
Abstract
Generalised pustular psoriasis (GPP), a severe neutrophilic skin disease characterised by the sudden and widespread eruption of superficial sterile pustules, remains a challenging disease with limited treatment options. The recent discovery of genetic mutations associated with GPP and advances in understanding of the molecular mechanisms of autoinflammation have resulted in identification of key cytokines that drive the development and progression of GPP. Accumulating evidence demonstrates that interleukin (IL)-36 acts as a central node cytokine by orchestrating the hyperactivation of key pro-inflammatory cytokines and stimulating immune cells, including neutrophilic accumulations, a unique feature of GPP skin lesions. These findings are paving the way for the discovery and development of novel targeted GPP therapeutics that block the IL-36 pathway and neutralise the pathogenic immunologic mechanisms and pro-inflammatory cytokines. This article provides an overview of the current evidence that supports the role of IL-36 as a central node cytokine in GPP pathogenesis.
Video Abstract (MP4 1789 KB)
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Generalised pustular psoriasis (GPP) is a severe neutrophilic skin disease characterised by the sudden and widespread eruption of superficial sterile pustules with or without systemic inflammation and is considered a distinct disease from plaque psoriasis |
Gene mutations in the interleukin (IL)-36 cytokine family, such as loss-of-function mutations in the IL-36 receptor antagonist and overexpression of IL-36, have been discovered in patients with GPP. Such mutations drive an inflammatory cascade leading to the activation and accumulation of immune cells, particularly neutrophils in the skin |
Advances in understanding of the underlying molecular mechanisms of GPP pathogenesis and clinical validation of novel pharmacological targets establish IL-36 as a central node that drives the development and progression of GPP |
Innovative therapies targeting IL-36 for the treatment of GPP and other autoinflammatory skin disorders, that were historically considered unmet medical needs, are in development |
Digital Features
This article is published with digital features, including a video abstract and a full Japanese translation, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.19552333.
Introduction
Generalised pustular psoriasis (GPP) is a severe neutrophilic skin disease characterised by the sudden and widespread eruption of superficial sterile pustules with or without systemic inflammation [1]. Patients with GPP may or may not have a history of plaque psoriasis. The clinical course of the disease can be relapsing or persistent. GPP flares can be life-threatening if not treated due to severe systemic complications [2].
GPP is considered a distinct disease from plaque psoriasis. Typically, psoriasis is classified into several subtypes: plaque, inverse, guttate, erythrodermic and pustular psoriasis. In addition to these subtypes, certain patients with psoriasis may experience psoriatic arthritis, an inflammatory arthritis that may be associated with arthritis of the joints, enthesitis, dactylitis, psoriasis and nail involvement [3]. GPP is a form of pustular psoriasis [4, 5]. Due to the association between GPP and plaque psoriasis, GPP is classified as GPP alone or GPP with plaque psoriasis [6]. Most GPP cases that are not associated with plaque psoriasis (GPP alone) are mainly due to the presence of homozygous or compound heterozygous mutations of IL36RN, which encodes the interleukin (IL)-36 receptor antagonist (IL-36Ra). A small number of patients with GPP with plaque psoriasis have IL36RN mutations [7]. According to the Japanese guidelines for the management and treatment of GPP, a definitive diagnosis of GPP can be established based on the recurrence of systemic symptoms and multiple sterile pustules that could merge, forming lakes of pus and neutrophilic subcorneal pustules that are characterised histopathologically as spongiform pustules of Kogoj [8].
With advances in understanding of the molecular basis of GPP pathogenesis, the term “autoinflammatory keratinisation diseases” has been recently proposed to refer to diseases with mixed pathologic mechanisms that involve autoinflammation and autoimmunity and are linked to IL-36Ra-related pustulosis, CARD14-mediated psoriasis, pityriasis rubra pilaris type V and familial keratosis lichenoides chronica. GPP without plaque psoriasis can be considered an autoinflammatory keratinisation disease [9, 10].
This article provides an overview of the role of IL-36 in the pathogenesis of GPP and its function as a central node that orchestrates the pathogenic role of downstream cytokines. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Treatment of GPP
There are no approved GPP-specific treatments in the USA and Europe. Furthermore, the evidence that supports the current therapies is not well established and is mainly based on case reports and small open-label non-randomised trials [2, 11]. In Japan, several biologics are currently approved for the treatment of GPP, including the tumour necrosis factor (TNF)-blocking agents adalimumab, infliximab and certolizumab pegol; the monoclonal antibodies that antagonise IL-17/IL-17R, secukinumab, brodalumab and ixekizumab; and the IL-23 inhibitors risankizumab and guselkumab. In Taiwan and Thailand, brodalumab is the only approved biologic [8, 12,13,14,15,16,17,18]. A summary of current treatment options for GPP in Japan is provided in Table 1.
