
Abstract
Dopamine replacement represents the standard therapy for Parkinson’s disease (PD), a common, chronic, and incurable neurological disorder; however, this approach only treats the symptoms of this devastating disease. In the search for novel disease-modifying therapies that target other relevant molecular and cellular mechanisms, Drosophila has emerged as a valuable tool to study neurodegenerative diseases due to the presence of a complex central nervous system, the blood–brain barrier, and a similar neurotransmitter profile to humans. Human PD-related genes also display conservation in flies; DJ-1β is the fly ortholog of DJ-1, a gene for which mutations prompt early-onset recessive PD. Interestingly, flies mutant for DJ-1β exhibit PD-related phenotypes, including motor defects, high oxidative stress (OS) levels and metabolic alterations. To identify novel therapies for PD, we performed an in vivo high-throughput screening assay using DJ-1β mutant flies and compounds from the Prestwick® chemical library. Drugs that improved motor performance in DJ-1ß mutant flies were validated in DJ-1-deficient human neural-like cells, revealing that zaprinast displayed the most significant ability to suppress OS-induced cell death. Zaprinast inhibits phosphodiesterases and activates GPR35, an orphan G-protein-coupled receptor not previously associated with PD. We found that zaprinast exerts its beneficial effect in both fly and human PD models through several disease-modifying mechanisms, including reduced OS levels, attenuated apoptosis, increased mitochondrial viability, and enhanced glycolysis. Therefore, our results support zaprinast as a potential therapeutic for PD in future clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD), a progressive neurodegenerative movement disorder [1], is characterized by resting tremor, muscular rigidity, bradykinesia postural instability, and other non-motor symptoms that worsen patients’ quality of life [2]. The appearance of these symptoms is caused by the selective loss of dopaminergic neurons in the substantia nigra pars compacta, which leads to a reduction in dopamine levels in the striatum [3], and the formation of intracellular protein aggregates called Lewy bodies in neurons [4]. Additional factors associated with PD progression include elevated oxidative stress (OS) levels, mitochondrial dysfunction, and metabolic alterations [5, 6]. Striatal dopamine replacement using dopamine precursors, dopamine agonists, or inhibitors of dopamine metabolism [7] currently represents the most commonly employed treatment for PD; however, this therapeutic approach only treats symptoms during early-stage disease and does not modify the underlying disease in the long term. Furthermore, the efficacy of this treatment strategy decreases after long-term administration, and it cannot counteract or delay PD progression. Therefore, we require novel therapies that target other disease-related molecular and cellular mechanisms in the hope of modifying the underlying disease [8, 9]. In this sense, multiple studies have evaluated compounds whose mechanisms of action may impact calcium transport, OS levels, mitochondrial function, autophagy, or energy metabolism as potential PD treatments [7, 10, 11]. Notably, the current PD drug development pipeline, based on ongoing clinical trials, involves therapeutics that treat both symptoms and the underlying disease [12].
Although most PD cases are sporadic and involve the contribution of environmental factors and aging, there also exist familial cases caused by mutations in specific genes [13]. Studies have identified clear links between sporadic and familial PD cases, which are clinically and pathologically indistinguishable except for age at onset [14]. Functional studies of genes involved in familial PD have led to a better understanding of the mechanisms and molecular pathways underlying PD pathophysiology that may govern/influence progressive neurodegeneration [15,16,17]. Mutations in one such gene, DJ-1 (also known as PARK7), associate with an early-onset recessive form of Parkinsonism [18]. DJ-1 was initially described as an oncogene, but additional functions for the DJ-1 protein include roles as an antioxidant via free-radical scavenging, a transcriptional regulator of antioxidant genes, a redox-dependent molecular chaperone, a modulator of mitochondrial function, a deglycase, and as a factor involved in proteolysis and metabolism [19,20,21,22]. Also, a study encountered an over-oxidized and inactive form of the DJ-1 protein in brains of sporadic PD patients [23], strongly suggesting that results obtained in animal and cell models of familial PD based on loss of DJ-1 function may also have relevance to the sporadic form of the disease [24].
Drosophila has recently emerged as an essential tool in the study of neurodegenerative diseases due to the presence of a complex central nervous system (CNS), a blood–brain barrier (BBB), and similar neurotransmitters to humans [25,26,27]. Furthermore, more than 70% of human disease-related genes display conservation in flies [28], including orthologs of genes involved in familial PD cases (e.g., DJ-1, PINK1, PRKN, or LRRK2) [29]. Flies harboring mutations in such genes exhibit PD-related phenotypes and have allowed the identification of potentially pathogenic mechanisms and modifiers of PD pathology through genetic or pharmacological approaches [30]. Our previous studies revealed that flies carrying mutations in DJ-1β (Drosophila ortholog of the human DJ-1 gene) exhibited shortened lifespans, motor defects, high OS levels, and hypersensitivity to OS-inducing toxins [31, 32]. Interestingly, we also discovered that supplementation with antioxidant compounds efficiently suppressed some of these phenotypes, thereby confirming Drosophila as an amenable model organism to identify and validate new drugs with therapeutic potential in PD patients [31,32,33]. Supporting this assumption, we subsequently performed a pilot screen to evaluate the effect of antioxidant, anti-inflammatory, and neuroprotective compounds in DJ-1β mutant flies. Excitingly, selected compounds that attenuated motor defects associated with loss of DJ-1β function also increased the viability of DJ-1-deficient human neuroblastoma cells subjected to OS conditions [34], thereby supporting the translatability of pharmacological studies carried out in Drosophila. The conservation of the BBB in flies increases the probability of encountering novel and relevant therapeutic compounds to treat PD and other human diseases [27, 35].
The present study aimed to identify drugs as novel treatment options for PD through an in vivo high-throughput screening (HTS) assay in a Drosophila PD model (DJ-1β inactivating mutation) using compounds from the Prestwick® Chemical Library (PCL). We identified drugs that attenuated motor defects in DJ-1β mutant flies and then further evaluated the potential neuroprotective effect of candidates in DJ-1-deficient human neuron-like cells. Among the compounds identified, zaprinast (ZAP) treatment displayed the most significant reduction in OS-induced cell death. We found that ZAP suppressed multiple disease phenotypes in fly and human cell PD models based on DJ-1 deficiency, and exhibited disease-relevant mechanisms of action. Thus, ZAP covers various aspects of translational cross-species and multi-phenotype modeling, as recommended to improve drug discovery in neurodegenerative diseases [36]. Taken together, our results support ZAP as a potentially interesting therapy for PD that could exert beneficial effects in future clinical trials.
Material and Methods
Drosophila Stocks
Fly stocks employed in this study were the DJ-1βex54 strain (referred to as DJ-1β) from the J. Chung laboratory [37] and the park25 strain (referred to as park) from the A. J. Whitworth laboratory [38]. Stocks and fly crosses were cultured using standard Drosophila feed at 25 ºC unless otherwise indicated.
