs and is downregulated upon minocycline treatment in a glaucoma rat model, TP53BP2 may represent a novel gene associated with POAG. Additional screening of the TP53BP2 gene in other familial and sporadic patients with POAG from different populations is required to confirm its involvement in the disease.
References
Quigley HA, Broman AT (2006) The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90(3):262–267. doi:10.1136/bjo.2005.081224
Hitchings RA (1992) Low tension glaucoma—its place in modern glaucoma practice. Br J Ophthalmol 76(8):494–496
Grosskreutz C, Netland PA (1994) Low-tension glaucoma. Int Ophthalmol Clin 34(3):173–185
Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK 2nd, Wilson MR et al (2002) The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol 120(6):701–713 discussion 829-730
Sheffield VC, Stone EM, Alward WL, Drack AV, Johnson AT, Streb LM, Nichols BE (1993) Genetic linkage of familial open angle glaucoma to chromosome 1q21-q31. Nat Genet 4(1):47–50. doi:10.1038/ng0593-47
Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden SL, Nishimura D, Clark AF et al (1997) Identification of a gene that causes primary open angle glaucoma. Science 275(5300):668–670
Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, Heon E, Krupin T et al (2002) Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 295(5557):1077–1079. doi:10.1126/science.1066901
Sarfarazi M, Child A, Stoilova D, Brice G, Desai T, Trifan OC, Poinoosawmy D, Crick RP (1998) Localization of the fourth locus (GLC1E) for adult-onset primary open-angle glaucoma to the 10p15-p14 region. Am J Hum Genet 62(3):641–652. doi:10.1086/301767
Wirtz MK, Samples JR, Rust K, Lie J, Nordling L, Schilling K, Acott TS, Kramer PL (1999) GLC1F, a new primary open-angle glaucoma locus, maps to 7q35-q36. Arch Ophthalmol 117(2):237–241
Pasutto F, Keller KE, Weisschuh N, Sticht H, Samples JR, Yang YF, Zenkel M, Schlotzer-Schrehardt U et al (2012) Variants in ASB10 are associated with open-angle glaucoma. Hum Mol Genet 21(6):1336–1349. doi:10.1093/hmg/ddr572
Monemi S, Spaeth G, DaSilva A, Popinchalk S, Ilitchev E, Liebmann J, Ritch R, Heon E et al (2005) Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum Mol Genet 14(6):725–733. doi:10.1093/hmg/ddi068
Suriyapperuma SP, Child A, Desai T, Brice G, Kerr A, Crick RP, Sarfarazi M (2007) A new locus (GLC1H) for adult-onset primary open-angle glaucoma maps to the 2p15-p16 region. Arch Ophthalmol 125(1):86–92. doi:10.1001/archopht.125.1.86
Mackay DS, Bennett TM, Shiels A (2015) Exome sequencing identifies a missense variant in EFEMP1 co-segregating in a family with autosomal dominant primary open-angle glaucoma. PLoS One 10(7):e0132529. doi:10.1371/journal.pone.0132529
Sarfarazi M, Akarsu AN, Hossain A, Turacli ME, Aktan SG, Barsoum-Homsy M, Chevrette L, Sayli BS (1995) Assignment of a locus (GLC3A) for primary congenital glaucoma (buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics 30(2):171–177
Stoilov I, Akarsu AN, Sarfarazi M (1997) Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet 6(4):641–647
Vincent AL, Billingsley G, Buys Y, Levin AV, Priston M, Trope G, Williams-Lyn D, Heon E (2002) Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet 70(2):448–460. doi:10.1086/338709
Melki R, Colomb E, Lefort N, Brezin AP, Garchon HJ (2004) CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J Med Genet 41(9):647–651. doi:10.1136/jmg.2004.020024
Wiggs JL, Kang JH, Yaspan BL, Mirel DB, Laurie C, Crenshaw A, Brodeur W, Gogarten S et al (2011) Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma in Caucasians from the USA. Hum Mol Genet 20(23):4707–4713. doi:10.1093/hmg/ddr382
Burdon KP, Macgregor S, Hewitt AW, Sharma S, Chidlow G, Mills RA, Danoy P, Casson R et al (2011) Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat Genet 43(6):574–578. doi:10.1038/ng.824
Pasquale LR, Loomis SJ, Kang JH, Yaspan BL, Abdrabou W, Budenz DL, Chen TC, Delbono E et al (2013) CDKN2B-AS1 genotype-glaucoma feature correlations in primary open-angle glaucoma patients from the United States. Am J Ophthalmol 155(2):342–353. doi:10.1016/j.ajo.2012.07.023
Li Z, Allingham RR, Nakano M, Jia L, Chen Y, Ikeda Y, Mani B, Chen LJ et al (2015) A common variant near TGFBR3 is associated with primary open angle glaucoma. Hum Mol Genet 24(13):3880–3892. doi:10.1093/hmg/ddv128