The Role of Cytokines in GPP Pathogenesis
The pathogenic mechanisms of GPP are not clear. Recent studies identified several mutations linked to GPP, including those in IL36RN, CARD14, AP1S3 and MPO (Table 2). These genes are involved in the regulation of key inflammatory and immune pathways, most notably the IL‐1/IL‐36–chemokines–neutrophil pathogenic axis [6, 19]. Additionally, a rare loss-of-function mutation was identified in SERPINA3, which encodes serine protease inhibitor A3 (serpin A3) that inhibits several proteases in patients with GPP. Specifically, the heterozygous deletion c.966delT/p.Tyr322Ter in two patients with GPP was confirmed by Sanger sequencing. This rare variant was significantly associated with GPP and may impact the inhibitory effect of serpin A3 on cathepsin G, which is involved in activation of IL-36 and downstream pro-inflammatory cytokines [20].
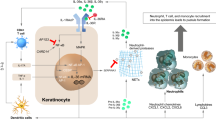
Recent findings further confirmed the role in GPP of sustained activation of IL-1 and IL-36, which induces neutrophil chemokine expression, infiltration and pustule formation [21]. The IL-36 subfamily of cytokines is part of the IL-1 superfamily. It is composed of three pro-inflammatory agonists, IL-36α, IL-36β and IL-36γ, which activate the IL-36 receptor (IL-36R), and one antagonist (IL-36Ra). IL-36 cytokines play a key role in signalling between epithelial cells, dendritic cells and neutrophils, which constitute the central cytokine node responsible for the initiation, continuation and exacerbation of inflammation [22].
The IL-1 ligand family members are present as a single cluster on human chromosome 2 and are likely to be formed through a series of gene duplications of the prototypical IL-1 family cytokine IL-1β. IL-36 is a member of the IL-1 family, which includes IL-1β, IL-1α, IL-36α, IL-36β, IL-36γ, IL-36Ra, IL-37, IL-38 and IL-1Ra (Table 3) [23]. These findings are supported by a study by Yamamoto et al., which demonstrated that the levels of serum cytokines IL-1β, IL-1Ra, IL-6, IL-10, IL-12p70, IL-18, IL-22, interferon (IFN)-γ and vascular endothelial growth factor were positively correlated with GPP clinical markers, including severity scores of GPP, white blood cell counts and serum C-reactive protein levels [24]. The activation of IL-36 cytokines requires proteolytic cleavage by proteases released from activated neutrophils. Cathepsin G, elastase and proteinase-3 differentially process and activate all three IL-36 family members. Therefore, neutrophil-derived proteases can amplify inflammation through the processing of extracellular cytokines (Fig. 1) [25]. These observations were further confirmed by a study by Clancy et al. that showed that proteases released by activated neutrophils into the extracellular space are potent modulators of IL-1α, IL-1β, IL-33, IL-36α, IL-36β and IL-36γ activation states. Thus, neutrophils play a key role in modulating inflammatory responses through the processing of multiple IL-1 family cytokines (Fig. 1) [26].

Several studies have established IL-36 as a central node in GPP pathogenesis. The sustained unopposed activation of IL-36R leads to activation of the transcription factor NFκB, which results in massive production of several key inflammatory cytokines, including CXCL8, TNF-α, IL-1 and IL-23 [21, 27,28,29]. Moreover, the serum and mRNA levels of IL-17 are substantially higher in pustular psoriasis [30]. The pathogenic role of IL-17A, the most potent member of the IL-17 family, was confirmed by the demonstrated efficacy of several IL-17A inhibitors in patients with GPP (Fig. 2). [15, 31].

IL-36 is a central node in GPP pathogenesis. IL-36 acts as a central node cytokine in the pathogenesis of GPP. The overexpression or unopposed activation of IL-36 due to IL36RN mutations results in hyperactivation of the IL-36 pathway in GPP [9, 10]. GPP, generalised pustular psoriasis; IFN, interferon; IL, interleukin; Th17, T-helper type 17; TNF, tumour necrosis factor
IL-36 Familial Mutations and GPP
The first observation of the familial incidence of GPP was described in 1972 by Landry and Muller. In this report, a patient, his maternal uncle, another uncle and grandmother experienced symptoms of GPP with disease onset early in childhood. The symptoms were aggravated by streptococcal infections [32]. More recently, a significant linkage to an interval of 1.2 megabases on chromosome 2q13-q14.1 and a homozygous missense mutation in IL36RN was identified in patients with GPP, which further confirms the clinical relevance of the IL-36 pathway in the pathogenesis of GPP [33]. Homozygous and heterozygous IL36RN mutations were also discovered in cases of impetigo herpetiformis, a rare pustular dermatosis that affects pregnant women and shares clinical and histologic features of GPP [6]. A recent case report by Sawabe et al. described the diagnosis of a patient with GPP with co-existing mutations in IL36RN and CARD14, suggesting that concurrent mutations in IL36RN and CARD14 may also be a predisposing factor of GPP [34]. Another report further confirmed the potential role of the IL-36 pathway. Exosome sequencing of five unrelated patients with GPP revealed that loss of function of IL36RN constitutes the genetic basis of GPP and implicates innate immune dysregulation in this severe episodic inflammatory disease, thereby highlighting IL-1 signalling as a potential therapeutic target [35].
