
Abstract
Background
Cleidocranial dysplasia (CCD) is a rare genetic disorder affecting bone and cartilage development. Clinical features of CCD comprise short stature, delayed ossification of craniofacial structures with numerous Wormian bones, underdeveloped or aplastic clavicles and multiple dental anomalies. Several studies have revealed that CCD development is strongly linked with different mutations in runt-related transcription factor 2 (RUNX2) gene.
Objective
Identification and functional characterization of RUNX2 mutation associated with CCD.
Methods
We performed genetic testing of a patient with CCD using whole exome sequencing and found a novel RUNX2 frameshift mutation: c.1550delT in a sporadic case. We also compared the functional activity of the mutant and wild-type RUNX2 through immunofluorescence microscopy and osteocalcin promoter luciferase assay.
Results
We found a novel RUNX2 frameshift mutation, c.1550delT (p.Trp518Glyfs*60). Both mutant RUNX2 and wild-type RUNX2 protein were similarly confined in the nuclei. The novel mutation caused abrogative transactivation activity of RUNX2 on osteocalcin promoter.
Conclusions
We explored a novel RUNX2 deletion/frameshift mutation in a sporadic CCD patient. This finding suggests that the VWRPY domain may play a key role in RUNX2 transactivation ability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cleidocranial dysplasia (CCD), also referred as a Scheuthauer syndrome, is a rare autosomal dominantly inherited disorder which is represented by various skeletal abnormalities (Lotlikar et al. 2018). The global CCD occurrence rate is usually one in every millions of newborns without gender preponderance (Offiah et al. 2019). Clinical features of CCD is diverse and involves short stature, delayed ossification of craniofacial structures with numerous Wormian bones, underdeveloped or aplastic clavicles, multiple dental defects such as teeth hypoplasia, delayed primary teeth exfoliation, prolonged eruption of permanent teeth and malocclusion (Konishi et al. 2019). Other skeletal abnormalities, such as hypoplastic iliac wings, distal phalanx dysplasia, knock-knees and malformations of spine can be also observed (Farrow et al. 2018). The CCD can be diagnosed prenatally, from early childhood till late adolescence and diagnostic tools ranges from ultrasound investigations (in prenatal period) to panoramic radiography, although early diagnosis is considered as aTkey problem in CCD management (Zeng et al. 2018).
Several studies have revealed that CCD development is strongly linked with different mutations affecting runt-related transcription factor 2 (RUNX2) gene (Jaruga et al. 2016; Xuan et al. 2008), which is a transcription factor involved in osteoblastic differentiation and skeletal morphogenesis. Human RUNX2 gene consisting 8 coding exons, is located in chromosome 6p21 (Levanon et al. 1994). RUNX2 gene comprises Q/A domain on N-terminal part, RUNT domain which is crucial for DNA-binding to a specialized motif and heterodimerization with core-binding factor subunit beta (CBFβ) (Zhang et al. 2017), as well as proline/serine/threonine-rich (PST) domain on its C-terminus (Yoshida et al. 2002). The core-binding factor subunit alpha-1 (CBFA1) protein, that RUNX2 gene encodes is expressed densely in skeletal structures and considered as a key transcription factor for numerous stages of osteogenesis (Sun et al. 2016). Noticeably, mice with targeted RUNX2 disruption have bone formation failure owing to osteoblast deficiency (Zhong et al. 2016). Moreover, it also has been suggested that RUNX2 represents crucial role in tooth development, and its transcriptional failure could promote dental lamina excess activation leading to supernumerary teeth with consequent effect on permanent teeth (Wen et al. 2020). Till recent years, almost 194 causative mutations in RUNX2 have been identified, and this number is still rising (Otto et al. 2002). However, RUNX2 mutations can be found only in two-third of patients with CCD, and 30%–40% of cases are triggered by novel mutations (Hordyjewska-Kowalczyk et al. 2019).