Drug Treatment and Climbing Assays
The effect of the 1120 compounds from the PCL, which are dissolved in 100% dimethyl sulfoxide (DMSO) at a final concentration of 5 mM, on the locomotor ability of DJ-1β mutant flies was evaluated. To do this, 40 L2 stage larvae were cultured in tubes (100 × 16 mm, SARSTEDT) with 1 ml of standard food containing 0.2% DMSO or supplemented with each compound at a final concentration of 10 µM. After eclosion, adult male and females flies were transferred to new tubes, and a climbing assay performed five days later using a protocol adapted from a previous study [34]. Briefly, flies were divided into 2–4 groups, depending on the amount of individuals hatching in that period. Subsequently, each group of flies was transferred to graduated plastic tubes, acclimated for 1 min, gently tapped down to the tube bottom, and allowed to climb for 10 s. This process was recorded and repeated four times for each group of flies. Climbing ability was determined as the average of the height reached by each group of flies. Compounds that improved locolomotor activity of flies with a P value below 0.05 were selected as positive candidates. All drugs were screened blindly. For further experiments, a new batch of zaprinast was obtained (Santa Cruz Biotechnology) and a 100 mM stock dissolved in DMSO was prepared.
We used a protocol adapted from a previous study for climbing assays in park mutants [38]. Briefly, 5 tubes (75 × 23.5 mm, SARSTEDT) with 80 L2 stage larvae each were cultured with 2.5 ml of standard food containing 0.1% DMSO or supplemented with 10 µM zaprinast. After hatching, homozygous park male flies were transferred to new tubes, and a climbing assay was performed 3 to 4 days later. Briefly, groups of 10–15 flies were acclimated for 1 min in new vials, gently tapped down to the tube bottom, and allowed to climb. We counted the number of flies that crossed a line drawn at 7 cm from the vial's bottom in 10 s. This process was recorded and repeated three times for each group of flies.
Cell Culture and Drug Treatment
DJ-1-deficient and pLKO.1 control SH-SY5Y neuron-like cells previously generated by our laboratory [34] were cultured in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) supplemented with 10% fetal bovine serum, 1% non-essential amino acids, and 100 mg/ml penicillin/streptomycin at 37 °C and 5% CO2. All cell culture materials were purchased from Biowest. Viability of cells treated with candidate compounds, the GPR35 antagonist CID2745687 and DMSO as vehicle was evaluated using an MTT (3-(4, 5-dimethylthiazol-2-yl)-2–5-diphenyltetrazolium bromide) assay, as previously described [22]. To determine whether CID2745687 was able to interfere with the neuroprotective effect of zaprinast, cells were pretreated for 2 h with different concentrations of the compound before the addition of zaprinast. Next, viability assays were carried out as described [22]
Quantification of Protein Carbonyl Group Formation and H2O2 Levels
Protein carbonylation and H2O2 levels were measured in 5-day-old DJ-1β mutant flies that were treated with the vehicle (0.1% DMSO) as control or treated with 10 μM zaprinast. Protein carbonyl groups were measured in fly extracts using 2,4-dinitrophenyl hydrazine derivatization in 96-well plates (Greiner 96-well plate, polypropylene) as previously described [22]. H2O2 levels were measured using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen) in fly extracts as previously described [34]. All experiments were carried out using three biological replicates and three technical replicates for each sample.
Western Blotting
Protein extraction and Western blots of pLKO.1 and DJ-1-deficient SH-SY5Y cells treated with 1 µM zaprinast and vehicle (0.1% DMSO) under OS conditions were carried out as previously described [22]. The primary antibodies used were anti-Akt, anti-phospho-Akt (Ser473), anti-JNK, and anti-phospho-JNK (Thr183/Tyr185) (1:1000, Cell Signaling). Secondary antibodies used were anti-rabbit or anti-mouse HRP-conjugated (1:5000, Sigma). Quantifications of protein levels were performed with an ImageQuant™ LAS 4000mini Biomolecular Imager (GE Healthcare), and images were analyzed with ImageJ software (NIH).
Mitochondrial Viability
Mitochondrial viability assays were performed using the MitoTracker™ Red FM (Invitrogen) fluorescence dye in DJ-1-deficient and control SH-SY5Y cells. Briefly, 50 × 104 cells were seeded on glass coverslips in P60 cell culture dishes and incubated overnight. Next, the cell culture medium was removed, and fresh medium supplemented with 1 μM zaprinast or 0.1% DMSO was added and cells incubated for 24 h. Subsequently, the cell culture medium was removed, and cells were incubated in 60 nM Mitotracker™ Red FM diluted in incomplete cell culture medium for 30 min. Finally, cells were washed three times with PBS, and coverslips were mounted onto microscope slides with Vectashield (mounting medium) with DAPI (Vector Laboratories). Fluorescence images were acquired using fluorescence microscopy (Leica DMI3000 B) and analyzed with ImageJ software (NIH).
Enzymatic Assays
The enzymatic activities of enolase (Eno; EC 4.2.1.11), phosphofructokinase (Pfk; EC 2.7.1.11), pyruvate kinase (Pk; EC 2.7.1.40), and hexokinase (Hk; EC 2.7.1.1) were measured using coupled enzymatic assays in extracts of pLKO.1 and DJ-1-deficient SH-SY5Y cells treated with 1 µM zaprinast or vehicle (0.1% DMSO) for 24 h as previously described [22]. All experiments were performed in triplicate.
Quantification of ATP Levels
ATP levels in SH-SY5Y cells were measured using the ATP Determination Kit (Invitrogen). One day prior to the assay, 10,000 pLKO.1 or DJ-1-deficient cells were seeded in a white 96-well plate and incubated for 24 h with 1 µM zaprinast or with 0.1% DMSO as vehicle medium. Subsequently, they were incubated with 50 µM of H2O2 for 3 h, and later 100 µl of the ATP assay mix was added. Luminescence intensity was measured using an Infinite 200 PRO reader (Tecan). An MTT assay was also performed in order to calculate cell viability. All experiments were performed in triplicate and results are expressed as relative luminescence intensity per cell viability.
Statistical Analyses
The significance of differences between means was assessed using a t-test when two experimental groups were analyzed. In experiments in which more than two experimental groups were used, the statistical analysis was made using the ANOVA test and Tukey’s post hoc test. Differences were considered significant when P < 0.05. Data are expressed as means ± standard deviation (s.d.).
Results
A High-Throughput Chemical Screen in DJ-1β Mutant Flies and Validation in DJ-1-Deficient Human Cells
HTS assays for drug discovery carried out in cell models do not account for anatomical integrated physiological factors that impact a drug’s ability to interact with its target or modify the response to target engagement. Results obtained using in vivo preclinical models provide a much more unambiguous indication of the potential pharmacological effects of a drug in humans [39, 40]. To identify novel treatments for PD, we performed an in vivo HTS assay using a Drosophila PD model (DJ-1β inactivation) and compounds from the PCL library, which are dissolved in 100% DMSO at a final concentration of 5 mM. The assay's rationale was that drugs able to suppress the motor deficits observed in DJ-1β mutant flies, a phenotype resembling a classic PD symptom [2], may also display benefits when tested in DJ-1-deficient cells [34]. Figure 1a provides an overview of the screening strategy used in this study.