The unopposed IL-36 signalling hyperactivation stimulates antigen-mediated and possibly pathogenic T-helper type 17 (Th17) responses in GPP, promoting antigen-driven Th17 responses, which in the absence of exogenous triggers may trigger autoimmune reactions [36]. The central role of the IL-36 pathway in driving the pathogenesis of GPP was further confirmed by the development of IL-36 pathway inhibitors (e.g. spesolimab and imsidolimab). Efficacy of the anti-IL36R monoclonal antibody spesolimab has been shown in an open-label proof-of-concept trial in seven patients with a GPP flare and in a randomised, placebo-controlled double-blind trial (Effisayil™ 1) in 53 patients with a GPP flare, suggesting that the IL-36 pathway may play a pathogenic role among patients with GPP of broad genetic backgrounds [37]. An interim analysis from an open-label trial of imsidolimab (also an anti-IL36R monoclonal antibody) in eight patients with GPP also supports IL-36 as a central player in the pathogenesis of GPP [38].
IL-36 as a Central Node in GPP Pathogenesis
Recent evidence demonstrated that IL-36 signalling plays a key role in the development of psoriatic lesions based on in vivo pre-clinical studies in transgenic and knockout mice. IL-36 knockout mice (IL-36R−/−:K5.Stat3C mice) had a downregulated gene expression of psoriasis-related cytokines, including IL-23/IL-17 axis cytokines. Additionally, IL-36-deficient keratinocytes were resistant to stimulation by d-glucan, which usually upregulates the expression of IL-36 members, S100A8, S100A9 and IL-17C, in vitro. These findings established that IL-36 signalling plays a role as a gatekeeper that potentially links innate immunity to the pathogenesis of psoriasis [39]. These observations were further supported by a study by Wang et al. [40], which showed that IL-36γ inhibits differentiation and induces inflammation of keratinocytes via the Wnt signalling pathway in psoriasis. These findings are clinically relevant. A retrospective immunohistochemical study of psoriatic lesions of 40 patients with psoriasis and matched controls demonstrated that IL-36γ was strongly expressed (in four or more layers) in the nuclei of suprabasal epidermal keratinocytes, while, in all controls, weak cytoplasmic expression was detected in the basal layer. Additionally, the elevated IL-36γ expression was significantly associated with psoriasis severity [41].
IL-36 cytokines play a central role in the recruitment and activation of neutrophils and Th17 cells in psoriatic skin. IL-36 cytokines induce chemokines, and cytokines that interfere with differentiation/cornification programmes in the epidermis and promote pathological angiogenesis and endothelial cell activation. IL-36 cytokines are upregulated in the psoriatic epidermis, and their expression is strongly induced by TNF-α and IL-17 [42]. These findings can be translated into the discovery of novel therapies for GPP. A small-molecule high-throughput screen identified A-552, but not the closely related family member IL-36α, as a potent antagonist of human IL-36γ. A-552 suppressed IL-36γ-induced responses in mouse and human disease models of psoriasis [43]. In addition to the pathogenic role of IL-36 in the skin, its potential role in the extracutaneous disease manifestations associated with GPP and plaque psoriasis was demonstrated in recent studies. Patients with GPP had a prominent IFN type-I signature, which correlates with abnormal IL-36 activity [44]. Pre-clinical in vivo studies in a mouse model of deficiency of IL-36 receptor antagonist (DITRA) also demonstrated an essential role for the IL-36 pathway in the pro-inflammatory responses in the skin and epithelial barrier function in the intestine [45].
Moreover, recent findings in mice showed that the activation of toll-like receptor 4 (TLR4) signalling is attenuated by IL-37, an antagonist of IL-1 family cytokines [46]. Shibata et al. demonstrated that, in IL36RN−/− mice (a model of autoinflammatory syndromes associated with DITRA), blocking TLR4 signalling using the TLR4 antagonist TAK-242 resulted in the resolution of the cutaneous, articular and hepatic autoinflammatory symptoms, which further highlights the central role of the IL-36 pathway and demonstrates a novel therapeutic strategy to treat GPP [46, 47].
