
Abstract
Introduction
The history of levothyroxine has been linked to advances in the treatment of thyroid disease and to date it is the standard therapy for the treatment of hypothyroidism. Bioequivalence studies are the most widely used method to demonstrate interchangeability, although controversy persists regarding the best design for this molecule declared as a narrow therapeutic index product in many countries. This study aimed to evaluate the pharmacokinetic profile of two formulations of levothyroxine to determine bioequivalence between them.
Methods
This two-period, randomized, crossover, blind study was conducted in 80 healthy volunteers, of both sexes, using a single levothyroxine dose of 600 μg with a washout period of 42 days. Blood sampling was performed at − 30 min, − 15 min, and 0 h pre-dose and 30 min, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, 24, and 48 h post-dose.
Results
A total of 78 subjects successfully completed both periods. There were no serious adverse events during the study and both formulations were well tolerated. Baseline correction of serum levothyroxine concentrations was performed before statistical analysis. The mean maximum plasma concentration of the test product (Levotiroxina MK®) was 57.49 ng/mL while for the reference product it reached 59.32 ng/mL. Importantly, both test and reference formulations reached maximum concentrations in plasma at about the same time. The areas under the pharmacokinetic curves with the test product showed AUC0−t of 1407.1 ng h/mL and the reference product 1394.3 ng h/mL. The bioequivalence statistical analysis showed that the 90% confidence interval (CI90%) of the ratio of test over reference formulation was within the bioequivalence margins of 90–111%. For Cmax, the test/reference ratio was 96.2% with CI90% of 91.6–100.9%, and for AUC0−t the test/reference ratio was 99.9 with CI90% of 93.3–107.0%.
Conclusions
Both formulations have the same pharmacokinetic profile and are bioequivalent in the narrow therapeutic index required by some health authorities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Hypothyroidism is a prevalent and chronic disease that affects a significant proportion of the population and requires lifelong treatment with levothyroxine. In many Latin American countries, the population has limited access to quality medications for treatment of chronic diseases |
Several formulations of levothyroxine from different sources are currently available in Latin American countries, although their efficacy is uncertain given its narrow therapeutic range. By carrying out a comparative bioavailability study we could show that a generic formulation of levothyroxine manufactured in Colombia is bioequivalent to the reference product, providing solid scientific evidence for its use in patients |
This study aimed to establish the bioequivalence in the narrow therapeutic range for a generic Levothyroxine formulation and the reference product |
What was learned from the study? |
This study evaluated and compared the bioavailability between two formulations of levothyroxine following international guidelines for bioequivalence demonstration |
Our results show that intersubject variability of levothyroxine increased when correcting pharmacokinetic curves for the endogenous component baseline |
In countries where levothyroxine is classified as a narrow therapeutic drug, the CI90% for bioequivalence demonstration is reduced to 0.9–1.11, instead of the conventional 0.8–1.25. Hence, a larger sample size is required to demonstrate bioequivalence between the test and reference formulations |
Introduction
The first therapies offered to patients with hypothyroidism were based on the provision of extracts derived from animal thyroid; indeed, therapies of porcine origin are still available in some countries [1, 2]. In 1955, a more soluble form of levothyroxine became available, levothyroxine sodium, allowing its use in clinical practice. It should be noted that the use of levothyroxine sodium was only popularized by doctors in the mid-1980s, once it was shown that monotherapy with it was superior in safety and efficacy to the combination of porcine origin of T4/T3 for return to the euthyroid state [2]. Since then, its use has been recommended as the first line in the management of hypothyroidism and it is now considered one of the most prescribed medications in the world [2, 3].
Levothyroxine sodium was first marketed in the USA under the name Synthroid®, manufactured at that time by Flint Pharmaceuticals. In 1995 Flint was acquired by Knoll Laboratories which in turn was acquired by Abbott Laboratories in 2001 [4]. Other levothyroxine sodium-containing products were developed by other pharmaceutical companies such as Merck in 1972, and registered around the world, in a time when regulations to produce levothyroxine were not particularly demanding. This was the situation until the end of the twentieth century, when several reports emerged on the lack of efficacy of some levothyroxine products at the end of their shelf life. As a result of investigations, the US Food and Drug Administration (FDA) increased the quality requirements for levothyroxine manufacturers of the marketed brands at that time [2]. Later, the FDA declared levothyroxine as a molecule with a narrow therapeutic index [5,6,7], requiring individualization of the dose because it presents risks for the clinical prognosis and control of patients with supra- and subtherapeutic concentrations [8]. Since then, a more restrictive control of levothyroxine-containing products was imposed (Fig. 1).