A High-throughput chemical screen and drug validation. a Scheme of the screening procedure used in the current study. Each screening stage (depicted by an arrow) led to several positive-hit compounds taken to the next stage. b Motor performance of DJ-1ß mutant flies obtained during the primary HTS for the 1120 evaluated drugs analyzed by climbing assays. Results are normalized to data obtained in DJ-1ß mutants treated with vehicle (DMSO). See “Material and Methods” section for details. c The viability of DJ-1-deficient cells was measured by MTT assays in the presence of OS (induced with 100 µM H2O2) and treated with the ten selected compounds. Results are normalized to data obtained in vehicle-treated DJ-1-deficient cells (DMSO). Error bars show s.d. from three independent biological replicates (*P < 0.05; **P < 0.01; ***P < 0.001)
First, we evaluated the effect of the 1120 PCL drugs on the locomotor ability of DJ-1β mutants by culturing L2 larvae in media supplemented with each compound at a concentration of 10 μM during development and 5 days after fly eclosion, and climbing assays were performed at that age (see “Material and Methods”). Among the drugs evaluated, 138 significantly improved the locomotor performance of DJ-1β mutant flies (Fig. 1a, b). We then evaluated the ability of these compounds to reduce OS-induced death in DJ-1-deficient SH-SY5Y cells [34]. We discovered that 19 of the 138 compounds identified in the screen performed in PD model flies displayed neuroprotective effects in DJ-1-deficient cells (validation step, Fig. 1a). We discarded 5 molecules after an in-depth literature review of their properties and adverse reactions given the description of Parkinsonism or motor dysfunctions among side effects in humans (Fig. 1A). At this point, we retested 8 drugs from the PCL not identified to be beneficial for PD model flies in the primary screen but with a mechanism of action similar to the 14 remaining drugs in the final list of potential therapeutic compounds (hit-expansion step, Fig. 1a). Finally, we carried out a revalidation step with the 22 leads in DJ-1-deficient cells and selected 10 candidate compounds that exhibited clear beneficial effects in cell viability assays (Fig. 1a, c).
Encouragingly, this list included two compounds (clonidine and bumetanide) currently under evaluation in Phase II clinical trials in PD patients (NCT03552068 and NCT03899324, respectively), which supports the relevance of results obtained in the HTS assay. We also identified meclizine, an FDA-approved antiemetic drug recently shown to attenuate PD-related phenotypes in fly and human cell models based on DJ-1 inactivation by increasing glycolysis [22]. Other compounds belonged to different chemical categories (such as phosphodiesterase (PDE) inhibitors or corticosteroids) prescribed for various therapeutic indications (anti-hypertensive, anti-inflammatory, or antibiotic compounds). As zaprinast (ZAP) reduced OS-induced death in DJ-1 mutant cells by the most significant degree (Fig. 1c), we selected this compound for further analyses. This drug is a phosphodiesterase (PDE) inhibitor, and can also activate the GPR35 orphan G-protein coupled receptor [41, 42]. Interestingly, it was previously reported that ZAP restores striatal long-term depression in mice transgenic for A53T-SNCA [43], and that it attenuated L-DOPA-induced dyskinesia in genetic and toxin-induced models of PD in mice and rats [44, 45] by inhibiting PDEs in both cases. Therefore, we assessed the effect of ZAP supplementation in our fly and human cell PD models to determine any disease-modifying potential.
Zaprinast Suppresses PD-Related Phenotypes in DJ-1ß and parkin Mutant Flies
The DJ-1 protein plays an essential role in the defense against OS through several pathways [46]. Accordingly, PD models based on DJ-1-deficiency present high OS levels [32, 33, 47], which are clearly associated with PD pathology [3]. We previously reported that DJ-1ß mutant flies exhibited increased levels of reactive oxygen species (ROS) and protein carbonylation (a consequence of high ROS levels) compared to control flies of the same age [32,33,34, 47]. Thus, we evaluated whether ZAP supplementation suppressed those phenotypes in DJ-1β mutant flies. Our results revealed that DJ-1ß mutants treated with ZAP during development and 5 days after eclosion presented a mild but significant reduction in H2O2 production (a component of the total ROS pool) compared to flies treated with vehicle (Fig. 2a). Consistently, we also observed a significant reduction in protein carbonylation after ZAP supplementation (Fig. 2b). Therefore, ZAP treatment in PD model flies based on DJ-1β deficiency was able to reduce OS levels, which have been shown to have a causative role in their motor deficits [34]. Of note, our preliminary results established that ZAP is also able to suppress motor defects in park mutant flies (Fig. 2c), another Drosophila model of familial PD [38]. Considering that no ortholog of human GPR35 does exist in the Drosophila genome, ZAP is probably exerting its beneficial effect in PD model flies through its PDE inhibitor activity. Indeed, Drosophila PDE1, PDE6, and PDE11 function were already shown to be sensitive to ZAP treatment [48]

Effect of zaprinast on PD model flies. a H2O2 levels in DJ-1β mutant flies treated with 10 μM ZAP were analyzed using the Amplex H2O2 Red Kit (Invitrogen). b Protein carbonylation levels in DJ-1ß mutants treated with 10 μM ZAP were analyzed by absorbance. In all cases, data were expressed as arbitrary units (a.u.) per mg of protein, and results were referred to data obtained in flies cultured in vehicle medium (DMSO). c Motor performance of y,w control flies and park mutant flies treated with vehicle (DMSO) or 1 µM ZAP was evaluated performing a climbing assay. Error bars show s.d. from at least three replicates and three independent experiments (*P < 0.05; ***P < 0.001)
Zaprinast Activates Akt Signaling and Downregulates the JNK Pathway in DJ-1-deficient human cells
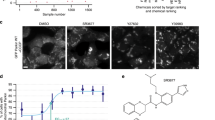
We next determined any neuroprotective effect of ZAP in DJ-1-deficient cells (as shown in Fig. 1c). First, we evaluated the ability of ZAP exposure to protect against the OS-induced death of DJ-1-deficient human neuroblastoma cells in a dose-dependent manner by pretreating them with ZAP (0.1–80 µM) and measuring viability via MTT assays. Our results demonstrated that ZAP significantly attenuated cell death at low concentrations (between 0.1 and 10 µM), with 1 µM the most effective (Fig. 3a). As shown in Fig. S1, ZAP did not affect viability in control cells at those concentrations.