Wiggs JL, Yaspan BL, Hauser MA, Kang JH, Allingham RR, Olson LM, Abdrabou W, Fan BJ et al (2012) Common variants at 9p21 and 8q22 are associated with increased susceptibility to optic nerve degeneration in glaucoma. PLoS Genet 8(4):e1002654. doi:10.1371/journal.pgen.1002654
Bailey JN, Loomis SJ, Kang JH, Allingham RR, Gharahkhani P, Khor CC, Burdon KP, Aschard H et al (2016) Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat Genet 48(2):189–194. doi:10.1038/ng.3482
Hodapp E PRI, Anderson DR. (1993) Clinical decisions in glaucoma. The CV Mosby Co;, St Louis
Mikelberg FS, Parfitt CM, Swindale NV, Graham SL, Drance SM, Gosine R (1995) Ability of the Heidelberg Retina Tomograph to detect early glaucomatous visual field loss. J Glaucoma 4(4):242–247
Wollstein G, Garway-Heath DF, Hitchings RA (1998) Identification of early glaucoma cases with the scanning laser ophthalmoscope. Ophthalmology 105(8):1557–1563. doi:10.1016/S0161-6420(98)98047-2
Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L et al (2001) ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 8(4):781–794
Bergamaschi D, Samuels Y, Jin B, Duraisingham S, Crook T, Lu X (2004) ASPP1 and ASPP2: common activators of p53 family members. Mol Cell Biol 24(3):1341–1350
Chatoo W, Abdouh M, Bernier G (2011) p53 pro-oxidant activity in the central nervous system: implication in aging and neurodegenerative diseases. Antioxid Redox Signal 15(6):1729–1737. doi:10.1089/ars.2010.3610
Culmsee C, Mattson MP (2005) p53 in neuronal apoptosis. Biochem Biophys Res Commun 331(3):761–777. doi:10.1016/j.bbrc.2005.03.149
Nickells RW (1999) Apoptosis of retinal ganglion cells in glaucoma: an update of the molecular pathways involved in cell death. Surv Ophthalmol 43:151–161
Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L et al (2001) ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 8(4):781–794. doi:10.1016/S1097-2765(01)00367-7
Wilson AM, Morquette B, Abdouh M, Unsain N, Barker PA, Feinstein E, Bernier G, Di Polo A (2013) ASPP1/2 regulate p53-dependent death of retinal ganglion cells through PUMA and Fas/CD95 activation in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience 33(5):2205–2216. doi:10.1523/JNEUROSCI.2635-12.2013
Wilson AM, Chiodo VA, Boye SL, Brecha NC, Hauswirth WW, Di Polo A (2014) Inhibitor of apoptosis-stimulating protein of p53 (iASPP) is required for neuronal survival after axonal injury. PLoS One 9(4):e94175. doi:10.1371/journal.pone.0094175
Chen XL, Bai YJ, Hu QR, Li SS, Huang LZ, Li XX (2016) Small interfering RNA targeted to ASPP2 promotes progression of experimental proliferative vitreoretinopathy. Mediat Inflamm. doi:10.1155/2016/7920631
Levkovitch-Verbin H, Waserzoog Y, Vander S, Makarovsky D, Piven I (2014) Minocycline upregulates pro-survival genes and downregulates pro-apoptotic genes in experimental glaucoma. Graefe’s archive for clinical and experimental ophthalmology Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 252(5):761–772. doi:10.1007/s00417-014-2588-4
Levkovitch-Verbin H, Kalev-Landoy M, Habot-Wilner Z, Melamed S (2006) Minocycline delays death of retinal ganglion cells in experimental glaucoma and after optic nerve transection. Arch Ophthalmol 124(4):520–526. doi:10.1001/archopht.124.4.520
Acknowledgements
This study was supported by the Stichting Blindenhulp, a Shaffer grant from the Glaucoma Research Foundation and the following foundations: Glaucoomfonds, Oogfonds, and Algemene Nederlandse Vereniging ter Voorkoming van Blindheid, which contributed through UitZicht.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study adhered to the principles of the Declaration of Helsinki and was approved by the Institutional Ethical Review Board of the Radboud University Medical Center in Nijmegen, the Netherlands.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Micheal, S., Saksens, N.T., Hogewind, B.F. et al. Identification of TP53BP2 as a Novel Candidate Gene for Primary Open Angle Glaucoma by Whole Exome Sequencing in a Large Multiplex Family. Mol Neurobiol 55, 1387–1395 (2018). https://doi.org/10.1007/s12035-017-0403-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0403-z