Timeline of events related to the discovery of thyroid hormones in the twentieth century and recently the bioequivalence guidelines of thyroxine-containing products. Dr. Edward Calvin Kendwall, Nobel Prize in Chemistry in 1950 for his discovery of cortisol, did not foresee the importance of his first discovery during his doctoral thesis in 1914 when he found a compound (later called thyroxine) with four iodine atoms in the thyroid gland [1]. In 1927 Harington and Barger synthesized the thyroid hormone T4, known as levothyroxine; however, it was very insoluble so it could not be used in clinical practice [2, 7]. Dr. Debora Doniach discovered thyroid autoimmunity. The USP pharmacopeia and the FDA have updated some technical specifications, such as the requirement of a new potency for the tablet throughout its shelf life. The FDA and USP established since 2009 the assay limits between 95% and 105% of the declared active principle, although other pharmacopeias such as the British (BP) maintain their limits between 90% and 110% [4, 9]. This new potency range forced manufacturers to reformulate composition and performance of the formulations developed before 2009 [2, 7, 9]
Levothyroxine formulations have been substantially modified during the last couple of decades. To ensure safety and efficacy of new formulations, changes in product composition and manufacturing conditions required evaluation of the bioequivalence of formulations before and after changes were implemented (SUPAC guidelines, FDA). Indeed, to comply with this new regulatory requirement, initial changes in formulation for Eutirox®, the brand name levothyroxine manufactured by Merck Germany, were evaluated in 216 healthy subjects that participated in a bioequivalence trial comparing formulations before and after modifications [9]. The most recent change in manufacturing of Eutirox® was also evaluated in a bioequivalence trial conducted in 44 healthy subjects (NCT03979274). Similarly, GSK conducted a bioequivalence trial on its brand name levothyroxine manufactured in India after a change in the source of the active pharmaceutical ingredient (API) (NCT01536678).
However, the prescription of generic levothyroxine faced opposition from medical associations such as the Association of Clinical Endocrinologists (AACE), the Endocrine Society, and the American Thyroid Association. These associations raised concerns on the validity of bioequivalence studies requested by the FDA to register generic versions of levothyroxine by questioning the interchangeability of branded with generic versions in the treatment of patients [10]. Thus, medical associations did not recommend switching among products containing levothyroxine, and, if needed, patients required monitoring of T3 and thyroid-stimulating hormone (TSH) levels. Similar concerns about interchangeability were raised in Europe by the European Thyroid Association (ETA) and Thyroid Federation International (TFI), complaining about the lack of reporting changes in formulations by manufacturers [11].
Such initial lack of endorsement by medical associations on the use of generic levothyroxine has been jointly battled by health authorities, health insurers, and institutions. The requirements for the execution of bioequivalence studies of levothyroxine-containing products have been updated by the FDA (2014) and European Medicines Agency (EMA) (2020). For instance, the EMA revised bioequivalence requirements for levothyroxine by narrowing the acceptance criteria to 90–111% for the area under the curve (AUC) pharmacokinetic parameter. These new guidelines call for larger subject samples to demonstrate bioequivalence of levothyroxine formulations. Two long-term studies comparing patients prescribed levothyroxine of different sources found no difference in treatment between Synthroid and generic versions that comply with bioequivalence requirements. Indeed, the FDA recently published a statement based on these data, reinforcing that approval of generic levothyroxine products should be based on a detailed, complex bioequivalence study design with narrower margin for allowable pharmacokinetic differences between the generic and brand-name products [12].
To date, levothyroxine is the most prescribed generic drug globally, and several generic versions have been developed and are marketed around the world. For a newly developed levothyroxine formulation, an accurate evaluation of its bioequivalence is of paramount importance, providing clinical evidence to sustain its safety, efficacy, and interchangeability with the reference, branded, product. In the present study we report the bioequivalence study of Levotiroxina MK® manufactured by Tecnoquímicas, Colombia. This product was reformulated to comply with new specifications as requested by the FDA and the local health authority in Colombia (INVIMA). For this reason, a bioequivalence study was carried out comparing the test product with the reference product, Eutirox® manufactured by Merck S.A.