Although we have known more about the clinical and functional characteristics of RUNX2 because of recent studies, there are still some potential unknown factors that urge further exploration. RUNX2, as the key contributory gene in CCD, is conceivable to be a therapeutic target for CDD. In current study, we explored a novel RUNX2 deletion mutation: c.1550delT (p.Trp518Glyfs*60) in a sporadic case and also showed its clinical features, possible pathogenesis and functional characteristics.
Materials and methods
Subjects
The subjects were examined in the Department of Endocrinology, Qilu Hospital of Shandong University, China. The peripheral blood samples were obtained for genetic testing from all participants, including 15 year-old boy with suspected CCD and his unaffected parents. We also carried out clinical and radiological examinations on patient, and collected his medical history.Current study was accepted by the ethics committee of QiLu hospital of Shandong University (ethical approval number KYLL-2019-2-111). All subjects in our study signed consent form voluntarily with the review of the ethical committee. The study methods were performed according to the ethical committee accepted guidelines.
Mutation analysis
The whole-exome sequencing (WES) was carried out on DNA from venous blood sample. Fragmentation of the genomic DNA, paired-end adaptor ligation, amplification and purification were implemented, and the all human exons together with 50 bp bases in their adjacent introns were captured by xGen® Exome Research Panel. The DNA library was performed post-capture amplification and purifying, and then arrayed by the Illumina HiSeq sequencing platform. All test and sequence analysis were supplied by the Beijing Fujun Gene Biotechnology Co., Ltd (Beijing, China).
In silico assays for a Runx2 frameshift
Using phyer2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index), the WT-RUNX2 and W518Gfs-RUNX2 structural conformation were comprehensively analyzed and predicted using the threading method and the heavy-head prediction method, respectively, and the WT-RUNX2 and W518Gfs-RUNX2 structural models were established as the reference instruction (Kelley et al. 2015). Additionally, SAVE5.0 3D Structure Viewer (https://saves.mbi.ucla.edu/) to visualize 3D structure has been used.
Cell culture and transfection
Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco’s modified Eagle’s medium basic (DMEM basic, Gbico, Grand Island, NY, USA, cat: C11995500BT) supplemented with 10% fetal bovine serum (FBS, Gbico, Grand Island, NY, USA, cat: 10,099,141), penicillin (100 IU/mL), and streptomycin (100 μg/mL) as formerly detailed (Hu et al. 2014; Wang et al. 2014). Cells transfection with the plasmids carrying needed genes by using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) was performed to investigate protein expression and other related studies.
Western blotting analysis
We used the human embryonic kidney (HEK) 293 cells to study the WT- RUNX2 and W518Gfs-RUNX2 expression as previously described. For western blotting analysis, the cells were seeded in 10 cm2 plates. One day later, the HEK293 cells were transfected with pcDNA3.1-GFP, pcDNA3.1-WT-RUNX2-GFP and pcDNA3.1 W518Gfs-RUNX2-GFP. After 48 h transfection, the cells were cultured and subjected to SDS–PAGE. Western blotting was performed with a rabbit polyclonal anti-GFP antibody (1:1000, proteintech, Wuhan, Hubei, China, cat: 50,430-2-AP) and a horse radish peroxidase-conjugated goat anti-rabbit IgG polyclonal antibody (1:10,000, Zhongshan Golden Bridge, Beijing, China, Cat: ZB-2301) as a second antibody. Signals were detected using a chemiluminescence kit (Millipore, California, USA, Cat: WBKLS0050). Each experiment was repeated three times.
Luciferase reporter assays
Previously, seeded the HEK-293 cells into 96-well culture plate 24 h before this assay, 10,000/well, 200 ul cell suspension, culture at 37 ℃, 5% CO2 (Sun et al. 2020). On the second day, pRL-TK and pGL3-basic-osteocalcin promoter plasmid has been added to each group, and each group were also transfected separately with different concentrations of pcDNA3.1, pcDNA3.1-WT-RUNX2, pcDNA3.1-W518Gfs-RUNX2 (0 ug, 1 ug, 2 ug). After 48 h, the follow-up dual-luciferase detection operation was performed. For details, we followed the dual-luciferase reporter assay system and analyzed the results.