Role of the IL-36 Pathway in Other Skin Disorders
The IL-1 family members IL-36α (IL-1F6), IL-36β (IL-1F8) and IL-36γ (IL-1F9) and the receptor antagonist IL-36Ra (IL-1F5) play a central role in regulating inflammatory skin activity. Furthermore, IL-36α-treated monocyte-derived dendritic cells (MO-DCs) enhanced allogeneic CD4+ T-cell proliferation, demonstrating that IL-36 can stimulate the maturation and function of DCs and drive T-cell proliferation. These data indicate that IL-36 cytokines actively propagate skin inflammation via activation of keratinocytes, antigen-presenting cells (APCs) and, indirectly, T cells [48]. IL-36 also seems to play a role in infectious dermatoses. Microbial triggers, especially Staphylococcus aureus infection, increase the production of pro-inflammatory IL-36 cytokines and initiate/promote the inflammation of skin lesions [29].
The enhanced expression of IL-36 family members has been shown to play a key role in the pathogenesis of Netherton syndrome, a rare autosomal recessive skin disease caused by loss-of-function mutations in SPINK5, which encodes lympho-epithelial Kazal-type-related inhibitor protein that results in the unopposed activity of epidermal kallikrein-related peptidases (KLKs), mainly KLK5, KLK7 and KLK14 [49].
In addition to its role in psoriasis, the role of IL-36 has been investigated in the pathogenesis of allergic contact dermatitis (ACD). Gene expression of all three IL-36 agonists, but not IL-36Ra, was enhanced in ACD-affected skin. In addition, an ex vivo model showed that the addition of recombinant IL-36Ra in skin explants causes the reduction of IL-36α, IL-36β and IL-36γ gene expression [50].
The role of neutrophil extracellular traps, web-like structures composed of neutrophil DNA, in the pathogenesis of psoriasis is not clear; however, recent studies suggest that neutrophil extracellular traps are induced in a psoriasis model of IL-36Ra-deficient mice, which suggests a potential role in the pathology of psoriasis-like lesions [51]. Mutations in MPO, which encodes the neutrophilic enzyme myeloperoxidase (MPO), that result in MPO deficiency in neutrophils and monocytes were found to be associated with GPP [6]. MPO-deficient mouse and human cells are characterised by altered neutrophil function and impaired clearance of neutrophils by monocytes (efferocytosis), promoting prolonged neutrophil persistence in inflamed skin [6]. These findings suggest a potential role in other neutrophil-driven diseases such as Sweet syndrome (acute febrile neutrophilic dermatosis) and pyoderma gangrenosum, as MPO deficiency has been described in a single individual with pyoderma gangrenosum [52]. The potential role of MPO mutations in other inflammatory skin conditions was further confirmed by the findings of MPO screening in conditions phenotypically related to GPP, which further revealed disease alleles in one subject with acral pustular psoriasis and in two individuals with acute generalised exanthematous pustulosis. A subsequent analysis of UK Biobank data demonstrated that the c.20312A>C and c.1705C>T (p.Arg569Trp) disease alleles were also associated with increased neutrophil abundance in the general population [53].
Animal studies also demonstrated a central role for IL-36Ra in wound healing. In IL36–/– mice, wound healing was delayed; however, treatment with a TLR4 antagonist normalised the impact on wound healing [54]. Moreover, in IL36–/– mice, cutaneous ischaemia–reperfusion injury was exacerbated; however, treatment with a TLR4 antagonist or PAD4 inhibitor normalised the impact on cutaneous ischaemia–reperfusion injury [55]. Additionally, recent studies in animal models suggest a potential role of IL-36 in the pathogenesis of ACD based on the finding that IL-36Ra deficiency might be involved in T-cell priming in the lymph nodes [56]. Further studies in the well-established 1-fluoro-2,4-dinitrobenzene mouse model of contact hypersensitivity also demonstrated that IL-36α, IL-36β and IL-36γ did not impact disease phenotype, and their role may be limited to the amplification of priming and/or inflammatory responses [56].
