
Abstract
One of the major challenges faced by oil extraction industry is the unstable behavior of asphaltene formation, yet not fully understood. The prediction of asphaltene formation depends on the small changes in chemical characteristics and composition of the crude oil. Consequently, the study of molecular structure and molecular properties such as density is of a great practical interest. Other properties become very complex to assess when the asphaltene fraction contains 105 different molecules. Average molecular parameters are used to obtain information about asphaltenes. The density of the asphaltenes can easily be calculated and, thus, can be used to evaluate predictive capacities of the average structure. This present work of molecular dynamic simulations was carried out to evaluate the asphaltene densities of an Australian oil field. Simulations of molecular dynamics are used to assess the average structure densities representing different asphaltenes in a specific oil field. These simulations assist in predicting the formation of asphaltene structural model. Comparatively, the experimental densities are higher than the calculated ones. However, the calculated values demonstrate the appropriate trend. The chemical structure shows essential accuracy, as evidenced by average structures, which can be used to estimate the densities qualitatively. Hence, systematic study was carried out on the basis of the effects of various structures on the calculated densities of asphaltene. The main characteristics of the molecules which yielded highest densities are those found with low hydrogen–carbon ratio and big condensed aromatic rings. Moreover, better and improved density values were produced by a recently developed group contribution method than simulations of molecular dynamics, which is still being lower than that of experimental values.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Natural asphaltene consists of a mixture of high-molecular-weight hydrocarbons with a chain of up to 150 carbon atoms. The color of the structure lies between black and black–brown with the properties of a viscous fluid and a distinct smell. There are two kinds of natural asphaltene. One is the solid form which deposits and is mixed with asphalt Rocks (Masson 2008). The other type comes out from the ground because of cracks resulting from the formative movements of the crust of the seismic movements (Yousefi 2008). The natural asphalt has a chemical structure and chemical composition similar to the asphalt of petroleum, but differs in physical properties (Alobaidy 2010). Bad rheological properties of natural asphalt make useless for industrial application. Chemically, the structures of asphalt have two parts (Villaneuva and Zanzolto 2008). The first one is asphaltene which is insoluble in normal heptane or normal hexane, and the other is maltene which soluble in aforementioned. In addition, asphaltene is composed of polyheterogeneous aromatic rings substituted by the nitrogen, oxygen, and sulfur atoms of high molecular weight. It is soluble in carbon disulfide (CS2). The maltene part consists of saturated hydrocarbons, non-polarized aromatics, and the resin (representing polarized aromatics) giving the molecules the properties of high adhesion. These adhesive properties result in force of attraction between the components of asphalt and, thus, affect the behavior of physical properties when used for industrial purposes (Havog 2010).
Asphaltenes are regarded as solids of high molecular weight. Benzene and toluene, being aromatic solvents, are soluble in asphaltene while insoluble in paraffinic solvents (Speight et al. 1985; Eskin et al. 2011). Resins show solubility in less molar mass aliphatic hydrocarbons (n-heptane) and aromatic solvents (benzene and toluene) while insolubility in ethyl acetate (Aguilar et al. 2013). The heavy portion of crude oil is considered to be resins and asphaltene, made up of polar molecules. Both the structures include heteroatoms (nitrogen, sulfur, and oxygen) and metals (iron, vanadium, and nickel). Nevertheless, asphaltene comprises of a greater amount of metals and heteroatoms of larger molar mass and aromaticity (Aguilar et al. 2013; Mullins et al. 2003).
The major problem faced by oil extraction industry is the unstable behavior of asphaltene formation, yet not fully predictable. The prediction of asphaltene formation depends on the small changes in chemical characteristics and composition of the crude oil. Consequently, the study of molecular structure and molecular properties such as density is of a great practical interest. Other properties become very complex to assess when the asphaltene fraction contains 105 different molecules. Average molecular parameters are used to obtain information about asphaltenes. The density of the asphaltenes can easily be calculated and, thus, can be used to evaluate predictive capacities of the average structure. The present work of molecular dynamic simulations was carried out to evaluate the asphaltene densities of an Australian oil field.
Materials and methods
Two extra-heavy Australian crude oils obtained from two different wells of the same reservoir, named as crude oil A (6.2° API) and crude oil B (7.2°API) with temperatures at 25 °C, were identified as n-C7 asphaltenes source. The n-C7 asphaltene content of crude oil A and B was 12.6 and 11.5 wt%, respectively. The asphaltene fraction was isolated from crude oil as n-heptane (Sigma-Aldrich St. Louis, MO, 99%), as described in the previous studies.
