
Abstract
Introduction
Objective observational studies have shown that basal metabolic rate (BMR) decreases in patients with Alzheimer’s disease (AD), but the causal relationship between BMR and AD has not been established. We determined the causal relationship between BMR and AD by two-way Mendelian randomization (MR) and investigated the impact of factors associated with BMR on AD.
Methods
We obtained BMR (n = 454,874) and AD from a large genome-wide association study (GWAS) database (21,982 patients with AD, 41,944 controls). The causal relationship between AD and BMR was investigated using two-way MR. Additionally, we identified the causal relationship between AD and factors related with BMR, hyperthyroidism (hy/thy) and type 2 diabetes (T2D), height and weight.
Results
BMR had a causal relationship with AD [451 single nucleotide polymorphisms (SNPs), odds ratio (OR) 0.749, 95% confidence intervals (CIs) 0.663–0.858, P = 2.40E-03]. There was no causal relationship between hy/thy or T2D and AD (P > 0.05). The bidirectional MR showed that there was also a causal relationship between AD and BMR (OR 0.992, Cls 0.987–0.997, NSNPs18, P = 1.50E−03). BMR, height and weight have a protective effect on AD. Based on MVMR analysis, we found that genetically determined height and weight may be adjusted by BMR to have a causal effect on AD, not height and weight themselves.
Conclusion
Our study showed that higher BMR reduced the risk of AD, and patients with AD had a lower BMR. Because of a positive correlation with BMR, height and weight may have a protective effect on AD. The two metabolism-related diseases, hy/thy and T2D, had no causal relationship with AD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Why carry out this study? |
Objective observational studies have shown that basal metabolic rate (BMR) decreases in patients with Alzheimer's disease (AD), but the causal relationship between BMR and AD has not been established |
In this study, we examined the bidirectional causal relationship between BMR and AD using a genetically informed method |
What was learned from the study? |
Higher BMR reduced the risk of AD, and patients with AD had a lower BMR |
Our study also revealed that height and weight may have an impact on AD by influencing basal metabolic rate |
The two metabolism-related diseases, hyperthyroidism (hy/thy) and type 2 diabetes, had no causal relationship with AD |
Introduction
Alzheimer’s disease (AD) is the primary contributor to dementia and the most prevalent cause of death [1,2,3]. AD significantly decreases the quality of life for individuals and poses a significant challenge to public health and society as a whole [4, 5]. Despite extensive research, the cause of sporadic AD remains elusive. To date, there is no established effective treatment, and there are no confirmed preventive measures available [6,7,8].
The basal metabolic rate (BMR) is considered a crucial indicator of minimal metabolism required to sustain life and is a significant component of total energy expenditure. BMR is influenced by various factors such as body weight, height and health status. Numerous studies suggest that metabolic dysfunction increases the risk of AD. For instance, metabolic dysfunction in both the body and brain can contribute to the development of AD. Impaired glucose metabolism in the brain has been linked to AD and may start several years before the onset of clinical symptoms, making it an intrinsic part of AD pathogenesis [9, 10].
A number of acquired factors increase the risk of developing AD. Among those factors are diabetes and obesity [11]. Both obesity and uncontrolled diabetes are characterized by an increased BMR [12]. Due to the possibility of a long incubation period between exposure and results, randomized controlled trials, the gold standard for causal reasoning, are not feasible [13]. Confounding factors, reverse causation and measurement errors can bias observational studies [14]. Besides, the relationship between BMR and AD has not been reported. In this study, we tried to identify the risk and causal relationship between BMR and AD. This may be of value for extending prevention and treatment strategies of AD.
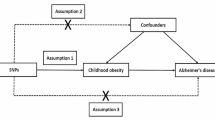
Mendelian randomization (MR) analysis is a gene-based analysis method that utilizes randomly assigned genetic variation to infer the causal effect of exposure on outcomes. MR utilizes genetic variants, such as single nucleotide polymorphisms (SNPs), as instrumental variables to reduce confounding bias in exposure and enhance causal inference of exposure-outcome associations [15, 16]. In MR analysis, SNPs, as genetic tools, must meet the following three basic assumptions: (1) the genetic variation must be truly associated with exposure; (2) genetic variation should not be associated with any confounding factors associated with exposure outcomes; (3) there is no direct link between genetic variation and outcomes [17] (Fig. 1). In this study, we determined the causal relationship between BMR and AD by two-way MR.