Statistics
Data were presented as the mean ± standard error of mean (SEM) from at least three independent experiments. Statistical comparisons were carried out using paired t-test, ANOVA or Bonferroni test with GraphPad Prism 6.0 (GraphPad Software, San Diego, California, USA). The protein bands from the western blot were quantified applying Image J software (National Institutes of Health, Bethesda, Maryland, USA). Each experiment was repeated at least in triplicate. Statistically significant differences were considered at p value < 0.05.
Results
Clinical manifestation
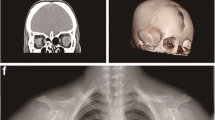
Among three participants of our study, diagnosis of CCD was confirmed only in 15-year old male proband. His past history and physical examinations has differed noticeably from his parents. On extraoral examination, typical features of CCD, such as midline depression of upper forehead, narrow and dropped shoulders and short stature were observed. His photographs were obtained and he was referred to radiographic investigations (Fig. 1).

Typical and radiological findings in the CCD patient. a Frontal facial view of patient representing midline depression of forehead and bilateral hypoplastic clavicles. b Hypoplasia of the clavicles abnormal facility in the opposing shoulders. c Panoramic radiography revealed primary teeth retention, numerous impacted permanent teeth in both maxilla and mandible. d Chest X-ray showed bilateral hypoplastic clavicles hypoplasia of iliac bones, wide symphysis pubis with a bell-shaped thoracic cavity and scoliosis. e, f No abnormalities were seen in the bones of the bilateral elbow joints, and the epiphyseal line showed closure
Oral manifestations and radiographic features
His panoramic radiograph showed mixed dentition with delayed exfoliation of deciduous teeth, retained and impacted permanent teeth. Aplasia of lateral two-thirds of right and one-third of left clavicles, cone shaped thorax, scoliosis and deformed ribs were found during chest radiography (Fig. 1).
Sequencing results and biochemical characterization of RUNX2 (c.1550delT) gene mutation
Sequencing analysis was implemented in the coding region of RUNX2 gene. RUNX2 (c.1550delT) mutant was detected in the proband, but not in his parents (Fig. 2A). The c.1550delT mutation was clustered in the terminal VWRPY of highly conserved PST domain (Fig. 2B–C). It is predicted that mutations at this site may change the protein activity (Fig. 2D).

Partial sequence diagram of RUNX2 and biochemical characterization of the RUNX2 (c.1061G > T) variant. A Partial sequence diagram of RUNX2. A heterozygous c.1550delT transition mutation is shown using an arrow (GenBank accession number: NM_001024630.4). This frameshift mutation resulted in changes in amino acid synthesis starting from amino acid Trp 518 (p.Trp518Glyfs). B RUNX2 structural domains. Mutations at the protein level are indicated below the PST domain. C Cross-species conservation of Trp518-RUNX2. D Protein structure prediction of the RUNX2 (WT and Trp518Glyfs). RUNX2 WT protein sequence is 518WRPY521 and the RUNX2 (c.1550delT) mutant sequence is 518GDHI521
Functional characterizations of RUNX2 (c.1550delT) gene mutation
Western blot analysis confirmed that RUNX2 (c.1550delT) mutant gene did not cause reduction in RUNX2 protein expression level (Fig. 3A, B). Some studies have illustrated that RUNX2 is predominantly located in the cell nucleus and little perinucleolar region (Javed et al. 2000; Young et al. 2007). In order to verify the RUNX2 mutation nuclear localization, we transfected both pGFP-RUNX2 and pGFP-RUNX2-Trp518Glyfs plasmids into HEK293T cells. The subcellular localization of the RUNX2 and the Trp518Glyfs mutation was observed by in situ immunofluorescence microscopy (Young et al. 2007). Both RUNX2 mutation and wild-type RUNX2 accumulated in the nuclei of HEK293T cells (Fig. 3C). This denoted that the subcellular compartmentation of the RUNX2 mutation was not affected. It has been reported in previous studies that RUNX2 is responsible for the transactivation of the osteoblast-specific osteocalcin gene in osseous cells (Zaidi et al. 2001). Luciferase assays demonstrated the RUNX2 transactivation activity and the transcriptional regulation of the osteocalcin promoter. The RUNX2 (c.1550delT) variant induced the osteocalcin promoter activity lower than RUNX2-WT (Fig. 3D).