Effect of zaprinast on viability and Akt/JNK pathway activities in DJ-1-deficient cells. a MTT assays measured the viability of DJ-1-deficient cells in the presence of OS (induced with 100 µM H2O2). Cells were either treated with vehicle (DMSO) or with ZAP (0.1–80 μM). Results were normalized to data obtained in vehicle-treated mutant cells (-). b, c Antibodies against Akt, p-Akt, JNK, and p-JNK were used to detect proteins of interest in DJ-1-deficient cells subjected to OS and treated with 1 μM ZAP by Western blot (upper panels). The relative ratios of p-Akt/Akt and p-JNK/JNK were analyzed by densitometry (lower panels). Results are referred to data obtained in vehicle-treated DJ-1-deficient cells and expressed as arbitrary units (a.u.). In all cases, error bars show s.d. from three independent experiments in which three biological replicates were used (*P < 0.05; ***P < 0.001)
Apoptosis has been identified as an important mechanism that leads to neuronal death in PD [49]. This process is highly regulated by the activity of several kinases like the pro-survival factor Akt and the pro-apoptotic factor JNK [50]. Previous studies revealed that DJ-1 knockdown in human neuroblastoma SH-SY5Y cells and rat adrenal pheochromocytoma PC12 cells decreased Akt phosphorylation, thus suppressing the Akt signaling pathway. Besides, it also increased JNK phosphorylation, thus overactivating JNK signaling pathway and promoting cell death [51, 52]. Analysis of Akt and JNK phosphorylation levels in our DJ-1-deficient cells confirmed the presence of similar molecular alterations (Fig. S2). Since ZAP showed a neuroprotective effect in DJ-1-deficient cells, we performed Western blot assays to determine whether pretreatments with 1 µM ZAP affected Akt or JNK phosphorylation levels in such cells. Phosphorylation of Akt at Ser473 and total Akt levels were measured in mutant cells pretreated with ZAP. Our results showed that ZAP supplementation was able to significantly increase Akt phosphorylation (Fig. 3b), which leads to its activation [51]. Besides, mutant cells pretreated with ZAP also displayed a significant reduction of JNK phosphorylation at Thr183 and Tyr185 (Fig. 3c), leading to its inactivation [51].
Taken together, these results support the neuroprotective effect of ZAP supplementation in DJ-1-deficient cells through the modification of Akt and JNK pathway activation. Previous studies reported that several PDE inhibitors exhibited anti-apoptotic properties through Akt pathway activation [53, 54]. Furthermore, neuroprotection mediated by pamoic acid, a potent GPR35 agonist, in stroke associates with an increase in Akt phosphorylation [55]; therefore, ZAP may exert its neuroprotective effect in PD model cells based on DJ-1 deficiency either by inhibiting PDEs or by activating GPR35.
Zaprinast Increases Mitochondrial Viability in DJ-1-Deficient Human Cells
Mitochondrial dysfunction has been related to PD pathogenesis [56]. Accordingly, previous studies have revealed that DJ-1 knockdown decreases active mitochondrial mass and alters mitochondrial morphology and function [51, 57, 58]. Mitochondrial alterations in DJ-1-deficient cells could be associated with the inhibition of Akt pathway and with JNK activation [51, 59], which have both been proposed as novel therapeutic targets for PD [51]. As ZAP supplementation led to Akt activation and JNK inhibition (Fig. 3b, c), we hypothesized that these changes might improve mitochondrial viability. We discovered that DJ-1 mutant cells displayed a significant decrease in the active mitochondrial mass compared to pLKO.1 control cells using the MitoTracker™ Red FM dye, as evidenced by a reduction in fluorescence intensity (Fig. 4). As expected, pretreatment of DJ-1-deficient cells with ZAP resulted in a significant increase in the number of viable mitochondria compared to vehicle-treated cells (Fig. 4), supporting the therapeutic potential of ZAP as a means to ameliorate PD-associated mitochondrial dysfunction.

Effect of zaprinast on mitochondrial activity in DJ-1-deficient cells. a Representative images of SH-SY5Y cells stained with MitoTracker Red FM, a specific mitochondrial dye, and the nuclear dye DAPI (blue) acquired via fluorescence microscopy. Cells stained were pLKO.1 control cells and DJ-1-deficient cells pretreated with vehicle (DMSO), and DJ-1-deficient cells treated with 1 µM ZAP. Scale bar, 50 µm. b Graphical representation of Mitotracker Red FM fluorescence quantification from a. At least ten images of each strain and condition were analyzed. Results are normalized to data obtained in vehicle-treated pLKO.1 control cells and expressed as arbitrary units (a.u.). Error bars show s.d. from three independent experiments in which three biological replicates were used (***P < 0.001)
Zaprinast Enhances the Activity of Key Glycolytic Enzymes in DJ-1-Deficient Human Cells
It has been recently shown that PD is also characterized by metabolic alterations [22, 60, 61]. In fact, lack of DJ-1 leads to an enhancement of the glycolytic pathway, probably as a means to counteract the reduction of ATP levels caused by mitochondrial dysfunction [22, 62]. Despite this, it has been reported that primary midbrain cultures from DJ-1 mice embryos still showed reduced ATP levels when compared to wild-type controls [63]. The glycolytic rate can be evaluated by measuring the activity of key enzymes involved in this pathway [64], and we recently reported that DJ-1-deficient cells presented higher activities of hexokinase (Hk), phosphofructokinase (Pfk), enolase (Eno), and pyruvate kinase (Pk) when compared to vehicle-treated cells [22]. Furthermore, several studies have demonstrated that enhancing glycolysis may represent a promising target for PD treatment [22, 65]. Therefore, we evaluated whether ZAP treatment affected glycolysis in DJ-1-deficient cells by quantifying the activity of these key glycolytic enzymes. Interestingly, our results demonstrated that ZAP supplementation led to a significant and robust increase in Hk and Eno activity, and to a mild increase of Pk activity in DJ-1-deficient cells (Fig. 5A). According to this, we found that this enhancement of glycolysis resulted in an increase in ATP levels in ZAP-treated DJ-1 mutant cells compared to those treated with vehicle (Fig. 5b). These results confirm our previous hypothesis that the increase in the glycolytic pathway is aimed to recover ATP levels [22] which are reduced in DJ-1-deficient cells when compared to controls (Fig. 5b). Our results also showed that ZAP treatment led to increase Eno and Pfk activities in control cells (Fig. S3), although no significant changes in ATP levels were found in such cells (Fig. 5b). Taken together, these results indicate that ZAP may contribute to enhance glycolysis and to restore ATP levels in PD model cells. A previous study had suggested that inhibition of cGMP PDE isoforms may control the ability of ZAP to increase insulin-mediated glucose uptake in skeletal muscles [66]; moreover, GPR35 has been also shown to promote glycolysis by activating Na/K-ATPase activity [67].