Directed acyclic graphs for Mendelian randomization. The directed acyclic graph presents the causal relationship among instrumental variable (SNPs), exposure (basal metabolic rate, BMR), outcome (Alzheimer's disease, AD) and confounding factors (G). Other letters indicate the direction of factors that can have an impact. ×, no influence
Methods
Genetic Variants Associated with BMR Were Obtained from GWAS
We used data from the UK Biobank, where 454,874 participants were tested for BMR and 9,851,867 SNPs were measured, which were detected in excerpt from the MR-Base platform[18]. All the participants of the datasets were of European origin. The data were analyzed and processed using the R package TwoSampleMR [18]. The extraction criteria for instrumental variables are (P < 5 × 10–8, R2 < 0.001, kb distance > 10,000). BMR in UK Biobank was calculated according to the Oxford equation in a unit of standard deviation (SD), with 1 SD = 1358.32 kilo-joule (KJ) [19]. Ethics approval and informed consent were not required for the present study, as they were obtained in the original studies. The original studies were conducted in compliance with the Declaration of Helsinki.
GWAS Summary Data on AD
The summary genetic statistics for AD were obtained from the International Genomics of Alzheimer’s Project (IGAP), comprised of four consortia, Alzheimer Disease Genetics Consortium (ADGC), Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE), The European Alzheimer’s Disease Initiative (EADI) and Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES). The dataset in IGAP was a GWAS metaanalysis of 46 case-control studies with approximately 10.5 million SNPs and 63,926 European individuals (21,982 AD cases and 41,944 cognitively normal controls) (available from the IEU GWAS database: https://gwas.mrcieu.ac.uk/) [20]. In the study population, the average age of patients with AD is 72.9 years old and that of cognitive function control group 72.4 years old, with some cases being anatomical or autopsy (Supplementary Material, Tables 3–4) [20]. Individuals with a high degree of relatedness were excluded from the analysis for all datasets, so our analysis primarily focuses on patients with sporadic AD, according to the criteria of the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the Alzheimer’s Disease and Related Disorders Association (ADRDA) [21, 22]. The screening criteria include “neuropsychological tests, advanced imaging, cerebrospinal fluid measurements, and other biological markers” [23]. AD is defined as a significant decline compared to previous levels at cognitive or/and behavioral (neuropsychiatric) aspects and involves impairment in at least two domains, such as memory, logic, visuospatial abilities and language functions. Refer to Table 1 for AD, BMR and other exposure-related information.
Other Datasets
The GWAS of standing height, weight, type 2 diabetes mellitus (T2D) and hyperthyroidism/thyrotoxicosis (hy/thy) is the same MR-Base platform excerpt with GWAS ID ukb-b-10787, ukb-b-12039, ebi-a-GCST010118 and ukb-b-20289, respectively [18].
Statistical Analysis
MR was performed using the TwoSampleMR (version 0.5.6) package in R (version 4.2.1). In the main analysis, we applied the inverse-variance weighted (IVW) MR method to estimate the associations between BMR and the risk of AD [24]. The IVW method only provides unbiased estimates when horizontal pleiotropy is balanced or absent [25]. Instrument strength is assessed by intensity and precision between genetic variation and exposure. For each SNP, the F statistic is calculated using the formula \(F={R}^{2}\times \frac{\mathrm{N}-2}{1-{R}^{2}}\) \({R}^{2}==2\times {\mathrm{beta}}^{2} \times \mathrm{ EAF}\times \frac{1-\mathrm{EAF}}{2}\times {\mathrm{beta}}^{2} \times \mathrm{EAF}\times (1-\mathrm{EAF})+2\times {\mathrm{SE}}^{2} \times \mathrm{N}\times \mathrm{ EAF }\times (1-\mathrm{EAF})\) (N represents the number of participants, EAF represents the effector allele frequency, and beta is the estimated effect of SNP) to assess its ability to independently predict outcomes. If the F statistic for the instrument-exposure association was significantly > 10, the likelihood of weak instrumental variable bias was low [15].