Discussion
GPP remains a challenging disease with few well-validated treatment options. The recent advances in understanding of the molecular mechanisms in inflammatory diseases and the characterisation of genetic mutations associated with GPP have demonstrated novel pharmacological targets to manage and treat GPP. Among the most notable mutations discovered in patients with GPP is the loss of function of IL36RN, a gene that encodes IL-36Ra. The unopposed activation of IL-36 pathway has established IL-36, a central node in the pathogenesis of GPP, as a critical pharmacological target for the treatment of GPP. Targeting IL-36 has been validated clinically in a proof-of-principle clinical trial of the anti-IL-36R monoclonal antibody spesolimab, which demonstrated efficacy within 1 week of treatment in patients with GPP flares [37]. An interim analysis of an open-label trial of imsidolimab (also an anti-IL36R monoclonal antibody) in eight patients with GPP also supports the central role of IL-36 in the pathogenesis of GPP [38].
Collectively, advances in understanding of the underlying molecular mechanisms of GPP pathogenesis and the clinical validation of novel pharmacological targets establish IL-36 as a central node that drives the development and progression of GPP (Fig. 2). The secretion of IL-36 by keratinocytes or inflammatory cells, and the subsequent stimulation of autocrine and paracrine pathways leads to an amplified autoinflammatory and autoimmune response mediated by several key cytokines, including IL-8, CXCL1, CXCL2, CCL20, IL-12, IL-1β, IL-23, IL-6 and TNF-α. These cytokines further activate T cells, leading to the secretion of IL-22, IL-17 and IFN-γ. Additionally, chemotaxis of activated neutrophils establishes GPP as a neutrophilic skin disorder as these further amplify the inflammatory mechanism through neutrophil-mediated cleavage and activation of pro-inflammatory cytokines.
The emerging role of the IL-36 pathway in other autoinflammatory skin disorders further confirms IL-36 as a central node that orchestrates a pathogenic autoinflammatory mechanism. Accumulating evidence implicates IL-36 cytokines in the activation of keratinocytes and APCs, and the proliferation of T cells [48]. These findings provide a plausible explanation for the susceptibility of a patient with IL36RN mutations to triggers of flares such as Staphylococcus aureus infections. IL-36 cytokines have been also found to play a role in the pathogenesis of the rare inflammatory skin disease Netherton syndrome [49]. Furthermore, IL-36 cytokines were found to play a potential role in the pathogenesis of ACD [50]. These discoveries may lead to the identification of novel treatments for ACD and rare conditions that are driven by neutrophil extracellular traps such as acute febrile neutrophilic dermatosis and pyoderma gangrenosum. Further characterisation of the regulatory mechanisms of IL-36 cytokines in wound healing mechanisms, such as the role of TLR4, may also lead to the development of novel pharmacological treatments beyond targeting IL-36R.
Conclusion
In summary, recent advances in understanding of the genetic and molecular basis of GPP pathogenesis, including the discovery of IL-36 as a central node, are driving the development of innovative treatments for GPP and other autoinflammatory skin disorders that were historically considered unmet medical needs.
Change history
03 December 2022
Caption (description) of the Supplementary file changed from “Supplementary file1” to “Supplementary file – Japanese translation”
03 December 2022
A peer-reviewed video abstract was retrospectively added to this publication.
References
Navarini AA, Burden AD, Capon F, et al. European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol. 2017;31(11):1792–9.
Gooderham MJ, Van Voorhees AS, Lebwohl MG. An update on generalized pustular psoriasis. Expert Rev Clin Immunol. 2019;15(9):907–19.
Leung YY, Ogdie A, Orbai AM, et al. Classification and outcome measures for psoriatic arthritis. Front Med (Lausanne). 2018;5:246.
Bachelez H. Pustular psoriasis: the dawn of a new era. Acta Derm Venereol. 2020;100(3):adv00034.
Benjegerdes KE, Hyde K, Kivelevitch D, Mansouri B. Pustular psoriasis: pathophysiology and current treatment perspectives. Psoriasis (Auckl). 2016;6:131–44.
Zhou J, Luo Q, Cheng Y, Wen X, Liu J. An update on genetic basis of generalized pustular psoriasis (review). Int J Mol Med. 2021;47(6):118.
Sugiura K, Oiso N, Iinuma S, et al. IL36RN mutations underlie impetigo herpetiformis. J Invest Dermatol. 2014;134(9):2472–4.
Fujita H, Terui T, Hayama K, et al. Japanese guidelines for the management and treatment of generalized pustular psoriasis: the new pathogenesis and treatment of GPP. J Dermatol. 2018;45(11):1235–70.
Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases. J Allergy Clin Immunol. 2017;140(6):1545–7.
Akiyama M, Takeichi T, McGrath JA, Sugiura K. Autoinflammatory keratinization diseases: an emerging concept encompassing various inflammatory keratinization disorders of the skin. J Dermatol Sci. 2018;90(2):105–11.
Hoegler KM, John AM, Handler MZ, Schwartz RA. Generalized pustular psoriasis: a review and update on treatment. J Eur Acad Dermatol Venereol. 2018;32(10):1645–51.