Methods
Study Design
An open-label, randomized, two-treatment, two-period and two-sequence, crossover, bioequivalence study was conducted in healthy volunteers of both sexes under fasting conditions. Volunteers were given a single 600-μg dose of levothyroxine sodium (six tablets of 100 μg) with a 42-day washout period between each treatment period. Subjects who met the following criteria were eligible: age 18–55 years, both sexes, a body mass index (BMI) between 18 and 30 kg/m2, and clinical and laboratory examinations in the range of normality. The number of volunteers was estimated according to the variability data reported for the innovative product [9]. Each volunteer received six 100-μg tablets of the test product (Levotiroxina MK® tablets 100 μg manufactured by Tecnoquímicas) and six tablets of the reference product (Eutirox® tablets 100 μg manufactured by Merck) in two periods. The clinical phase was carried out at the facilities of the DominguezLab Biopharmaceutical Research Center (Paraná, Argentina), where all the volunteers were admitted and overnight fasting for at least 10 h before the dosing was guaranteed. All participating volunteers were Argentine natives of Hispanic-Latino descent. Blood samples were collected at − 30 min, − 15 min, and 0 h prior to dosing and 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, 24, and 48 h post-dose. The clinical trial was monitored by the study sponsor (Tecnoquímicas) (Table 1).
Analytical Methods
A sensitive and specific validated high-performance liquid chromatographic with tandem mass spectrometry (HPLC–MS/MS) method for quantitation of levothyroxine in human serum was developed using an internal isotopically labeled standard. A UFLC chromatograph (Shimadzu) coupled to a Qtrap 5500 mass spectrometer (ABSciex) was used. Plasma proteins were precipitated with acetonitrile, and the chromatographic separation was carried out using a Hypersil gold 50 × 2.1 mm column (Thermo Fisher), with a particle size of 1.9 mm, using a mixture as mobile phase 80% methanol and 20% 10 mM ammonium acetate, with the addition of 0.1% formic acid. Levothyroxine-13C6 (Toronto Research Chemicals) was used as an internal standard. The levothyroxine quantification technique was validated for the parameters of sensitivity, selectivity, linearity, accuracy, and precision (within-run and between-run), matrix effect, and stability considering acceptance criteria recommended in national and international regulations.
Statistical Analysis
Bioequivalence was determined on the basis of the log-transformed pharmacokinetic parameters AUC0−t and Cmax with baseline correction for both formulations, i.e., subtracting the average T4 in blood obtained in three pre-dose blood samples (before ingesting the drugs) from each serum measurement. The ratio of the geometrical least squares means for the log-transformed pharmacokinetics parameters was analyzed and the 90% confidence interval was determined. The bioequivalence limits (0.90–1.11) for AUC0−t and Cmax were established by applying the Schuirmann test. Those statistical procedures that did not exceed the 5% level of significance and among those with the lowest risk of erroneously rejecting the equivalence between formulations were accepted. The analysis of variance (ANOVA) evaluated the effect of the sequence and the individuals on AUC0−t and Cmax for each formulation. Phoenix® WinNonlin® version 8.2 software (Certara USA, Inc., Princeton, NJ) was used.
The study and its execution were carried out following the ethical guidelines and principles for research in human beings and were approved by the institutional ethics committee (CEIID). All the volunteers received complete information of the methodology and objectives of the study and gave their informed consent. This trial was registered at ClinicalTrials.gov (NCT04573907). The study was approved by the following CEIID members Gonzalo Battauz, Lucia Grippo, Paula Denise Ludi, Alejandro Robles, and Dr. Cornalo Romanela Tamara. This study was in compliance with the Declaration of Helsinki 1964 and its later amendments.
Results
Clinical Phase
Of the participating volunteers who started the study, 78 completed the two treatment periods, registering one withdrawal for personal reasons and an exclusion due to a positive dose of drugs of abuse for a subject before admission to the second period of hospitalization. The median age and BMI of the volunteers participating in the bioequivalence study were 25.7 years and 24.7, respectively. There were no serious adverse events or voluntary withdrawals due to side effects during the study, and both formulations were well tolerated (Fig. 2).