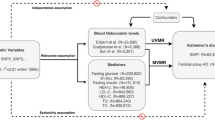
The heterogeneity among genetic instruments was evaluated by Cochran’s Q test [26]. If heterogeneity existed, a random-effect IVW model was used [27, 28]; otherwise, a fixed-effect IVW model was used. Other MR methods used as sensitivity analysis included weighted median and MR-Egger, using the “TwoSampleMR” R package, as well as MR-pleiotropy residual sum and outlier (MR-PRESSO) (with 10,000 simulations performed), using the “MR-PRESSO” R package [29]. MR-egger regression with bootstrap standard error is performed in the MR analysis to produce pleiotropic-robust causal estimates [30]. If at least 50% of the variables are valid instruments, then estimates based on median (weighted) are unbiased [31]. In addition, multiple sensitivity analyses were performed to verify the robustness of the MR test, such as MR-Egger intercept test and leave-one-out analysis. MR-Egger intercept analysis is used to evaluate directional pleiotropy. When its P value is > 0.05, directionality pleiotropy is not present. Cochran’s Q statistics were computed to test the presence of heterogeneity. We also used a two-way MR analysis method to detect whether the exposure to the outcome had a reverse causal relationship. The statistical analysis process is shown by Fig. 2. Leave-one-out sensitivity analysis did not lead to an appreciable change in the MR estimates (Supplementary Material).
Results
There was a Causal Relationship Between Alzheimer’s Disease and Basal Metabolic Rate
We used a two-sample MR method to analyze BMR with AD outcomes. The SNPs associated with BMR and AD are listed in Supplementary Tables 5 and 6. We also counted the F value of each SNP and calculated that the average F value was 81.6 and minimum value 29.9. This showed that the tool variable was not weak. In the sensitivity analysis, the two samples were heterogeneous, but there was no horizontal pleiotropy, so the results of weighted media analysis and IVW analysis were more suitable, and the P value of both detection methods was < 0.05, indicating a significant difference. The value of OR showed that each SD increase in BMR reduced the risk of AD (OR 0.752, 95% CI 0.664–0.851), while the risk of AD decreased with increasing height and weight, possibly because height and weight contribute to BMR (Table 2). This suggests that a high BMR has a protective effect on AD.
Interestingly, our reverse testing revealed that an increased risk of AD had a diminished impact on BMR (beta = − 0.7%, 95% CI − 0.011 to 0.003) (Table 3). When using MR-PRESSO detection, abnormal values were detected, which may have a greater impact on horizontal pleiotropy. After correction, it was found that the outlier-corrected P value was still < 0.05, and the “distortion test” detected P > 0.05, which mean outliers had no influence on the results.
There Was No Causal Relationship Between Hyperthyroidism or Type 2 Diabetes Mellitus and Alzheimer’s Disease
Both type 2 diabetes (T2D) and hyperthyroidism/thyrotoxicosis (hy/thy) affected BMR, so we investigated the relationship between the two diseases and AD. Results showed that neither disease had a causal relationship with AD (P > 0.05) (Table 4). BMI is associated with BMR but has been shown to be unrelated to AD risk [32]. Therefore, the screening criteria for SNPs for both diseases as exposure were the same as for BMR.
We performed an MR analysis of AD and two fundamental factors associated with BMR, body weight and height. Both weight and height decreased the risk of AD in two-sample MR analysis and reduced the risk of developing AD by influencing BMR in MVMR analysis.
Discussion
Summary
In this study, we performed univariate MR analysis using genetic variation as a non-confounding agent to explore the BMR on AD. The causal relationship was studied between disease or phenotype associated with BMR. By applying four complementary single-variable MR methods with different potential assumptions, we found evidence suggesting that a higher genetic susceptibility to BMR can reduce the risk of AD. The results of the MVMR analysis showed that height and weight may have an effect on AD by altering BMR. Furthermore, we identified some of the diseases that affect BMR, T2D and hy/thy had no causal relationship with AD. This suggested that, although the two diseases increased BMR, they may also be related to confounding factors that reduce the risk of AD. Through two-way MR analysis, we found that there is a direct causal relationship between the decline in BMR and patients with AD (the same direction of beta in the four methods). This suggested that a decrease of BMR in normal people might increase the risk of AD (Table 5).
