
Abstract
Genotyping individuals using forensic or non-invasive samples such as hair or fecal samples increases the risk of allelic amplification failure (dropout) due to the low quality and quantity of DNA. One way to decrease genotyping errors is to increase the number of replicates per sample. Here, we have developed the software SNP+ to estimate the dropout probability and the subsequent required number of replicates to obtain the reliable genotype with probability 95%. Moreover, the software predicts the minor allele frequency and compares two competing models assuming equal or allele-specific dropout probabilities by Bayes factor. The software handles data from one SNP to high density arrays (e.g., 100,000 SNPs).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Single nucleotide polymorphisms (SNPs) are biallelic markers largely abundant in most genomes and with low mutation rate (~ 10−9 per generation) (Brumfield et al. 2003; Morin et al. 2004). SNPs can be associated to diseases, susceptibility to environmental factors or quantitative trait locus (Erichsen and Chanock 2004; Amos et al. 2008; Casellas et al. 2008; Nickels et al. 2013). In forensic medicine, SNPs can be useful to identify individuals from non-invasive samples by using short amplicons (Sobrino et al. 2005). However, individual identification using degraded samples with less than 100 copies of gDNA may cause genotyping errors (Giardina et al. 2009; von Thaden et al. 2020). Allelic amplification failure, or dropout is the most common error caused by stochastic effects of the PCR reaction (Taberlet and Luikart 1999). To reduce dropout ratio, multiplex pre-amplification or increased replicates per sample could be performed (Bellemain and Taberlet 2004; Sastre et al. 2009). However, both solutions increase time and cost for genotyping individuals. In order to reduce genotyping errors using non-invasive samples without cost, we decided to develop a software (SNP+) to predict the dropout probability of each SNP from a sample of replicated genotypes. Moreover, two alternative parametrizations were compared by a Bayes factor to check for within-SNP homogeneous dropout probability against different dropout probabilities for each allele.
Material and methods
The SNP+ software analyzes each SNP independently, taking as a starting point a vector y of n genotypes ordered by individual (m) and replicates within individual (y’ = [y’1 y’2 … y’m]), where n1 is the number of replicates for the first individual, and n = n1 + n2 + … + nm. Assuming two alleles, A and B, the Bayesian joint posterior distribution generalizes to
and focuses on estimating the allele frequency (fA), as well as the dropout probability for allele A (εA) or B (εB). Taking a particular genotype yi with possible outcomes AA, AB, BB and missing genotype (miss.), its Bayesian likelihood is computed as
where
And \(p\left( {{\text{AA}}} \right) = f_{{\text{A}}}^{{2}} ,p\left( {{\text{AB}}} \right) = {2}f_{{\text{A}}} \left( {{1} - f_{{\text{A}}} } \right)\), and \(p\left( {{\text{BB}}} \right) = f_{{\text{B}}}^{{2}}\). Note that the model assumes that a BB individual cannot be genotyped as AA (the probability of false alleles is zero). A priori distributions for fA, εA and εB were assumed flat between 0 and 1.
For each SNP, the model was solved by a Metropolis–Hastings sampling process (Metropolis et al. 1953) with 500,000 iterations after a burn-in period of 10,000 iterations. Two alternative parameterizations (\(\varepsilon_{{\text{A}}} = \varepsilon_{{\text{B}}} vs.\varepsilon_{{\text{A}}} \ne \varepsilon_{{\text{B}}}\)) were compared by Bayes factor (Kass and Raftery 1995). The minimum number of within-individual replicates required to predict the reliable genotype with probability 95% was calculated as log(0.05)/log(εA). All these procedures have been implemented in the SNP+ software, available at http://www.casellas.info/software.html.
Results and discussion
The program generates the following text delimited output files:
-
(1)
Summary table of the probability of error, confidence interval, replications, and Bayes factor of all the SNPs (output file example in Fig. 1).
-
(2)
SNP-by-SNP report of dropout probabilities with their confidence intervals, minimum number of replicates, Bayer factor comparing a single dropout probability against two independent dropout probabilities.
-
(3)
Predicted genotypes for each individual and SNP, and probability of error, if any.
-
(4)
Pairwise comparison between individuals and the probability to have an identical genotype.
-
(5)
SNP-by-SNP report of the minor allele frequency (MAF) and probability of identity (PI).

Output file generated by the SNP+ software for each analyzed SNP, its alleles, allele-specific or joint dropout probability (and minimum number of replicates to guarantee a 95% genotype probability), and the Bayes factor comparing the models with allele-specific and joint dropout probability
The software has been extensively tested on simulated data with appealing results. Figure 2 illustrated SNP+ ability to detect allele-specific departures in dropout probabilities, as well as the increase in statistical relevance (i.e., Bayes factor) as estimated dropout differences increase. The U-shaped scatter plot misidentified a common dropout probability in less than 3% of the simulated data sets, and this percentage reduced below 1% with seven replicates per sample (results not shown). Sample size does not modify the dropout probability but the accuracy of the (dropout probability) estimate we obtain with SNP+.