The n-C7 asphaltene samples were named according to the crude oil employed, such as asphaltene A (AspA) and asphaltene B (AspB). The elemental analyses and molecular weight of both n-C7 asphaltene samples are shown in Table 1. For the adsorption experiments, toluene (MerkK GaG, Germany, 99.5%) was used for re-solubilizing n-C7 asphaltenes extract in various mixtures of n-heptane + toluene (Heptol) at 0 (pure toluene), 20 (Heptol 20), and 50 v/v% (Heptol 50). Desorption experiments were conducted using different Heptol mixtures.
Simulation details
Molecular dynamics and mechanics were performed to calculate the average structural densities of different asphaltenes. The molecular dynamic and mechanic techniques adopted were built in Discover 2.9, an Amorphous Cell of Molecular Simulations and Insight II commercial software programs. The calculations incorporated the consistent valence force field (CVFF). Previously, this field force was used to demonstrate the resin and asphaltene behavior of aggregation and the amphiphiles activity as asphaltene stabilizers. Atoms charges are designated according to the CVFF database which remained unchanged during calculations.
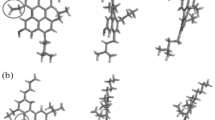
Figure 1 shows the selected average asphaltene structures. These structures relate to asphaltenes separated from crude oil in Australia from various origins while applying the standard technique IP-143. Based on 1 h NMR, these structures were produced according to the previously described procedure. Six structures (CN, MO29, MO21, MG27, BC6, and BC5) of asphaltene were associated with stable crude oil. Four structures (VG3, FU1, BO7, CO2) with unstable crude oil, while rest of the two are associated with asphaltene extracted from deposits of solid taken out from the field, considering that one of the wells was the source of the solids.
Many former studies also reported these structures. Furthermore, ruthenium ion-catalyzed oxidation of the immature asphaltene was carried out to calculate the experimental data about the two different structures and densities. One of the two structures is reported to be Athabasca asphaltene (Azinfar et al. 2017) and the second proposed structure is associated with asphaltene of Arab crude oil structure. The comparison of these structures with that shown in Fig. 1 is described by the reduced number of rings per nuclei of aromatic. Moreover, for the structures under consideration, various molecules were utilized to analyze the methodology employed which does not indicate particularly any asphaltene portion, and also the effects of various structural parameters on the density calculations.

Asphaltene samples’ average structures
The procedure followed to get the optimized average structures for every asphaltene is as follows: minimization of the constructed model potential energy, after that the determination of molecular dynamics constant temperature NVT of 10 ps. The calculation of molecular dynamics used in the setup was in order of five arrangements from lowest energy, followed by the reduction of potential energy so as to decrease the energy even further.
Five amorphous structures were generated using these five molecules based on best five conformers (BC5, MO21, CN, DTJ, and FUI). The hydrogen-to-carbon (H/C) ratio and density (F) correlation established on the experimental data were calculated for the initial density of these structures.
The following is the correlation:
This correlation included materials related to crude oil-containing processed and virgin crude oils, achieved using the experimental data.
The former procedure was followed to obtain the five amorphous structures which were further relaxed to reduce energy while exposing these structures to simulated annealing, i.e., 5000 ps cycles repeated five times from 200 to 500 K, adopting the molecular dynamics (NVT). The structure was reduced again at the end of every annealing cycle, aiming to stop the minima of metastable regional high energy. The purpose of this procedure was to stop the metastable local high-energy minima.
Molecular dynamics for isothermal-isobaric (NPT) were operated for 300 ps at 298 K. Every NPT operation started along 50 ps of an equilibration phase, i.e., 50,000 steps per time steps (1 fs). Densities’ data were compiled along with the final 100 ps. Figure 2 shows the calculation procedure of the scheme (Fig. 3).

Calculation of densities by molecular simulation

Past production, crude oil, LPG and condensate resources, and other resources of Australian crude oil (GeoScience, 2018. http://www.ga.gov.au/scientific-topics/energy/resources/petroleum-resources/oil)
Density measurement
A standard glass pycnometer was used to measure asphaltene density. To displace the fluid, n-heptane was used, while the capacity of pycnometer was measured as 10 cm3. The following steps were taken in the procedure: the pycnometer was introduced with a sample weight of 0.50–0.55 g. Afterwards, the addition of 9–9.5 cm3n-heptane was done while removing the air bubbles trapped by ultra-sonication technique. The n-heptane was filled in the pycnometer followed by immersion and thermostated at 25 °C in a water bath. The stopper is pushed tightly in place. After some time, pycnometer is taken out and weighed before drying. In the next step, the volume of asphaltene is determined. The volume of n-heptane was calculated by determining density and mass at operating temperature reported previously. The ratio of calculated density and mass is used to determine the asphaltene density. The calculations were precise to a figure of the second decimal. The absolute derivations were found to be 0.02 g/cm3.