Comparison to Previous Findings
Previous observational studies have shown that there is a correlation between human height and AD. For men, height in the highest quartile [> 179.7 cm (70.75 in)] had a 59% lower risk of developing AD that in the lowest quartile [< 169.5 cm (66.75 in)], controlling for year of birth and education (P = 0.03). For women without an APOE ε4 allele, increasing height was associated with lower risk for AD (OR 0.88; P = 0.01) [33]. This is consistent with our analysis where we speculate that genetically determined higher height may reduce the risk of AD by increasing BMR based on MVMR analysis.
Body height and weight affect BMR through body surface area. Our results showed that heavier weight reduced the risk of developing AD by influencing BMR in MVMR analysis. At present, there is no strong evidence that weight is directly linked to AD, although a low body weight may be found in AD. Some studies have shown that weight loss occurs in patients in the middle and late stages of AD [34], but other studies suggest that weight loss may occur before cognitive dysfunction [35]. This may be due to the decrease in trace elements in the body during weight loss, which increases the risk of developing AD [36,37,38]. The decrease in leptin during weight loss can also lead to a decline in cognitive function [39]. Longitudinal cohort studies have also shown that weight gain reduces the risk of AD [40].
Abnormal thyroid function and some metabolic diseases, for example, T2D, will affect the BMR, which is usually one of their diagnostic indicators. There is evidence to suggest people with T2D may be at a higher risk of developing AD [41,42,43,44], and some studies have found that treating diabetes may also help to slow the progression of AD [45, 46]. Some research suggests that people with T2D may have an increased risk of developing AD due to the effects of high blood sugar on the brain [47, 48]. High levels of blood sugar can damage blood vessels and nerve cells in the brain, which may contribute to the development of AD [49, 50]. However, the relationship between T2D and AD is complex and not fully understood. Our results suggest that there is no causal relationship between T2D and that observational studies may be due to confounders.
There is a link between hormonal imbalance and neurodegenerative diseases, e.g., clinical studies have found that some patients with AD have Cushing's syndrome (CS). CS is caused by the long-term release of excessive glucocorticoids (GCs) from the adrenal glands, which subsequently impairs brain function and induces dementia [51]. Changes in plasma thyroid hormone (TH) levels in patients with hyperthyroidism may affect neuronal function [52, 53]. Generally, the risk of thyroid dysfunction is markedly increased in older people [54]. Behavior, cognition, cerebral blood flow and glucose consumption in AD are associated with TH deficiency, but whether there is a causal relationship between thyroid dysfunction and AD is inconclusive [55]. Our results suggest that hyperthyroidism is not causally related to AD.

Bidirectional Mendelian randomization flowchart in this study
BMR represents the energy expenditure necessary to maintain basic physiological functions, including cardiac activity, respiration, conduction of nerve impulses, ion transmembrane transport and metabolic activity [56, 57]. Until now, there have been no research reports on the correlation between BMR and AD. Our present study indicated that an increase in genetically determined BMR may reduce the risk of AD.
It is well known the brain makes up only a small part of our body's total mass, but it is the largest source of energy expenditure, accounting for > 20% of total oxygen metabolism. Neurons are the most energetic cells in the brain, consuming up to 75–80% of the oxygen in the brain [58]. This in itself suggests that neurons are highly sensitive to disruptions in glucose metabolism and mitochondrial dysfunction [59]. The gradual decline in metabolic efficiency during aging renders neurons more vulnerable to toxic damage [60, 61]. If changes in energy metabolism may occur, the vulnerability of these neurons may increase before the appearance of clinical symptoms, leading to the loss of neurons [62]. In humans, low brain metabolism can cause cognitive impairment [63]. We also provide the possibility of this causal relationship here.
Currently, accumulating studies have shown that abnormal glucose metabolism in the brain is an early event before the pathological features of Aβ deposition in AD [64,65,66,67,68]. Numerous studies highlight the negative effects of Aβ on mitochondrial function, and mitochondrial dysfunction is observed in APP and APP/PS1-based transgenic mouse models [69,70,71,72,73]. Thus, metabolic abnormalities are also considered to be the driving factors and hallmarks of AD [65, 66]. We macroscopically identify the causal relationship between energy metabolism and AD from the perspective of BMR first, which has certain significance for prevention and treatment AD.