Functional characterization of the RUNX2 (c.1061G > T) variant. A Protein expression of RUNX2 (WT and Trp518Glyfs). B The histogram of the RUNX2 protein expression level analysis. n.s., denotes RUNX2 Trp518Glyfs compare with the empty denotes p > 0.05. C Nuclear localization of WT and mutant RUNX2. D Luciferase results of HEK293 cells were transfected with each RUNX2 expression vector (WT and Trp518Glyfs). *, denotes RUNX2 WT plasmid compare with the empty plasmid, p < 0.05; #, denotes RUNX2 Trp518Glyfs compare with RUNX2 WT, p < 0.05, n = 6
Discussion
Human RUNX2 gene comprises Q/A domain on N-terminal part, RUNT domain, as well as PST domain on its C-terminus. More noticeably, RUNT domain include NLS on its C-terminus, and this part has been reported to be important for the protein nuclear transportation (Ryoo et al. 2010). It also has been found that PST domain, to be more specific, its nuclear matrix targeting sequence (NMTS)-associated subnuclear foci is engaged in both activation of downstream factors such as osteocalcin gene and subnuclear localization of RUNX2 (Zaidi et al. 2001).Similarly, Q/A domain also participates in transactivation activity of RUNX2 target genes (Kauffenstein et al. 2016).
In our study, we found a novel RUNX2 (c.1550delT) mutation,causing frameshift starting from 518 codon, eventually led to aberrant VWRPY domain in C-terminus. The protein sequence of RUNX2 WT 518WRPY521 is replaced by 518GDHIEIPQQWPSGIWGPHPTRINIYIYRESAYICISISYLQSAYFLEDFSFTHSVMILQP577. The local spatial structure of the protein is changed because the local secondary structure of the protein changes from alpha helix to random coil. Stop codon of RUNX2 WT is TGA at gene site 1566. Stop codon of c.1550delT is TAA at gene site 1734. Considerably, neither RUNX2 protein expression, nor its subcellular distribution was impaired in current study, which might be explained by intact RUNT domain. Several studies have proposed that osteocalcin promoter activation is VWRPY-dependent (Qin et al. 2017). In the present study, abnormal VWRPY domain led to aberrant downstream activation of osteocalcin promoter that further supported previous findings. Zaidi et al. suggested that VWRPY is not crucial for RUNX2 nuclear retention (Zaidi et al. 2001). Since we have not found defective nuclear retention of RUNX2, our findings approved the point of Zaidi et al.
Currently, more than 48 phenotypic characteristics of CCD have been registered by OMIM (Qin et al. 2017). Although, numerous previous studies aiming to reveal genotype–phenotype association have been performed, controversy related to this point is still exist (Quack et al. 1999; Zhang et al. 2010). Additionally, some studies failed to find association between RUNX2 mutation and the severity of CCD (Lou et al. 2009). Moreover, threshold level of RUNX2 mutation that initiates CCD remains unidentified (Xu et al. 2017). Although CCD is considered as autosomal dominant disease, as stated in recent studies, it can be present in a sporadic pattern almost in 30% of cases (Huang et al. 2013). Notably, the genetic testing for possible mutations revealed that both of patient’s parents had normal RUNX2 in our study, suggesting that mutation occurred de novo. Although VWRPY is not hotspot for mutations and aberrant VWRPY did not lead to impaired RUNX2 protein synthesis, subcellular distribution in present research, it precipitated CCD with classical phenotype. This might be explained by possible influence of other determinants on CCD phenotype.
To conclude, we explored a novel RUNX2 deletion/frameshift mutation in a sporadic CCD patient. This finding emphasizes on crucial role of VWRPY domain in RUNX2 transactivation ability. Further studies are awaited to explore more RUNX2 mutations for revealing potential attributors as well as genotype–phenotype association.