Aromaticity determination
The aromaticity of carbon was calculated using a 13C nuclear magnetic resonance. Spectrometer (Brucker ACP-400) was set at a frequency of 100.614 MHz to determine the spectra. Afterward, samples were mixed in CD2Cl2 and 0.2M of Cr-III acetylacetonate was added. To suppress the overhauser effect, an opposite gated decouple method was followed.
Results and discussion
Since 1965, several offshore oil and gas fields were explored in Australia. After the confirmation of reservoirs being commercially viable, field development programs were immediately started.
Experimental densities
Experimental densities calculated for the chosen asphaltene are shown in Fig. 1, comprising a Venezuelan coal sample utilized as a reference. A comparison was made between the obtained experimental values to the same values produced by various methods for coal and materials related to crude oil (Fig. 4). The similarity of densities with reported ones was found by this comparison through employing more sophisticated approach, despite being marginally on the lower side. As previously reported, the preferred samples consisted of asphaltene produced from stable/unstable crude oils and deposits. The nC7 insoluble/toluene and soluble materials are regarded as asphaltenes.

Obtained experimental densities and similar sample results comparison
Since polyaromatics density is closely linked to molecular topology, the relationship among the structural characteristics and density of asphaltene is worth evaluating. In Fig. 5a, the experimental densities are presented as a function of hydrogen-to-carbon ratio, while Fig. 5b shows the aromaticity of asphaltene. Asphaltene obtained from stable crude oil is certainly isolated from asphaltene produced from unstable crude oils and solid deposits in these figures. The results show that, by comparing asphaltenes produced from stable and unstable crude oils, the later one exhibited higher density, reduced H/C ratios, and larger aromaticities. This trend has been observed in the previous studies for various samples.

Experimental densities as a function of aH/C ratio. b Aromaticity of asphaltene
Calculated densities
Figure 1 indicates that, for each average structure, there are five of independent amorphous configuration generated, i.e., BC5, MO21, CN, DTJ, and FUI. This figure was used to estimate the densities according to the procedure previously described.
The densities described here were calculated as the mean of five set of amorphous structures. For all the studied cases, the absolute deviations found were nearly 0.02 g/cm3. The results show that hardly any of the evaluated average molecules consisted of heteroatoms, as shown in Fig. 1. Figure 6 shows the plotting of calculated densities against experimental densities as a function for assigned and unassigned heteroatoms. If calculations and experimental results are similar, the solid line in the graph shows the location of the point. By comparing the experimental and calculated densities, it was revealed that the values of calculated densities were considerably less than the value of experimental densities. The calculated results conform to every set of values, although the dispersion derived results most probably from the uncertainty of experimental data (0.02 g/cm3). This indicates the right trend, i.e., increases with increasing experimental values. The introduction of heteroatomic functionalities seems to improve the matching between the calculated and experimental values. This feature is to be explained comprehensively in the following section. Coal volumetric analysis indicates that simulation methods are to be employed to optimize the coal’s average models, although the present work shows that minor structural variations may cause significant changes in calculated densities. Based on the previous results, an initial study was established on overlene molecule structural changes. Various structural modifications were done on overlene molecules for determining the effect on calculated density. Using the method already mentioned, various molecules were examined and calculated density as shown in Fig. 6. By this figure, two trends can be observed.

Comparison between experimental and calculated densities through molecular
-
1.
Increasing the alkyl appendages length produces reduced calculated densities (from molecule A to molecule E) leaving out the C molecule.
-
2.
Molecule F, G or E, H of different isomers can significantly yield different densities. The effect of various structural aspects on asphaltene densities was analyzed systematically, on the foundation of this initial work.
Effect of hydrogen imperfection
In this section, two different models are being used: an amphoteric fraction representative structure confirmed earlier and a speculative structure of asphaltene with the same H/C ratio and molecular weight. These two molecules were altered to reduce their H/C ratio. To this extent, the structures were gradually eliminated by methylene and methyl groups and after every step and the density was calculated. In addition, the generated structure density was plotted as a function of the hydrogen–carbon ratio at each step. It was observed that higher the density, lower the H/C ratio.