Imafuku S, Honma M, Okubo Y, et al. Efficacy and safety of secukinumab in patients with generalized pustular psoriasis: a 52-week analysis from phase III open-label multicenter Japanese study. J Dermatol. 2016;43(9):1011–7.
Saeki H, Nakagawa H, Ishii T, et al. Efficacy and safety of open-label ixekizumab treatment in Japanese patients with moderate-to-severe plaque psoriasis, erythrodermic psoriasis and generalized pustular psoriasis. J Eur Acad Dermatol Venereol. 2015;29(6):1148–55.
Sano S, Kubo H, Morishima H, Goto R, Zheng R, Nakagawa H. Guselkumab, a human interleukin-23 monoclonal antibody in Japanese patients with generalized pustular psoriasis and erythrodermic psoriasis: efficacy and safety analyses of a 52-week, phase 3, multicenter, open-label study. J Dermatol. 2018;45(5):529–39.
Wilsmann-Theis D, Schnell LM, Ralser-Isselstein V, et al. Successful treatment with interleukin-17A antagonists of generalized pustular psoriasis in patients without IL36RN mutations. J Dermatol. 2018;45(7):850–4.
Yamasaki K, Nakagawa H, Kubo Y, Ootaki K, Japanese Brodalumab Study G. Efficacy and safety of brodalumab in patients with generalized pustular psoriasis and psoriatic erythroderma: results from a 52-week, open-label study. Br J Dermatol. 2017;176(3):741–51.
McKeage K, Duggan S. Risankizumab: first global approval. Drugs. 2019;79(8):893–900.
Choon SE, Lebwohl MG, Marrakchi S, et al. Study protocol of the global Effisayil 1 Phase II, multicentre, randomised, double-blind, placebo-controlled trial of spesolimab in patients with generalized pustular psoriasis presenting with an acute flare. BMJ Open. 2021;11(3):e043666.
Sugiura K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants. J Dermatol Sci. 2014;74(3):187–92.
Frey S, Sticht H, Wilsmann-Theis D, et al. Rare Loss-of-function mutation in SERPINA3 in generalized pustular psoriasis. J Invest Dermatol. 2020;140(7):1451–5.
Johnston A, Xing X, Wolterink L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol. 2017;140(1):109–20.
Murrieta-Coxca JM, Rodriguez-Martinez S, Cancino-Diaz ME, Markert UR, Favaro RR, Morales-Prieto DM. IL-36 cytokines: regulators of inflammatory responses and their emerging role in immunology of reproduction. Int J Mol Sci. 2019;20(7):1649.
Rivers-Auty J, Daniels MJD, Colliver I, Robertson DL, Brough D. Redefining the ancestral origins of the interleukin-1 superfamily. Nat Commun. 2018;9(1):1156.
Yamamoto M, Imai Y, Sakaguchi Y, Haneda T, Yamanishi K. Serum cytokines correlated with the disease severity of generalized pustular psoriasis. Dis Markers. 2013;34(3):153–61.
Henry CM, Sullivan GP, Clancy DM, Afonina IS, Kulms D, Martin SJ. Neutrophil-derived proteases escalate inflammation through activation of IL-36 family cytokines. Cell Rep. 2016;14(4):708–22.
Clancy DM, Sullivan GP, Moran HBT, et al. Extracellular neutrophil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep. 2018;22(11):2937–50.
Goldstein JD, Bassoy EY, Caruso A, et al. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci Alliance. 2020;3(6):e202000688.
Song HS, Kim SJ, Park TI, Jang YH, Lee ES. Immunohistochemical comparison of IL-36 and the IL-23/Th17 axis of generalized pustular psoriasis and acute generalized exanthematous pustulosis. Ann Dermatol. 2016;28(4):451–6.
Buhl AL, Wenzel J. Interleukin-36 in infectious and inflammatory skin diseases. Front Immunol. 2019;10:1162.
Yilmaz SB, Cicek N, Coskun M, Yegin O, Alpsoy E. Serum and tissue levels of IL-17 in different clinical subtypes of psoriasis. Arch Dermatol Res. 2012;304(6):465–9.
Plachouri KM, Chourdakis V, Georgiou S. The role of IL-17 and IL-17 receptor inhibitors in the management of generalized pustular psoriasis. Drugs Today (Barc). 2019;55(9):587–93.
Landry M, Muller SA. Generalized pustular psoriasis. Observations on the course of the disease in a familial occurrence. Arch Dermatol. 1972;105(5):711–6.
Marrakchi S, Guigue P, Renshaw BR, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365(7):620–8.