Effect of zaprinast on the activity of glycolytic enzymes and on ATP levels in DJ-1-deficient cells. a The activity of hexokinase (Hk), phosphofructokinase (Pfk), enolase (Eno), and pyruvate kinase (Pk) in DJ-1-deficient cells treated with 1 µM ZAP under OS condition induced with 50 µM H2O2. Results were normalized to data obtained in vehicle-treated cells (DMSO). In all cases, error bars show s.d. from three replicates and three independent experiments (*P < 0.05; **P < 0.01; ***P < 0.001). b ATP levels in DJ-1-deficient cells in the presence of OS (induced with 50 µM H2O2) treated with vehicle (DMSO) or with 1 μM ZAP were analyzed using the ATP Determination Kit (Invitrogen). Results were normalized to data obtained in vehicle-treated pLKO.1 cells (DMSO). Error bars show s.d. from three independent experiments in which three biological replicates were used (**P < 0.01; ***P < 0.001)
The GPR35 Antagonist CID2745687 Diminishes the Neuroprotective Effect of Zaprinast in DJ-1-Deficient Human Cells
As previously mentioned, ZAP is a PDE inhibitor but also a GPR35 agonist. While PDE inhibitors have been already proposed as possible PD therapies [68], very little is known about the effect of GPR35 agonists in PD models. Therefore, we decided to evaluate whether GPR35 activation could exert any neuroprotective effect in our human cell PD model. First, we confirmed by RT-qPCR analyses that the GPR35 gene was mildly expressed in SH-SY5Y cells (data not shown), as reported in [69]. Subsequently, we tested whether supplementation with kynurenine (KYN) could affect the viability of DJ-1-deficient cells. KYN is the precursor of kynurenic acid (KYNA) [70], which may represent the endogenous GPR35 ligand [71]. Consistently, our results showed that KYN treatment displayed a mild but significant neuroprotective effect in DJ-1-deficient cells (Fig. 6a), thus indicating that GPR35 agonists may represent potential candidates for PD treatment. Therefore, we hypothesized that ZAP could exert its neuroprotective effect in PD model cells through GPR35 activation. To confirm this assumption, we tested whether CID2745687 (methyl-5-[(tert-utylcarbamothioylhydrazinylidene)methyl]-1-(2,4-difluorophenyl)pyrazole-4-carboxylate), the only well-characterized GPR35 antagonist [72], could interfere with ZAP-mediated neuroprotection. We found that viability of ZAP-treated DJ-1-deficient cells was significantly reduced when pretreated with increasing concentrations of CID2745687 (Fig. 6b). In contrast, viability was not affected by CID2745687 treatment in DJ-1-deficient or control cells (Fig. S4). In summary, our results indicate that ZAP is able to reduce degeneration of PD model cells in part through GPR35 activation thus suggesting that GPR35 agonists could be considered as possible PD therapies.

Effect of kynurenic acid and CID2745687 in DJ-1-deficient cells. a MTT assays measured the viability of DJ-1-deficient cells in the presence of OS (induced by 100 µM H2O2). Cells were either treated with vehicle (DMSO) or with 10 µM kynurenic acid. b MTT assays measured the viability of DJ-1-deficient cells in the presence of OS (induced with 100 µM H2O2) treated with 1 µM ZAP. Cells were either pretreated with different concentrations of CID2745687 (0.1–10 µM) or with vehicle (-). Results were normalized to data obtained in vehicle-treated mutant cells. Error bars show s.d. from three independent biological replicates (*P < 0.05; ***P < 0.001)
Discussion
As PD remains an incurable disease and the fastest growing neurological disorder globally [73], we urgently need to develop novel, effective therapeutic strategies. With this aim, we performed an HTS assay using an in vivo Drosophila PD model to evaluate the effect of a range of compounds on a behavioral phenotype. To our knowledge, this study represents the first HTS assay carried out in adult flies that evaluates motor performance. We screened the PCL, a library that contains 1120 compounds most of them approved by regulatory agencies (FDA and EMA), and tested for bioavailability and safety in humans. Hence, our results may allow drug repurposing, a new trend in drug discovery by which new therapeutic applications for an existing drug are found, accelerating their potential for use in PD patients [74]. We identified ten compounds that improved locomotor activity in DJ-1ß mutant flies and increased viability in DJ-1-deficient cells subjected to OS. We focused on the non-commercialized drug ZAP as the most efficient candidate compound regarding improvements to the viability of PD model cells. We also identified compounds that reduced motor defects in DJ-1β mutant flies but had no effect on the viability of DJ-1-deficient cells during the validation step. While some could represent false positives, others could target other PD-relevant cell types, such as glial cells [75]; therefore, different validation assays with additional PD model cells may confirm their therapeutic potential.
ZAP is an inhibitor of the PDE1, 5, 6, 9, and 11 isoforms and a GPR35 agonist [41, 42], and studies have highlighted the expression of both PDE1 and GPR35 in the midbrain [68, 69]. PDE inhibitors have been proposed as PD therapies, evaluated in many preclinical studies, and patented as possible anti-PD drugs [68]. These compounds act by repressing the degradation of cAMP and/or cGMP, cyclic nucleotides involved in many CNS processes [68, 76]. Interestingly, alterations in cAMP and cGMP synthesis/degradation may lead to the onset of age-related diseases, including PD [76]. One of the multiple functions of both cyclic nucleotides is the regulation of the apoptosis-related Akt and JNK signaling pathways [53, 68, 77]. Here, we report that ZAP exerted a beneficial function in DJ-1-deficient SH-SY5Y cells by reducing stress-induced apoptosis through the activation of Akt and inhibition of JNK signaling [51, 52]. Therefore, these results agree with the function of ZAP as a PDE inhibitor. Indeed, its beneficial effect in PD model flies is probably exerted by this mechanism since no GPR35 orthologs are present in the Drosophila genome.
In contrast, we know relatively little about the GPR35 orphan receptor and its relation to PD, and ZAP could be acting not only as a PDE inhibitor but also as a GPR35 activator in our PD model human cells Studies have suggested a potential role for GPR35 in regulating neuronal excitability and synaptic release [78, 79] and controlling inflammation and the immune system [72, 80]. ZAP also inhibits N-type calcium channels through GPR35 activation [81], which have been found hyperactivated and overexpressed in an α-syn mouse model and linked to axonal degeneration [82]. These findings along with our results with ZAP led us to hypothesize that the pharmacological activation of GPR35 could represent a novel therapeutic approach to PD. Supporting this assumption, we found that KYN, the precursor of the endogenous GPR35 ligand KYNA [71], displayed a mild but significant neuroprotective effect in DJ-1-deficient cells. Consistently, we found that pretreatment of PD model cells with the bona fide GPR35 antagonist, CID2745687, was able to partially abolish the neuroprotective effect of ZAP in such cells. Taken together, these results indicate that GPR35 agonists may represent potential candidates for PD treatment. Interestingly, studies have demonstrated alterations to the KYN pathway (a reduction of KYNA levels) in PD patients with L-DOPA-induced dyskinesia [83]. Furthermore, while endogenous KYNA exhibits neuroprotective activity, its metabolites present neurotoxicity [84]; therefore, the KYN pathway may represent a promising target in the search for PD treatments. Accordingly, the inhibition of enzymes related to the degradation of KYNA also inhibits neurodegeneration in fly models of diseases, including PD [85].