Interestingly, the values of density achieved for amphoteric molecule (A) are consistently below than those evaluated for asphaltene model (B), which represents differences associated with molecular topological characteristics like connectivity, locations of chain, and ring size.
Effect of the poly-aromatic nuclei size
Poly-aromatic nuclei size is considered as one of the major differences among the molecules. However, the molecular weight, H/C ratio, and content of heteroatom are the same in both the models. Therefore, it is supposedly possible that density differences might be, at least, moderately ascribed to differences in ring sizes of poly-aromatic compounds. Poly-aromatic rings including amphoteric model are significantly smaller than the asphaltene model being used.
Study of various structural modification effects on molecular calculated densities: two more structures were studied containing poly-aromatic rings, relatively small in size. Athabasca asphaltene corresponds to the first structure, while the second structure to an isolated asphaltene extracted from a combination of Arabian crude oil. These structures were acquired from ruthenium ion-catalyzed oxidation.
According to these structures, density values were estimated as 1.03 g/cm3 for Athabasca and 1.00 g/cm3 for the Arabian mixture. However, the experimental density of Athabasca asphaltenes, in references, was found to be 1.16 g/cm3. On the contrary, average structures density results with designated heteroatoms (Table 1) differ from 1.06 to 1.17 g/cm3. Bigger poly-aromatic rings are present in these structures when compared with Arabian and Athabasca asphaltenes. By comparing both sets, the values show the existence of bigger poly-aromatic nuclei which tends to increase the structure densities.
Effect of the connectivity
Asphaltene from Athabasca and Arabian mixture were treated as samples. Four molecules were extracted by splitting both molecules. Afterward, the calculations for both sets were made. Both the set of molecules yields 1.00 g/cm3 from using this calculation. The results show no significant density differences for the molecules considered as a whole (1.00–1.03 g/cm3) or the parts of the set in the evaluations (1.00 g/cm3).
Effect of heteroatoms
Considering a few molecules here, the calculations of densities were done for both assigned and unassigned heteroatoms. The comparison in Fig. 7 shows that the addition of heteroatoms to the structure of molecule results in a minor rise in the trend for the calculated density. Consequently, this effect may be associated with the introduction of heavy atoms in the molecule and increasing the molecular interactions. Certainly, this final feature is related directly to a location inside the molecule and the functionality type.

Effect of heteroatoms on density (calculated)
Molecular dynamics comparison with group contribution technique employed to calculate poly-aromatic density
Currently, a new technique is devised for molar volume calculation of hydrocarbons consisting of condensed hydrocarbons of poly-aromatic. The application of this technique has successfully explained the molar volume of those heavy hydrocarbons which behave as a liquid with sticky nature or crystalline solid at 298 K. The results obtained clearly indicate excellent behavior compared to other similar results.
According to researchers, this recent group contribution method is appropriate in poly-aromatic hydrocarbons case with up to number seven aromatic rings. It is also used to calculate asphaltene density. The suggested technique uses basic structural data to produce the molecule molar volume as a total individual group of structural improvements that build the molecule.
The provided hydrocarbon of molar volume according to this technique is denoted by Vm:
where Nt, Nar, Nnr, Nac, and Nai represent the carbon total number, aromatic rings number, naphthenic rings number, aromatic conjunction atoms number, and the number carbons in aromatic inner, respectively.
The comparison among two sets of densities calculated by group contribution technique and molecular dynamics is shown. In case of similar results yielded by experiments and calculations, the solid line represents the dropping point. As observed, the values of densities produced by the method of group contribution are higher than the molecular simulation method, however, still below the experimental values. The technique involving group contribution which dominates the technique involving molecular dynamics is because of the development of group contribution method on a broad range of various molecules. Therefore, in certain aspects, the results obtained illustrate an average of isomers that the technique cannot differentiate. For example, Eq. (2) cannot distinguish among molecules with a similar amount of aliphatic chains and different aromatic locations or among molecules with a similar amount of aliphatic carbons but altered chain lengths. However, different calculated densities can be achieved by different isomers when the molecular dynamics technique is employed. In addition, for every calculation, only one molecule was treated. This provides a natural convenience for a technique like a group contribution technique, in which various molecules are averaged over a significant number. Expectations for improved calculated densities are made by introducing additional molecules. For example, a mixture of various molecules describes an asphaltene sample.