Sawabe Y, Hayashi K, Kamata M, et al. Case of generalized pustular psoriasis with coexisting mutations in IL36RN and CARD14. J Dermatol. 2019;46(10):e368–70.
Onoufriadis A, Simpson MA, Pink AE, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89(3):432–7.
Arakawa A, Vollmer S, Besgen P, et al. Unopposed IL-36 activity promotes clonal CD4(+) T-cell responses with IL-17A production in generalized pustular psoriasis. J Invest Dermatol. 2018;138(6):1338–47.
Bachelez H, Choon SE, Marrakchi S, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med. 2019;380(10):981–3.
AnaptyBio. IMSIDOLIMAB. https://www.anaptysbio.com/pipeline/imsidolimab/. 2020. Accessed 8 Dec 2021.
Ohko K, Nakajima K, Kataoka S, Takaishi M, Sano S. IL-36 signaling is essential for psoriatic inflammation through the augmentation of innate immune responses. J Invest Dermatol. 2019;139(6):1400–4.
Wang W, Yu X, Wu C, Jin H. IL-36gamma inhibits differentiation and induces inflammation of keratinocyte via Wnt signaling pathway in psoriasis. Int J Med Sci. 2017;14(10):1002–7.
Farag AGA, Marea AH, Al-Sharaky DR, Al Bahat SM. Interleukin 36 gamma in psoriasis patients an immunohistochemical study. J Egypt Women’s Dermatol Soc. 2018;15(3):172–8.
Madonna S, Girolomoni G, Dinarello CA, Albanesi C. The significance of IL-36 hyperactivation and IL-36R targeting in psoriasis. Int J Mol Sci. 2019;20(13):E3318.
Todorovic V, Su Z, Putman CB, et al. Small molecule IL-36gamma antagonist as a novel therapeutic approach for plaque psoriasis. Sci Rep. 2019;9(1):9089.
Catapano M, Vergnano M, Romano M, et al. IL-36 promotes systemic IFN-I responses in severe forms of psoriasis. J Invest Dermatol. 2020;140(4):816–26.
Hovhannisyan Z, Liu N, Khalil-Aguero S, et al. Enhanced IL-36R signaling promotes barrier impairment and inflammation in skin and intestine. Sci Immunol. 2020;5(54):eaax1686.
Shibata A, Sugiura K, Furuta Y, Mukumoto Y, Kaminuma O, Akiyama M. Toll-like receptor 4 antagonist TAK-242 inhibits autoinflammatory symptoms in DITRA. J Autoimmun. 2017;80:28–38.
Fukushima H, Iwata Y, Watanabe S, et al. TAK-242 ameliorates contact dermatitis exacerbated by IL-36 receptor antagonist deficiency. Sci Rep. 2020;10(1):734.
Foster AM, Baliwag J, Chen CS, et al. IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol. 2014;192(12):6053–61.
Gouin O, Barbieux C, Leturcq F, Bonnet-des-Claustres M, Petrova E, Hovnanian A. Transgenic kallikrein 14 mice display major hair shaft defects associated with desmoglein 3 and 4 degradation, abnormal epidermal differentiation, and IL-36 signature. J Invest Dermatol. 2020;140(6):1184–94.
Mattii M, Ayala F, Balato N, et al. The balance between pro- and anti-inflammatory cytokines is crucial in human allergic contact dermatitis pathogenesis: the role of IL-1 family members. Exp Dermatol. 2013;22(12):813–9.
Watanabe S, Iwata Y, Fukushima H, et al. Neutrophil extracellular traps are induced in a psoriasis model of interleukin-36 receptor antagonist-deficient mice. Sci Rep. 2020;10(1):20149.
Haskamp S, Bruns H, Hahn M, et al. Myeloperoxidase modulates inflammation in generalized pustular psoriasis and additional rare pustular skin diseases. Am J Hum Genet. 2020;107(3):527–38.
Vergnano M, Mockenhaupt M, Benzian-Olsson N, et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease. Am J Hum Genet. 2021;108(4):757.
Saito K, Iwata Y, Fukushima H, et al. IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells. Sci Rep. 2020;10(1):14772.
Tanaka Y, Iwata Y, Saito K, et al. Cutaneous ischemia-reperfusion injury is exacerbated by IL-36 receptor antagonist deficiency. J Eur Acad Dermatol Venereol. 2021. https://doi.org/10.1111/jdv.17767.
Zaladonis A, Zhang X, Manupipatpong KK, Kalaiselvan S, Alvarez P, Jensen LE. Interleukin-36 (IL-36) system in the 1-fluoro-2,4-dinitrobenzene (DNFB) mouse model of allergic contact dermatitis. Allergy. 2020;75(8):2078–81.