This study also demonstrated that ZAP exerted its beneficial effect in PD models through different mechanisms. Mitochondrial alterations and high OS levels play an important role in PD development as well as in other neurodegenerative diseases [86, 87] and are strongly related to energy metabolism [60, 88]. Recent studies have highlighted the role of metabolic alterations in PD [22, 60, 61], with many genes involved in familial PD functionally linked to mitochondria (e.g., DJ-1, PRKN, or PINK1) [56]. Mitochondrial dysfunction leads to increased OS levels and reduced ATP production in PD models, which are counteracted by an increased glycolytic rate [22, 89, 90]. Interestingly, increasing glycolysis has been recently described as a potential therapeutic strategy for PD [22, 65]. Here, we confirmed the reduction in mitochondrial viability in DJ-1-deficient cells [51], which may increase OS accompanied by alterations in energy metabolism [88]. We demonstrated that ZAP supplementation increases mitochondrial viability in DJ-1-deficient cells, which in turn would cause a reduction in OS levels [90]. Furthermore, DJ-1-deficient cells pretreated with ZAP also displayed enhanced glycolysis, which led to an increase in ATP levels. As a result, ZAP exerted a protective effect in fly and human cell PD models based on DJ-1 deficiency by intervening in several cellular alterations with a pivotal role in PD.
Despite current efforts to encounter novel treatments for PD and the proposal of a considerable amount of compounds as potential therapeutics [29, 91], PD remains an incurable disease [73]. Candidate compounds obtained in preclinical models often fail when evaluated in clinical trials due to various factors, such as disease and patient heterogeneity, inadequate trial design, inappropriate endpoints, and poor patient selection. Our knowledge regarding PD physiopathology also remains incomplete [91, 92], thereby contributing to the selection of non-optimal candidate compounds in preclinical studies. For this reason, the identification of compounds that target multiple phenotypes in several disease models may allow for improved drug discovery in neurodegenerative diseases [36]. This study demonstrated that ZAP exerts its beneficial function in PD models through different disease-modifying mechanisms in PD models based on DJ-1 dysfunction. Given the discovery of over-oxidized and inactive DJ-1 protein in sporadic PD patients [23], we hypothesize that therapeutic compounds identified in DJ-1 models could find use in individuals with sporadic PD.
In summary, we identify ZAP as a potential therapeutic compound for PD using an HTS assay in a Drosophila model. PD is a multifactorial disorder [93]; therefore, a multi-therapy approach may be required to treat PD patients efficiently. In this sense, ZAP represents a promising drug, given its function through both PDEs and GPR35 [41, 42], thus widening the therapeutic landscape. Although our results clearly demonstrate that the neuroprotective effect of ZAP in PD model cells is reduced by pretreatments with the GPR35 antagonist CID2745687, further studies are required to validate GPR35 agonists as novel therapies for PD. This study also validates Drosophila as a valuable model organism with huge potential in the drug discovery field, which may lead to the identification of novel therapies for PD and other human diseases.
References
Andrejack J, Mathur S. What People with Parkinson’s Disease Want. J. Parkinsons. Dis. IOS Press BV; 2020. p. S5–10.
Prell T, Witte OW, Grosskreutz J. Biomarkers for Dementia, Fatigue, and Depression in Parkinson’s Disease. Front Neurol. 2019;10:1–13.
Dauer W, Przedborski S. Parkinson’s disease: Mechanisms and models. Neuron. Cell Press; 2003. p. 889–909.
Scott L, Dawson VL, Dawson TM. Trumping neurodegeneration: Targeting common pathways regulated by autosomal recessive Parkinson’s disease genes. Exp. Neurol. Academic Press Inc.; 2017. p. 191–201.
Kaur R, Mehan S, Singh S. Understanding multifactorial architecture of Parkinson’s disease: pathophysiology to management. Neurol Sci. Springer Milan; 2019;40:13–23.
Anandhan A, Jacome MS, Lei S, et al. Metabolic Dysfunction in Parkinson’s Disease: Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. Brain Res. Bull. NIH Public Access; 2017. p. 12–30.
Athauda D, Foltynie T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat. Rev. Neurol. Nature Publishing Group; 2015. p. 25–40.
Oertel WH. Recent advances in treating Parkinson’s disease. F1000Research. Faculty of 1000 Ltd; 2017;6:260.
Stoker TB, Torsney KM, Barker RA. Emerging treatment approaches for Parkinson’s disease. Front. Neurosci. Frontiers Media S.A.; 2018. p. 693.
Camilleri A, Vassallo N. The Centrality of mitochondria in the pathogenesis and treatment of Parkinson’s disease. CNS Neurosci. Ther. Blackwell Publishing Ltd; 2014. p. 591–602.
M. Wilkins H, K. Morris J. New Therapeutics to Modulate Mitochondrial Function in Neurodegenerative Disorders. Curr Pharm Des. Bentham Science Publishers Ltd.; 2017;23:731–52.
McFarthing K, Buff S, Rafaloff G, Dominey T, Wyse RK, Stott SRW. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2020. J. Parkinsons. Dis. IOS Press; 2020. p. 757–74.
Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. Academic Press Inc.; 2020. p. 104782.
Niemann N, Jankovic J. Juvenile parkinsonism: Differential diagnosis, genetics, and treatment. Park. Relat. Disord. Elsevier Ltd; 2019. p. 74–89.
Chai C, Lim K-L. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr Genomics. Bentham Science Publishers; 2013;14:486–501.
Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Prim. Nature Publishing Group; 2017;3:1–21.
Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. Lancet Publishing Group; 2020. p. 170–8.
Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. American Association for the Advancement of Science; 2003;299:256–9.
Hijioka M, Inden M, Yanagisawa D, Kitamura Y. DJ-1/PARK7: A New Therapeutic Target for Neurodegenerative Disorders. Biol Pharm Bull Pharm Bull. 2017;40:548–52.
Sharma N, Rao SP, Kalivendi S V. The deglycase activity of DJ-1 mitigates α-synuclein glycation and aggregation in dopaminergic cells: Role of oxidative stress mediated downregulation of DJ-1 in Parkinson’s disease. Free Radic Biol Med. Pergamon; 2019;135:28–37.
Larsen SB, Hanss Z, Krüger R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. Springer; 2018;373:21–37.
Solana-Manrique C, Sanz FJ, Ripollés E, et al. Enhanced activity of glycolytic enzymes in Drosophila and human cell models of Parkinson’s disease based on DJ-1 deficiency. Free Radic Biol Med. Elsevier Inc.; 2020;158:137–48.
Repici M, Giorgini F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J Clin Med. MDPI AG; 2019;8:1377.
Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SMM. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev. Hindawi Limited; 2013;2013:683920.
Martin CA, Krantz DE. Drosophila melanogaster as a genetic model system to study neurotransmitter transporters. Neurochem. Int. Elsevier Ltd; 2014. p. 71–88.
McGurk L, Berson A, Bonini NM. Drosophila as an in vivo model for human neurodegenerative disease. Genetics. Genetics; 2015;201:377–402.
Hindle SJ, Bainton RJ. Barrier mechanisms in the Drosophila blood-brain barrier. Front. Neurosci. Frontiers Media S.A.; 2014.
Tsuda L, Lim Y-M. Alzheimer’s Disease Model System Using Drosophila. Springer, Singapore; 2018. p. 25–40.