Implications for Policy and Practice
Our study shows that there was a causal relationship between the decrease in BMR and increased risk of AD. Therefore, the degree of AD risk prediction could be judged by examining the basal metabolism and BMR. The risk of AD decades later could be predicted by the interval of BMR at young age. And the pathological burden of AD could be alleviated by increasing basal metabolism (such as exercise, increasing muscle mass, etc.) in the daily care of patients with AD.
Strengths and Limitations
This study comprehensively investigated the causal relationship between BMR and AD under a bidirectional MR design based on genetic tools selected from the current larger case load of AD and the largest BMR scale of causation. A series of sensitivity analyses were used to control pleiotropic bias and verify the robustness of the MR results. However, this still has certain limitations. First, we involved some heterogeneity in Cochran’s Q statistics during bidirectional MR analysis. To solve this problem, we chose the IVW random-effects method that provides robustness to heterogeneity and weighted media as our main MR method—random effects can avoid bias in results due to heterogeneity. Weighted median robust MR results are available in predictions where heterogeneity is present but no horizontal pleiotropy. Second, we detected potential pleiotropic effects in the MR-PRESSO test. For this, we increased confidence in the results by removing anomalies, and after removing outliers, the original IVW analysis was found. The causal effects identified with the weighted media analysis did not change due to outliers, which increases the reliability of the results. Third, the leave-one-out analysis method failed to identify individual SNPs that had biased effects on IVW. This indicated the reliability of the results. Finally, our findings suggested that elevated basal metabolism has a protective effect on AD, but we also found that both disorders do not lower AD risk during the analysis of diseases associated with elevated BMR. However, by MVMR analysis, both genetic phenotypes that are closely related to BMR for height and weight have some causal relationship with AD, which may be BMR, which has the effect of acting as an intermediary factor. This suggested that predicting AD by BMR alone still had certain drawbacks, for example, an increase in BMR in a diabetic patient with a reduced risk of AD may also result in other AD risk factors increasing morbidity.
The disadvantage of MR analysis is that current genome-wide association studies (GWAS) for diseases or large sample sizes are still insufficient. For example, the APOE ε4 allele remains the strongest genetic risk factor for sporadic Alzheimer’s disease [20]. Thyroid disease and type 2 diabetes both share common mechanisms with AD [74], and it is of great significance to study the causal relationship between diseases in the absence of horizontal pleiotropy. However, due to the lack of large samples and race-specific population studies in current GWAS or genetic scoring studies, we were unable to find appropriate data in the database to separately analyze the direct causal relationships of patients with AD carrying the APOE ε4 allele and other diseases.
Conclusion
In conclusion, we demonstrated that higher BMR had a significant effect on lower morbidity of AD. The elevated basal metabolism determined by innate genetic factors can reduce the risk of AD. This could be used as a potential indicator of the incidence of AD. We still need to further determine the specific mechanisms underlining how BMR affects the AD causal pathway and explore the specific relationship among these mechanisms.
Data availability
All the data used in this study were obtained from public databases, and the corresponding sources of the data have been cited in the manuscript.
References
TahamiMonfared AA, Byrnes MJ, White LA, Zhang Q. Alzheimer’s disease: epidemiology and clinical progression. Neurol Therapy. 2022;11(2):553–69. https://doi.org/10.1007/s40120-022-00338-8.
2022 Alzheimer's disease facts and figures. Alzheimer's Dementia. 2022;18(4):700–89. https://doi.org/10.1002/alz.12638
Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomed. 2019;14:5541–54. https://doi.org/10.2147/ijn.S200490.
Kępka A, Ochocińska A, Borzym-Kluczyk M, Chojnowska S, Skorupa E, Przychodzeń M, Waszkiewicz N. Healthy food pyramid as well as physical and mental activity in the prevention of Alzheimer’s disease. Nutrients. 2022. https://doi.org/10.3390/nu14081534.
Tatulian SA. Challenges and hopes for Alzheimer’s disease. Drug Discovery Today. 2022;27(4):1027–43. https://doi.org/10.1016/j.drudis.2022.01.016.
Koutsodendris N, Nelson MR, Rao A, Huang Y. Apolipoprotein E and Alzheimer’s disease: findings, hypotheses, and potential mechanisms. Ann Rev Pathol. 2022;17:73–99. https://doi.org/10.1146/annurev-pathmechdis-030421-112756.