Pharmacokinetic curve with the average at each sampling point of all volunteers with the test and reference formulation A without the baseline subtraction and B with the baseline subtraction (subtracted from the average of T4 at zero point)
Bioanalytical Phase
The bioanalytical phase was carried out at the DominguezLab Biopharmaceutical Research Center (Paraná, Argentina) between December 2019 and February 2020. Twenty-seven analytical runs were conducted to quantify the 2720 study samples and their corresponding incurred samples. A 1-day analytical run consisted of:
-
(a)
A calibration curve of concentrations from 30 to 360 ng/mL
-
(b)
Three initial quality controls (low, medium, and high)
-
(c)
Bioequivalence study samples from a volunteer, period 1 and period 2
-
(d)
Three final quality controls (low, medium, and high) to accept the samples from the volunteer
Steps b, c, and d were repeated depending on the number of volunteers tested on the same day, usually 3–5 volunteers.
The mean baseline-corrected serum concentration allowed one to estimate the changes in blood concentrations of this thyroid hormone for each formulation administered. Figure 2 shows the mean pharmacokinetics curves obtained for both test and reference products in all 78 treated subjects and pre-dose baseline-subtracted values. These curves showed a similar behavior both for test and reference products, reaching comparable Tmax and Cmax values. Analysis of the individual Cmax and AUC0−t reached in plasma indicated that subject response to both products was equally variable, and no consistent differences were observed.
Statistical Phase
The average pharmacokinetic parameters for each formulation are presented in Table 2. Both formulations displayed a similar concentration–time profile with no differences observed for any of the parameters evaluated. The mean maximum plasma concentration of the test product was 57.49 ng/mL while for the reference product it reached 59.32 ng/mL. Importantly, both test and reference formulations reached maximum concentrations in plasma at about the same time, with Tmax of 4.19 h and 3.73 h, respectively. Areas under the pharmacokinetic curves also reached similar values for both formulations, with the test product showing AUC0−t of 1407.1 ng h/mL and the reference product 1394.3 ng h/mL.
The result of the ANOVA showed no significant differences for the sequence, treatment period, or formulation effects as causes of variability. An intrasubject variability of 18.27% was observed for Cmax and an intrasubject variability of 26.19% for AUC.
The bioequivalence statistical analysis showed that the 90% confidence interval (CI90%) of the ratios of test over reference formulation for the two pharmacokinetic parameters evaluated in this study were within the bioequivalence margins of 90–111% (Table 3). For Cmax, the test/reference ratio was 96.2% with CI90% of 91.6–100.9%, and for AUC0−t the test/reference ratio was 99.9 with CI90% of 93.3–107.0%. Post hoc statistical power for all three parameters was higher than 80%.
Discussion
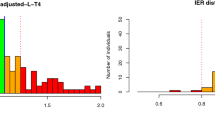
Levothyroxine sodium is a hormone replacement medicine indicated in patients with deficient thyroid hormone production or for TSH suppression due to a history of thyroid cancer [2]. In the body, thyroid hormone concentration is tightly controlled; intake of small doses of exogenous levothyroxine may cause strong changes in T4 and T3 levels, which in turn could cause profound physiological consequences. To optimize therapy, treatment with levothyroxine requires dose escalation and periodic adjustments [10]. It is then not surprising that certain health authorities like the Food and Drug Administration (USA), Health Canada, Paul Erlich Institute (Germany), and Agence nationale de sécurité du médicament et des produits de santé (ANSM, France) consider levothyroxine as a molecule with a narrow therapeutic index (NTI) (Fig. 3). As such, the requirements regarding the maximum differences in pharmacokinetic parameters (Cmax and AUC) that can be considered safely have been increased [13]. Instead of the commonly accepted bioequivalence margins of 80–125%, for NTI drugs the bioequivalence margin is restrained to 90–111%. The reason for such a restriction arises, in part, from the observation that these NTI drugs have low within-subject variability coefficients (usually below 10%). Accordingly, the estimated sample size for demonstrating bioequivalence in a two-sequence, two-period crossover study requires testing in a reasonable number of volunteers, usually 24 up to 36 volunteers.