The calculated densities can be further improved in two ways: first, partly removing the effect of various isomers; second, improvement in packing can be made by the existence of various sizes of molecules in similar volume. An essential part is played by the last effect for correlation of low calculated densities in the current work by applying molecular dynamics. New progress in discovering these hypotheses is made. As shown in Fig. 8, the calculated values of both sets correlate well with the correlation coefficient (0.848). This illustrates that the graphical description and molecular parameters provide qualitative correct information. Thus, information acquired predicts trends in the values of density including alternative thermodynamic properties.

A comparison of calculated densities using group contribution and molecular simulation
Currently, it is obvious from the results that calculation of precise values of properties cannot be done by average structures for asphaltenes by simulations of the molecule. Furthermore, the molecular dynamic simulations can be developed as a tool to improve average structure method by fitting the calculated properties. Moreover, simulations for molecular dynamics need improvement in an average structure in terms of testing various isomers and adjustment of calculated to experimental properties. Consequently, density is considered a potential parameter for adjusting its experimental determination which is easy and dependent on molecular structural characteristics.
Group contribution technique was found to provide better results than that of molecular simulations that carried out. Nonetheless, it is essential to note that, in a few standard pressure–temperature conditions, each group contribution produces only one value of a specific property. Contrarily, if an optimized method is introduced for developing molecular simulations for average structures, a broad range of thermo-physical properties can be determined under different conditions. This present study is one step towards the development of an advanced methodology to link thermo-physical properties and molecular structure of asphaltene.
Conclusion
Higher densities, lower H/C ratios, and high aromaticities are attributed to asphaltenes from crude oil of unstable nature and deposits, as compared with stable crude oil asphaltenes. The obtained results indicated that the chemical data supplied by average structures and molecular parameters show the essential accuracy that can be used to calculate the densities qualitatively.
Asphaltene density estimation, treating average structures through simulations of molecular dynamics, though liable to huge errors, still provides results that have the precise range and qualitatively accurate. Furthermore, the asphaltene average structure affected the modification of density by this technique and thus proves to be a significant tool to enhance the representation of a condensed structure. The low ratio and large rings of condensed poly-aromatic are associated with the highest densities yielded by the average structure of asphaltenes.
Using molecular dynamics, the large variations established between calculated and experimental densities can be mostly ascribed to the particular molecule to define asphaltene which results in poor packing leading to lower densities. To enhance the linking between experimental and calculated densities, a calculation based on group contribution was examined. Finally, a significant role of asphaltene density on asphaltene solubility was observed.
References
Aguilar JIS, Neto JSG, Almeida SM, Mansur CRE (2013) Evaluation of the influence of polyoxide-based surfactants on the separation process of model emulsions of asphaltenes using the FTIR-ATR technique. J Appl Polym Sci 128:1390–1397
Alobaidy R (2010) Msc. Thesis in chemistry, collage science-Anbar, Chap. 1, p 2
Azinfar B, Haddadnia A, Zirrahi M, Hassanzadeh H, Abedi J (2017) Effect of asphaltene on phase behavior and thermophysical properties of solvent/bitumen systems. J Chem Eng Data 62(1):547–557
Eskin D, Ratulowski J, Akbarzadeh K, Pan S (2011) Modeling asphaltene deposition in turbulent pipeline flows. Can J Chem Eng 89:421–441
Havog KL (2010) Asphalt category analysis and hazard characterization. In: The American Petroleum Institute Petroleum HPV Testing Group, USA, July 14, pp 8, 9–12
Masson JF (2008) Energy Fuel 122:2637–2640
Mullins OC, Groenzin H, Buch L, Gonzalez EB, Andersen SI, Galeana CL (2003) Molecular size of asphaltene fractions obtained from residuum hydrotreatment. Fuel 82:1075–1084
Speight JG, Werrick DL, Gould KA, Overfield RE, Rao BML, Savage DW (1985) Molecular weight and association of asphaltenes: a critical review. Revue de I’Institut Francais du Petrole 40(1):51–56
Villaneuva A, Ho S, Zanzolto L (2008) Asphalt modification with used Lubrieatoind. Can J Civ Eng 35:148
Yousefi AA (2008) The thermal rheological behavior progcolor count 1-45-55. http://www.ga.gov.au/scientific-topics/energy/resources/petroleum-resources/oil
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Elkahky, S., Lagat, C., Sarmadivaleh, M. et al. A comparative study of density estimation of asphaltene structures using group contribution methods and molecular dynamic simulations for an Australian oil field. J Petrol Explor Prod Technol 9, 2699–2708 (2019). https://doi.org/10.1007/s13202-019-0641-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13202-019-0641-x