Data availability
All data generated or analysed during this study are included in this published article.
References
Farrow E, Nicot R, Wiss A, Laborde A, Ferri J (2018) Cleidocranial dysplasia: a review of clinical, radiological, genetic implications and a guidelines proposal. J Craniofac Surg 29:382–389
Hordyjewska-Kowalczyk E, Sowinska-Seidler A, Olech EM, Socha M, Glazar R, Kruczek A, Latos-Bielenska A, Tylzanowski P, Jamsheer A (2019) Functional analysis of novel RUNX2 mutations identified in patients with cleidocranial dysplasia. Clin Genet 96:429–438
Hu QX, Dong JH, Du HB, Zhang DL, Ren HZ, Ma ML, Cai Y, Zhao TC, Yin XL, Yu X et al (2014) Constitutive Galphai coupling activity of very large G protein-coupled receptor 1 (VLGR1) and its regulation by PDZD7 protein. J Biol Chem 289:24215–24225
Huang Y, Song Y, Zhang C, Chen G, Wang S, Bian Z (2013) Novel RUNX2 frameshift mutations in Chinese patients with cleidocranial dysplasia. Eur J Oral Sci 121:142–147
Jaruga A, Hordyjewska E, Kandzierski G, Tylzanowski P (2016) Cleidocranial dysplasia and RUNX2-clinical phenotype-genotype correlation. Clin Genet 90:393–402
Javed A, Guo B, Hiebert S, Choi JY, Green J, Zhao SC, Osborne MA, Stifani S, Stein JL, Lian JB et al (2000) Groucho/TLE/R-esp proteins associate with the nuclear matrix and repress RUNX (CBF(alpha)/AML/PEBP2(alpha)) dependent activation of tissue-specific gene transcription. J Cell Sci 113(Pt 12):2221–2231
Kauffenstein G, Tamareille S, Prunier F, Roy C, Ayer A, Toutain B, Billaud M, Isakson BE, Grimaud L, Loufrani L et al (2016) Central role of P2Y6 UDP receptor in arteriolar myogenic tone. Arterioscler Thromb Vasc Biol 36:1598–1606
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858
Konishi KI, Mizuochi T, Yanagi T, Watanabe Y, Ohkubo K, Ohga S, Maruyama H, Takeuchi I, Sekine Y, Masuda K et al. (2019) Clinical Features, Molecular Genetics, and Long-Term Outcome in Congenital Chloride Diarrhea: A Nationwide Study in Japan. J Pediatr 214: 151–157 e6
Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y (1994) AML1, AML2, and AML3, the human members of the runt domain gene-family: cDNA structure, expression, and chromosomal localization. Genomics 23:425–432
Lotlikar PP, Creanga AG, Singer SR (2018) Clinical and radiological findings in a severe case of cleidocranial dysplasia. BMJ Case Rep 2018: bcr2018226671
Lou Y, Javed A, Hussain S, Colby J, Frederick D, Pratap J, Xie R, Gaur T, van Wijnen AJ, Jones SN et al (2009) A Runx2 threshold for the cleidocranial dysplasia phenotype. Hum Mol Genet 18:556–568
Offiah AC, Vockley J, Munns CF, Murotsuki J (2019) Differential diagnosis of perinatal hypophosphatasia: radiologic perspectives. Pediatr Radiol 49:3–22
Otto F, Kanegane H, Mundlos S (2002) Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat 19:209–216
Qin XY, Jia PZ, Zhao HX, Li WR, Chen F, Lin JX (2017) Novel mutation of cleidocranial dysplasia-related frameshift runt-related transcription factor 2 in a sporadic chinese case. Chin Med J (engl) 130:165–170
Quack I, Vonderstrass B, Stock M, Aylsworth AS, Becker A, Brueton L, Lee PJ, Majewski F, Mulliken JB, Suri M et al (1999) Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet 65:1268–1278
Ryoo HM, Kang HY, Lee SK, Lee KE, Kim JW (2010) RUNX2 mutations in cleidocranial dysplasia patients. Oral Dis 16:55–60
Sun M, Zhou X, Chen L, Huang S, Leung V, Wu N, Pan H, Zhen W, Lu W, Peng S (2016) The regulatory roles of MicroRNAs in bone remodeling and perspectives as biomarkers in osteoporosis. Biomed Res Int 2016:1652417