Confidence interval of 90% (CI90%) to declare bioequivalence of levothyroxine for different health authorities in the world. In Canada CI90% for AUC is extended to 0.9–1.12. France and Colombia have an CI90% of 0.9–1.11 for both pharmacokinetic parameters. In Germany and Canada this CI90% is demanded only for AUC. *Countries where it has been declared as having a narrow therapeutic index (NTI)
For levothyroxine, however, since bioequivalence requires comparability of baseline-subtracted pharmacokinetic parameters, variability has been reported in some cases to exceed 20% for intrasubject coefficient of variation [2]. Hence, imposing a 90–111% bioequivalence margin for levothyroxine is such a tight constraint that it requires sampling of at least 70 volunteers to evaluate bioequivalence in a two-sequence, two-period crossover study (Fig. 3) [9]. To mitigate these findings, the FDA has allowed generic drug developers to expand bioequivalence margins for NTI drugs beyond the 90–111% when variability exceeds a certain threshold (intrasubject CV higher than 10%), thus proposing a four-period, two-sequence replicate design to demonstrate bioequivalence. However, expansion of bioequivalence margins in NTIs is not yet widely accepted, and certain regulatory agencies only accept scaling bioequivalence limits for highly variable drugs (intrasubject CV higher than 30%). Consequently, to date there is no global consensus on the ideal design for a levothyroxine bioequivalence study. In this work, we conducted a randomized, two-period, two-sequence, crossover study in 78 healthy volunteers, with a washout period of at least five half-lives. The methodological design followed previous designs employed by Merck to determine the bioequivalence of the new Eutirox® formulation in 2017 and 2019, adjusted to the new FDA potency specification [13].
The dose used in the study of 600 μg of levothyroxine allowed one to identify the exogenous supply of this synthetic hormone and to differentiate it from the endogenous production. This high dose of levothyroxine can generate additional intrasubject variability especially in blood samples taken after 12 h by the self-regulatory system of the hypothalamic-pituitary-thyroid axis. For this reason, it was considered that the sampling up to 48 h corresponds better to the changes inherent to each formulation of levothyroxine supplied, since it is the calculated time that its complete passage through the digestive tract lasts, as suggested by the FDA in its guidance [6].
The baseline averaged for T4 allowed one to estimate the changes in blood concentrations of this thyroid hormone for each formulation. The measurement of free T3 and TSH in parallel was not performed in the execution of this bioequivalence study [13] because of the high variability and late changes in the reported measurements of TSH levels [14] and because the measurement of T3 depends on its peripheral conversion from T4 by intracellular deiodases and is associated with biological differences inherent to each individual and not to the formulations under study [15]. Unlike the free T4 quantification methods used in clinical practice for the follow-up and diagnosis of hypothyroid patients with immunoassay techniques, the use of a mass spectrometer coupled to a chromatograph (LC/MS–MS) guaranteed greater sensitivity for the quantification of low concentrations of free thyroid hormone T4 in serum [16].
The results obtained in the present study showed that the formulation of Tecnoquímicas is bioequivalent to Eutirox® (manufactured by Merck) and falls in the range required for molecules with a narrow therapeutic index. Both formulations presented an adequate tolerability profile without any adverse events.
Conclusions
As a narrow therapeutic index molecule in many countries, comparative in vivo pharmacokinetic studies for levothyroxine require larger sample sizes than those usually used in bioequivalence studies. Bioequivalence and interchangeability were demonstrated for the two studied formulations of levothyroxine that meet the requirements for a 90% confidence interval between 0.9 and 1.11 for the pharmacokinetic parameters Cmax and AUC.
References
Slater S. The discovery of thyroid replacement therapy. Part 2: The critical 19th century. J R Soc Med. 2011;104(2):59–63.
Mateo RCI, Hennessey JV. Thyroxine and treatment of hypothyroidism: seven decades of experience. Endocrine. 2019;66(1):10–7. https://doi.org/10.1007/s12020-019-02006-8.
Jacqueline Jonklaas SD. Levothyroxine prescriptions trends may indicate a downtrend in prescribing. Ther Adv Endocrinol Metab. 2020;11:1–14.
Abou-Taleb BA, Nounou MI, Khalafallah N, Khalil S. Effect of batch age on potency and dissolution of levothyroxine sodium tablets: impact of BP and USP monograph differences on dissolution results. Drug Dev Ind Pharm. 2018;44(11):1762–9. https://doi.org/10.1080/03639045.2018.1496446.