Zhou LL, Georgakopoulos JR, Ighani A, Yeung J. Systemic monotherapy treatments for generalized pustular psoriasis: a systematic review. J Cutan Med Surg. 2018;22(6):591–601.
Haustein UF, Rytter M. Methotrexate in psoriasis: 26 years’ experience with low-dose long-term treatment. J Eur Acad Dermatol Venereol. 2000;14(5):382–8.
Yan K, Xu W, Huang Y, et al. Methotrexate restores the function of peripheral blood regulatory T cells in psoriasis vulgaris via the CD73/AMPK/mTOR pathway. Br J Dermatol. 2018;179(4):896–905.
Uva L, Miguel D, Pinheiro C, et al. Mechanisms of action of topical corticosteroids in psoriasis. Int J Endocrinol. 2012;2012:561018.
Fujisawa T, Suzuki S, Mizutani Y, et al. Granulocyte and monocyte adsorption apheresis for generalized pustular psoriasis: therapeutic outcomes in three refractory patients. Ther Apher Dial. 2015;19(4):336–41.
Fu LW, Vender R. Systemic role for vitamin D in the treatment of psoriasis and metabolic syndrome. Dermatol Res Pract. 2011;2011:276079.
Wong T, Hsu L, Liao W. Phototherapy in psoriasis: a review of mechanisms of action. J Cutan Med Surg. 2013;17(1):6–12.
Ibbotson SH. A perspective on the use of NB-UVB phototherapy vs PUVA photochemotherapy. Front Med (Lausanne). 2018;5:184.
Morita A, Yamazaki F, Matsuyama T, et al. Adalimumab treatment in Japanese patients with generalized pustular psoriasis: results of an open-label phase 3 study. J Dermatol. 2018;45(12):1371–80.
Wang WM, Jin HZ. Biologics in the treatment of pustular psoriasis. Expert Opin Drug Saf. 2020;19(8):969–80.
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–61.
Sugiura K, Takemoto A, Yamaguchi M, et al. The majority of generalized pustular psoriasis without psoriasis vulgaris is caused by deficiency of interleukin-36 receptor antagonist. J Invest Dermatol. 2013;133(11):2514–21.
Sugiura K, Muto M, Akiyama M. CARD14 c.526G>C (p.Asp176His) is a significant risk factor for generalized pustular psoriasis with psoriasis vulgaris in the Japanese cohort. J Invest Dermatol. 2014;134(6):1755–7.
Mercurio L, Morelli M, Scarponi C, et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018;9(11):1104.
Cai Y, Xue F, Quan C, et al. A critical role of the IL-1beta-IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol. 2019;139(1):146–56.
Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol. 2009;124(6):1141–9.
Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015;42(6):991–1004.
Barbier L, Ferhat M, Salame E, et al. Interleukin-1 family cytokines: keystones in liver inflammatory diseases. Front Immunol. 2019;10:2014.
Acknowledgements
Funding
The author did not receive payment related to the development of the manuscript. Boehringer Ingelheim funded the journal’s Rapid Service Fees..
Medical Writing Assistance
Yasser Heakal, PhD, of OPEN Health Communications (London, UK), provided medical writing, editorial and formatting support, which was contracted and funded by Boehringer Ingelheim. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Authorship
The author meets the criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE), drafted the manuscript and made the decision to submit the manuscript for publication.
Author Contributions
KS: Concept, design, and writing of the manuscript. The author read and approved the final manuscript.
Compliance with Ethics Guidelines
This article is based on previously conducted studies as well as personal experience and does not contain any new studies with human participants or animals performed by the author.
Disclosures
The author has received consulting and/or speaker fees from AbbVie GK, Amgen Inc., Bristol Myers Squibb, Eisai Co., Ltd., Eli Lilly Japan, Janssen Pharmaceutical, Kaken Pharmaceutical CO., Ltd., Kyowa Hakko Kirin Co., Ltd., LEO Pharma Japan, Maruho Co., Ltd., Minophagen Pharmaceutical Co., Ltd., Nippon Boehringer Ingelheim Co., Ltd., Nippon Zoki Pharmaceutical Co., Ltd., Novartis Pharma, TAIHO Pharmaceutical Co., Ltd., Tsumura Co., Ltd., Sanofi, Sato Pharmaceutical Co., Ltd., Sun Pharmaceutical Industries, Torii Pharmaceutical Co., Ltd. and UCB Pharma.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Sugiura, K. Role of Interleukin 36 in Generalised Pustular Psoriasis and Beyond. Dermatol Ther (Heidelb) 12, 315–328 (2022). https://doi.org/10.1007/s13555-021-00677-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-021-00677-8