Solana-Manrique C, Moltó MD, Calap-Quintana P, Sanz FJ, Llorens JV, Paricio N. Drosophila as a model system for the identification of pharmacological therapies in neurodegenerative diseases. Insights into Hum Neurodegener Lessons Learn from Drosoph. Springer Singapore; 2019. p. 433–67.
Paricio N, Muñoz-Soriano V. Drosophila models of Parkinson’s disease: Discovering relevant pathways and novel therapeutic strategies. Parkinsons. Dis. Hindawi Limited; 2011.
Lavara-Culebras E, Paricio N. Drosophila DJ-1 mutants are sensitive to oxidative stress and show reduced lifespan and motor deficits. Gene. Elsevier; 2007;400:158–65.
Casani S, Gómez-Pastor R, Matallana E, Paricio N. Antioxidant compound supplementation prevents oxidative damage in a Drosophila model of Parkinson’s disease. Free Radic Biol Med. Pergamon; 2013;61:151–60.
Lavara-Culebras E, Muñoz-Soriano V, Gómez-Pastor R, Matallana E, Paricio N. Effects of pharmacological agents on the lifespan phenotype of Drosophila DJ-1β mutants. Gene. Elsevier; 2010;462:26–33.
Sanz FJ, Solana-Manrique C, Muñoz-Soriano V, Calap-Quintana P, Moltó MD, Paricio N. Identification of potential therapeutic compounds for Parkinson’s disease using Drosophila and human cell models. Free Radic Biol Med. Pergamon; 2017;108:683–91.
Konieczny P, Artero R. Drosophila SMN2 minigene reporter model identifies moxifloxacin as a candidate therapy for SMA. FASEB J. John Wiley and Sons Inc.; 2020;34:3021–36.
Homberg JR, Kyzar EJ, Stewart AM, et al. Improving treatment of neurodevelopmental disorders: recommendations based on preclinical studies. Expert Opin Drug Discov. Taylor and Francis Ltd; 2016;11:11–25.
Park J, Sung YK, Cha GH, Sung BL, Kim S, Chung J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005;361:133–9.
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. National Academy of Sciences; 2003;100:4078–83.
Aldewachi H, Al-Zidan RN, Conner MT, Salman MM. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering. 2021;8:30.
Krentz AJ, Weyer C, Hompesch M. Translational research methods in diabetes, obesity, and nonalcoholic fatty liver disease: A focus on early phase clinical drug development. Transl. Res. Methods Diabetes, Obesity, Nonalcoholic Fat. Liver Dis. A Focus Early Phase Clin. Drug Dev. Springer International Publishing; 2019.
Medina AE. Therapeutic utility of phosphodiesterase type I inhibitors in neurological conditions. Front. Neurosci. Frontiers Media SA; 2011.
Taniguchi Y, Tonai-Kachi H, Shinjo K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. FEBS Lett; 2006;580:5003–8.
Tozzi A, Costa C, Siliquini S, et al. Mechanisms underlying altered striatal synaptic plasticity in old A53T-α synuclein overexpressing mice. Neurobiol Aging. Neurobiol Aging; 2012;33:1792–9.
Picconi B, Bagetta V, Ghiglieri V, et al. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain. Oxford University Press; 2011;134:375–87.
Solís O, Espadas I, Del-Bel EA, Moratalla R. Nitric oxide synthase inhibition decreases l-DOPA-induced dyskinesia and the expression of striatal molecular markers in Pitx3-/- aphakia mice. Neurobiol Dis. Academic Press Inc.; 2015;73:49–59.
Raninga P V., Di Trapani G, Tonissen KF. The multifaceted roles of DJ-1 as an antioxidant. Adv Exp Med Biol. Springer New York LLC; 2017. p. 67–87.
Yang J, Kim MJ, Yoon W, et al. Isocitrate protects DJ-1 null dopaminergic cells from oxidative stress through NADP+-dependent isocitrate dehydrogenase (IDH). PLoS Genet. Public Library of Science; 2017;13.
Day JP, Dow JAT, Houslay MD, Davies S-A. Cyclic nucleotide phosphodiesterases in Drosophila melanogaster. Biochem J. Portland Press Ltd; 2005;388:333.
Erekat NS. Apoptosis and its Role in Parkinson’s Disease. Park Dis Pathog Clin Asp. Codon Publications; 2018. p. 65–82.
Ambacher KK, Pitzul KB, Karajgikar M, Hamilton A, Ferguson SS, Cregan SP. The JNK- and AKT/GSK3β- Signaling Pathways Converge to Regulate Puma Induction and Neuronal Apoptosis Induced by Trophic Factor Deprivation. Hetman M, editor. PLoS One. Public Library of Science; 2012;7:e46885.
Zhang XL, Wang ZZ, Shao QH, et al. RNAi-mediated knockdown of DJ-1 leads to mitochondrial dysfunction via Akt/GSK-3ß and JNK signaling pathways in dopaminergic neuron-like cells. Brain Res Bull. Elsevier Inc.; 2019;146:228–36.
Dhanasekaran DN, Premkumar Reddy E. JNK-signaling: A multiplexing hub in programmed cell death. Genes and Cancer. Impact Journals LLC; 2017. p. 682–94.
Peixoto CA, Nunes AKS, Garcia-Osta A. Phosphodiesterase-5 inhibitors: Action on the signaling pathways of neuroinflammation, neurodegeneration, and cognition. Mediators Inflamm. Hindawi Publishing Corporation; 2015.
Joshi R, Kadeer N, Sheriff S, Friend LA, James JH, Balasubramaniam A. Phosphodiesterase (PDE) inhibitor torbafylline (HWA 448) attenuates burn-induced rat skeletal muscle proteolysis through the PDE4/cAMP/EPAC/PI3K/Akt pathway. Mol Cell Endocrinol. Elsevier Ireland Ltd; 2014;393:152–63.
Sharmin O, Abir AH, Potol A, et al. Activation of GPR35 protects against cerebral ischemia by recruiting monocyte-derived macrophages. Sci Rep. Nature Research; 2020;10:1–13.
Park JS, Davis RL, Sue CM. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. Current Medicine Group LLC 1; 2018.
Andres-Mateos E, Perier C, Zhang L, et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. National Academy of Sciences; 2007;104:14807–12.
Krebiehl G, Ruckerbauer S, Burbulla LF, et al. Reduced Basal Autophagy and Impaired Mitochondrial Dynamics Due to Loss of Parkinson’s Disease-Associated Protein DJ-1. Petrucelli L, editor. PLoS One. Public Library of Science; 2010;5:e9367.
Heslop KA, Rovini A, Hunt EG, et al. JNK activation and translocation to mitochondria mediates mitochondrial dysfunction and cell death induced by VDAC opening and sorafenib in hepatocarcinoma cells. Biochem Pharmacol. Elsevier Inc.; 2020;171:113728.
Anandhan A, Jacome MS, Lei S, et al. Metabolic Dysfunction in Parkinson’s Disease_ Bioenergetics, Redox Homeostasis and Central Carbon Metabolism. 2017;
Shukla AK, Ratnasekhar C, Pragya P, et al. Metabolomic Analysis Provides Insights on Paraquat-Induced Parkinson-Like Symptoms in Drosophila melanogaster. Mol Neurobiol. Humana Press Inc.; 2016;53:254–69.