Predicted dropout probabilities on simulated data sets with 50 individuals and 5 replicate genotypes per individual. Genotypes were simulated under Hardy–Weinberg equilibrium (0.5 allele frequency) and dropout probabilities for each allele were 0.1 and 0.2, respectively. The Bayes factor compared the model with allele-specific dropout probabilities against the same dropout probability for both alleles
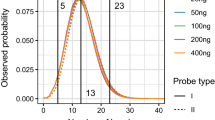
Moreover, we have used SNP+ to evaluate two panels using Open Array® technology (Thermo Fisher Scientific Inc). We analyzed 22 fecal samples and 114 hair samples from Iberian brown bears (Ursus arctos) using first a 120 SNP panel (data prepared for publication but not submitted). To decrease the cost of the analysis, we selected 60 SNPs out of 120 SNPs with the lowest dropout probabilities, and we repeated our analysis with SNP+ using 164 fecal samples and 173 hair samples. All samples were replicated four times, and about 25% and 20% of low-quality DNA fecal and hair samples, respectively (call rate < 25%) were not included in both analyses. Figure 3 shows the relative frequencies of the dropout probability for the four studies. The dropout probability was clearly low after selecting the panel of 60 SNPs using two types of non-invasive samples, on average 0.05 (studies C and D; 60 SNPs) versus 0.2 (studies A and B; 120 SNPs). In terms of variability and distribution mode, the study that obtains lower dropout probabilities is the study C, after SNP selection. The study with the highest probability of dropout is the study B probably because hair samples were hair-trapping collected and therefore, not all samples contained roots or enough hair quantity to obtain high DNA quality. To summarize, SNP+ calculates the dropout likelihood, the Bayes factor, PI and MAF, and can be used to select the best arrays from low density arrays up to high density arrays, avoiding those SNPs that require many replicates because they lead to error. Moreover, SNP+ shows the number of replicates needed per sample to reach a 95% of genotyping reliability per SNP.

Histograms showing the relative frequencies (%) of dropout probability in bear samples using the SNP+ software in four cases (“X” = average dropout probability)
References
Amos CI, Wu X, Broderick P et al (2008) Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet 40(5):616–622. https://doi.org/10.1038/ng.109
Bellemain E, Taberlet P (2004) Improved noninvasive genotyping method: application to brown bear (Ursus arctos) faeces. Mol Ecol Notes 4(3):519–522. https://doi.org/10.1111/j.1471-8286.2004.00711.x
Brumfield R, Beerli PA, Nickerson D et al (2003) The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol Evol 18:249–256. https://doi.org/10.1016/S0169-5347(03)00018-1
Casellas J, Varona L, Muñoz G et al (2008) Empirical Bayes factor analyses of quantitative trait loci for gestation length in Iberian × Meishan F2 sows. Animal 2(2):177–183. https://doi.org/10.1017/S1751731107001085
Erichsen HC, Chanock SJ (2004) SNPs in cancer research and treatment. Br J Cancer 90(4):747–751. https://doi.org/10.1038/sj.bjc.6601574
Giardina E, Pietrangeli I, Martone C et al (2009) Whole genome amplification and real-time PCR in forensic casework. BMC Genomics 10:159. https://doi.org/10.1186/1471-2164-10-159
Kass RE, Raftery AE (1995) Bayes factors. J Am Stat Assoc 90(430):773–795. https://doi.org/10.1080/01621459.1995.10476572
Metropolis N, Rosenbluth AW, Rosenbluth MN et al (1953) Equation of state calculations by fast computing machines. J Chem Phys 21(6):1087–1092. https://doi.org/10.1063/1.1699114
Morin P, Luikart G, Wayne RK (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216. https://doi.org/10.1016/j.tree.2004.01.009
Nickels S, Truong T, Hein R et al (2013) Evidence of gene-environment interactions between common breast cancer susceptibility loci and established environmental risk factors. PLoS Genet 9(3):e1003284. https://doi.org/10.1371/journal.pgen.1003284
Sastre N, Francino O, Lampreave G et al (2009) Sex identification of wolf (Canis lupus) using non-invasive samples. Conserv Genet 10(3):555–558. https://doi.org/10.1007/s10592-008-9565-6
Sobrino B, Brión M, Carracedo A (2005) SNPs in forensic genetics: a review on SNP typing methodologies. Forensic Sci Int 154(2–3):181–194. https://doi.org/10.1016/j.forsciint.2004.10.020
Taberlet P, Luikart G (1999) Non-invasive genetic sampling and individual identification. Biol J Lin Soc 68(1–2):41–55. https://doi.org/10.1111/j.1095-8312.1999.tb01157.x
von Thaden A, Nowak C, Tiesmeyer A et al (2020) Applying genomic data in wildlife monitoring: development guidelines for genotyping degraded samples with reduced single nucleotide polymorphism (SNP) panels. Mol Ecol Resour 20(3):662. https://doi.org/10.1111/1755-0998.13136
Acknowledgements
This work was funded by the LoupO project (EFA354/19) of the European Interreg Program V-A Spain-France-Andorra (POCTEFA 2014-2020). We are grateful to two anonymous reviewers for helpful comments on the manuscript.
Funding
Open Access Funding provided by Universitat Autonoma de Barcelona. Funding was provided by Interreg (EFA354/19).
Author information
Authors and Affiliations
Contributions
Conceptualisation: NS, JC, AM. Formal analysis: JC. Methodology: NS, JC. Resources: NS. Validation: NS, JC. Writing—original draft: NS Writing—review and editing: NS, JC, AM.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
For the present study no animal was captured or euthanized.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sastre, N., Mercadé, A. & Casellas, J. SNP+ to predict dropout rates in SNP arrays. Conservation Genet Resour 15, 113–116 (2023). https://doi.org/10.1007/s12686-023-01309-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-023-01309-3