Junior EA, Duarte Lf, Marques PR, Tosetti D, De Souza Sf, Pereira R, Calafatti SA. Levothyroxine bioequivalence study: determination in healthy volunteers by microparticle enzyme immunoassay. Glob J Med Res. 10(2):58–61.
FDA. Draft guidance on levothyroxine sodium. 2014. https://www.accessdata.fda.gov/scripts/cder/psg/index.cfm. Accessed 16 Nov 2022.
European Medicine Agency, Levothyroxine tablets 12.5 mcg, 25 mcg, 50 mcg, 75 mcg, 100 mcg and 200 mcg (and additional strengths within the range) product-specific bioequivalence. 2020. https://www.ema.europa.eu/en/documents/scientific-guideline/levothyroxine-tablets-125-mcg-25-mcg-50-mcg-75-mcg-100-mcg-200-mcg-additional-strengths-within-range_en.pdf. Accessed 16 Nov 2022.
Levothyroxine sodium abuse. React. Wkly. 1425, 24 (2012). https://doi.org/10.2165/00128415-201214250-00082.
Gottwald-Hostalek U, Uhl W, Wolna P, Kahaly GJ. New levothyroxine formulation meeting 95–105% specification over the whole shelf-life: results from two pharmacokinetic trials. Curr Med Res Opin. 2017;33(2):169–74. https://doi.org/10.1080/03007995.2016.1246434.
Gaitonde DY, Rowley KD, Sweeney LB. Hipotiroidism: an update. Am Fam Physician. 2012;86(3):244–51.
Fliers E, Demeneix B, Bhaseen A, Brix TH. European Thyroid Association (ETA) and Thyroid Federation International (TFI) joint position statement on the interchangeability of levothyroxine products in EU countries. Eur Thyroid J. 2018;7(5):238–42.
Real-world evidence from a narrow therapeutic index product (levothyroxine) reflects the therapeutic equivalence of generic drug products, US Food Drug Administration, 2017. https://www.fda.gov/drugs/news-events-human-drugs/real-world-evidence-narrow-therapeutic-index-product-levothyroxine-reflects-therapeutic-equivalence. Accessed 16 Nov 2022.
Salvatore Benvenga AC. Levothyroxine formulations: pharmacological and clinical implications of generic substitution. Adv Ther. 2019;36:59–71.
van der Spoel E, Roelfsema F, van Heemst D. Within-person variation in serum thyrotropin concentrations: main sources, potential underlying biological mechanisms, and clinical implications. Front Endocrinol (Lausanne). 2021;12(February):1–13. https://doi.org/10.3389/fendo.2021.619568.
Larsen PR, Zavacki AM. Role of the iodothyronine deiodinases in the physiology and pathophysiology of thyroid hormone action. Eur Thyroid J. 2012;2012:232–42.
Wang D, Stapleton HM. Analysis of thyroid hormones in serum by liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2010;397(5):1831–9.
Acknowledgements
To all people involved in the execution of this study.
Funding
This study was funded by Tecnoquímicas, Colombia, including the journal’s Rapid Service Fee and Open Access Fee.
Author Contributions
All authors contributed to the development of this study. IA, MJ, CB wrote the manuscript. MPR performed statistical analysis. All authors were involved in the study design, data analysis and critical review of the manuscript and gave their approval for its publication.
Disclosures
Maria Juliana Cruz and Isaac Arbeláez declare that they are employees of Tecnoquímicas. The other authors do not declare any conflict of interest.
Compliance with Ethics Guidelines
The study and its execution were carried out following the ethical guidelines and principles for research in human beings and were approved by the institutional ethics committee (CEIID). All the volunteers received complete information of the methodology and objectives of the study and gave their informed consent. This trial was registered at ClinicalTrials.gov (NCT04573907). The study was approved by the following CEIID members Gonzalo Battauz, Lucia Grippo, Paula Denise Ludi, Alejandro Robles, and Dr. Cornalo Romanela Tamara. This study was in compliance with the Declaration of Helsinki 1964 and its later amendments.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bertoncini, C.W., Palacios, M.J.C., Fritz, M.C. et al. Levothyroxine Bioequivalence Study and Its Narrow Therapeutic Index: Comparative Bioavailability Results Between Two Formulations Available in Latin America. Adv Ther 40, 1644–1654 (2023). https://doi.org/10.1007/s12325-022-02352-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-022-02352-6