Requejo-Aguilar R, Lopez-Fabuel I, Jimenez-Blasco D, Fernandez E, Almeida A, Bolaños JP. DJ1 represses glycolysis and cell proliferation by transcriptionally upregulating pink1. Biochem J. NIH Public Access; 2015;467:303.
Chen R, Park H-A, Mnatsakanyan N, et al. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. Nature Publishing Group; 2019;10.
Teslaa T, Teitell MA. Techniques to monitor glycolysis. Methods Enzymol. Academic Press Inc.; 2014. p. 91–114.
Cai R, Zhang Y, Simmering JE, et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J Clin Invest. American Society for Clinical Investigation; 2019;129:4539–49.
Genders AJ, Bradley EA, Rattigan S, Richards SM. cGMP phosphodiesterase inhibition improves the vascular and metabolic actions of insulin in skeletal muscle. Am J Physiol - Endocrinol Metab. American Physiological Society Bethesda, MD; 2011;301:E342–50.
Schneditz G, Elias JE, Pagano E, et al. GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump. Sci Signal. American Association for the Advancement of Science; 2019;12:9048.
Nthenge-Ngumbau DN, Mohanakumar KP. Can Cyclic Nucleotide Phosphodiesterase Inhibitors Be Drugs for Parkinson’s Disease? Mol. Neurobiol. Springer; 2018. p. 822–34.
The Human Protein Atlas [Internet]. Available from: https://www.proteinatlas.org/
Fukuwatari T. Possibility of amino acid treatment to prevent the psychiatric disorders via modulation of the production of tryptophan metabolite kynurenic acid. Nutrients. MDPI AG; 2020.
Cosi C, Mannaioni G, Cozzi A, et al. G-protein coupled receptor 35 (GPR35) activation and inflammatory pain: Studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology. Pergamon; 2011;60:1227–31.
Quon T, Lin L-C, Ganguly A, Tobin AB, Milligan G. Therapeutic Opportunities and Challenges in Targeting the Orphan G Protein-Coupled Receptor GPR35. Cite This ACS Pharmacol Transl Sci. 2020;2020:812.
Vacca VM. Parkinson disease. Nursing (Lond). NLM (Medline); 2019;49:24–32.
Gasparini F, Di Paolo T. Drug repurposing: Old drugs, new tricks to fast track drug development for the brain. Neuropharmacology. Elsevier Ltd; 2019. p. 1–3.
Domingues A V., Pereira IM, Vilaça-Faria H, Salgado AJ, Rodrigues AJ, Teixeira FG. Glial cells in Parkinson´s disease: protective or deleterious? Cell. Mol. Life Sci. Springer Science and Business Media Deutschland GmbH; 2020. p. 5171–88.
Kelly MP. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell. Signal. Elsevier Inc.; 2018. p. 281–91.
Zhang J, Wang Q, Zhu N, et al. Cyclic AMP inhibits JNK activation by CREB-mediated induction of c-FLIPL and MKP-1, thereby antagonizing UV-induced apoptosis. Cell Death Differ. Cell Death Differ; 2008;15:1654–62.
Mackenzie AE, Milligan G. The emerging pharmacology and function of GPR35 in the nervous system. Neuropharmacology. Elsevier Ltd; 2017. p. 661–71.
Reggio PH, Shore DM. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front Pharmacol. Frontiers Media S.A.; 2015;6.
Divorty N, Mackenzie AE, Nicklin SA, Milligan G. G protein-coupled receptor 35: an emerging target in inflammatory and cardiovascular disease. Front Pharmacol. Frontiers Media S.A.; 2015;6:41.
Guo J, Williams DJ, Puhl HL, Ikeda SR. Inhibition of N-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J Pharmacol Exp Ther. J Pharmacol Exp Ther; 2008;324:342–51.
Sgobio C, Sun L, Ding J, Herms J, Lovinger DM, Cai H. Unbalanced calcium channel activity underlies selective vulnerability of nigrostriatal dopaminergic terminals in Parkinsonian mice. Sci Rep. Nature Publishing Group; 2019;9.
Havelund JF, Andersen AD, Binzer M, et al. Changes in kynurenine pathway metabolism in Parkinson patients with L-DOPA-induced dyskinesia. J Neurochem. Blackwell Publishing Ltd; 2017;142:756–66.
Venkatesan D, Iyer M, Narayanasamy A, Siva K, Vellingiri B. Kynurenine pathway in Parkinson’s disease—An update. eNeurologicalSci. Elsevier B.V.; 2020. p. 100270.
Breda C, Sathyasaikumar K V., Idrissi SS, et al. Tryptophan-2,3-dioxygenase (TDO) inhibition ameliorates neurodegeneration by modulation of kynurenine pathway metabolites. Proc Natl Acad Sci U S A. National Academy of Sciences; 2016;113:5435–40.
Grünewald A, Kumar KR, Sue CM. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. Elsevier Ltd; 2019. p. 73–93.
Pozo Devoto VM, Falzone TL. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? DMM Dis. Model. Mech. Company of Biologists Ltd; 2017. p. 1075–87.
Bolaños JP, Moro MA, Lizasoain I, Almeida A. Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: Therapeutic implications. Adv. Drug Deliv. Rev. Elsevier; 2009. p. 1299–315.
Requejo-Aguilar R, Bolaños JP. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radic. Biol. Med. Elsevier Inc.; 2016. p. 123–37.
Giachin G, Bouverot R, Acajjaoui S, Pantalone S, Soler-López M. Dynamics of human mitochondrial complex I assembly: Implications for neurodegenerative diseases. Front. Mol. Biosci. Frontiers Media S.A.; 2016. p. 43.
Elkouzi A, Vedam-Mai V, Eisinger RS, Okun MS. Emerging therapies in Parkinson disease — repurposed drugs and new approaches. Nat. Rev. Neurol. Nature Publishing Group; 2019. p. 204–23.
Athauda D, Foltynie T. Challenges in detecting disease modification in Parkinson’s disease clinical trials. Park. Relat. Disord. Elsevier Ltd; 2016. p. 1–11.
Pang SYY, Ho PWL, Liu HF, et al. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. BioMed Central Ltd.; 2019. p. 1–11.
Acknowledgements
We are grateful to Dr. Chung and Dr. A. J. Whitworth for providing fly stocks and to Stuart P. Atkinson for English editing.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work was supported by the University of Valencia [grant number UV-INV-AE17-702300 to N.P.].
Author information
Authors and Affiliations
Contributions
Disclosure forms provided by the authors are available with the online version of this article.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sanz, F.J., Solana-Manrique, C., Torres, J. et al. A High-Throughput Chemical Screen in DJ-1β Mutant Flies Identifies Zaprinast as a Potential Parkinson’s Disease Treatment. Neurotherapeutics 18, 2565–2578 (2021). https://doi.org/10.1007/s13311-021-01134-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01134-2