Sun Y, Zhang D, Ma ML, Lin H, Song Y, Wang J, Ma C, Yu K, An W, Guo S et al. (2020) Optimization of a peptide ligand for the adhesion GPCR ADGRG2 provides a potent tool to explore receptor biology. J Biol Chem 296:100174
Wang HM, Dong JH, Li Q, Hu Q, Ning SL, Zheng W, Cui M, Chen TS, Xie X, Sun JP et al (2014) A stress response pathway in mice upregulates somatostatin level and transcription in pancreatic delta cells through Gs and beta-arrestin 1. Diabetologia 57:1899–1910
Wen Q, Jing J, Han X, Feng J, Yuan Y, Ma Y, Chen S, Ho TV, Chai Y (2020) Runx2 regulates mouse tooth root development via activation of WNT inhibitor NOTUM. J Bone Miner Res 35:2252–2264
Xu W, Chen Q, Liu C, Chen J, Xiong F, Wu B (2017) A novel, complex RUNX2 gene mutation causes cleidocranial dysplasia. BMC Med Genet 18:13
Xuan D, Li S, Zhang X, Hu F, Lin L, Wang C, Zhang J (2008) Mutations in the RUNX2 gene in Chinese patients with cleidocranial dysplasia. Ann Clin Lab Sci 38:15–24
Yoshida T, Kanegane H, Osato M, Yanagida M, Miyawaki T, Ito Y, Shigesada K (2002) Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am J Hum Genet 71:724–738
Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee SH, Yang X, Xie R, Javed A, Underwood JM et al (2007) Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature 445:442–446
Zaidi SK, Javed A, Choi JY, van Wijnen AJ, Stein JL, Lian JB, Stein GS (2001) A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J Cell Sci 114:3093–3102
Zeng L, Wei J, Zhao N, Sun S, Wang Y, Feng H (2018) A novel 18-bp in-frame deletion mutation in RUNX2 causes cleidocranial dysplasia. Arch Oral Biol 96:243–248
Zhang C, Zheng S, Wang Y, Zhao Y, Zhu J, Ge L (2010) Mutational analysis of RUNX2 gene in Chinese patients with cleidocranial dysplasia. Mutagenesis 25:589–594
Zhang X, Liu Y, Wang X, Sun X, Zhang C, Zheng S (2017) Analysis of novel RUNX2 mutations in Chinese patients with cleidocranial dysplasia. PLoS One 12:e0181653
Zhong Z, Niu P, Wang M, Huang G, Xu S, Sun Y, Xu X, Hou Y, Sun X, Yan Y et al (2016) Targeted disruption of sp7 and myostatin with CRISPR-Cas9 results in severe bone defects and more muscular cells in common carp. Sci Rep 6:22953
Funding
This work was supported by the Graduate Education Innovation Program of Cheeloo College of Medicine, Shandong University (Numbers: 2020Y11) and the National Natural Science Foundation of China (Grant Numbers: 81400814). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
JR and XH conceived and designed the study. FH, FL and YS contributed partly to sample collection and genotype data. NZ and XZ contributed reagents and materials. JY and SW collected data and performed the analyses. LG and BO interpreted the data and wrote the paper. All authors have read and approved the manuscript. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
All authors declare that they have no competing interests.
Ethics approval and consent to participate
All case information were collected with informed consent of the patient’s parents. Prior to genotyping and analysis, all samples were stripped of personal identifiers (if any existed).
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gong, L., Odilov, B., Han, F. et al. Identification a novel de novo RUNX2 frameshift mutation associated with cleidocranial dysplasia. Genes Genom 44, 683–690 (2022). https://doi.org/10.1007/s13258-022-01229-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-022-01229-w