Tan W, Qi L, Hu X, Tan Z. Research progress in traditional Chinese medicine in the treatment of Alzheimer’s disease and related dementias. Front Pharmacol. 2022;13: 921794. https://doi.org/10.3389/fphar.2022.921794.
Hindle A, Singh SP, Pradeepkiran JA, Bose C, Vijayan M, Kshirsagar S, Sawant NA, Reddy PH. Rlip76: an unexplored player in neurodegeneration and Alzheimer’s disease? Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23116098.
Kang S, Lee YH, Lee JE. Metabolism-centric overview of the pathogenesis of Alzheimer’s disease. Yonsei Med J. 2017;58(3):479–88. https://doi.org/10.3349/ymj.2017.58.3.479.
An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, Chia CW, Egan JM, Ferrucci L, Troncoso J, et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dementia. 2018;14(3):318–29. https://doi.org/10.1016/j.jalz.2017.09.011.
Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012. https://doi.org/10.1101/cshperspect.a006239.
Caron N, Peyrot N, Caderby T, Verkindt C, Dalleau G. Energy expenditure in people with diabetes mellitus: a review. Front Nutr. 2016;3:56. https://doi.org/10.3389/fnut.2016.00056.
Jones DS, Podolsky SH. The history and fate of the gold standard. Lancet. 2015;385(9977):1502–3. https://doi.org/10.1016/S0140-6736(15)60742-5.
Lawlor DA, Smith GD, Bruckdorfer KR, Kundu D, Ebrahim S. Those confounded vitamins: what can we learn from the differences between observational versus randomised trial evidence? Lancet. 2004;363(9422):1724–7. https://doi.org/10.1016/S0140-6736(04)16260-0.
Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:601. https://doi.org/10.1136/bmj.k601.
Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. JAMA. 2017;318(19):1925–6. https://doi.org/10.1001/jama.2017.17219%JJAMA.
Dimou NL, Tsilidis KK. A primer in Mendelian randomization methodology with a focus on utilizing published summary association data. Methods Mol Biol (Clifton, NJ). 2018;1793:211–30. https://doi.org/10.1007/978-1-4939-7868-7_13.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018. https://doi.org/10.7554/eLife.34408.
Henry CJ. Basal metabolic rate studies in humans: measurement and development of new equations. Public Health Nutr. 2005;8(7a):1133–52. https://doi.org/10.1079/phn2005801.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30. https://doi.org/10.1038/s41588-019-0358-2.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dementia. 2011;7(3):263–9. https://doi.org/10.1016/j.jalz.2011.03.005.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–44. https://doi.org/10.1212/wnl.34.7.939.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & dementia : the journal of the Alzheimer's Association 2011, 7(3):270–279. https://doi.org/10.1016/j.jalz.2011.03.008.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–74. https://doi.org/10.1093/ije/dyw220.
Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet. 2018;27(R2):R195–208. https://doi.org/10.1093/hmg/ddy163.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. 2017;36(11):1783–802. https://doi.org/10.1002/sim.7221.
Julian TH, Glascow N, Barry ADF, Moll T, Harvey C, Klimentidis YC, Newell M, Zhang S, Snyder MP, Cooper-Knock J, et al. Physical exercise is a risk factor for amyotrophic lateral sclerosis: convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine. 2021;68: 103397. https://doi.org/10.1016/j.ebiom.2021.103397.
Li R, Chen Y, Zhao A, Huang L, Long Z, Kang W, Yin Y, Tong S, Guo Y, Li S. Exploring genetic association of insomnia with allergic disease and asthma: a bidirectional Mendelian randomization study. Respiratory Res. 2022;23(1):84. https://doi.org/10.1186/s12931-022-02009-6.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8. https://doi.org/10.1038/s41588-018-0099-7.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25. https://doi.org/10.1093/ije/dyv080.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14. https://doi.org/10.1002/gepi.21965.
Nordestgaard LT, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Body mass index and risk of Alzheimer’s disease: a Mendelian randomization study of 399,536 individuals. J Clin Endocrinol Metab. 2017;102(7):2310–20. https://doi.org/10.1210/jc.2017-00195.
Petot GJ, Vega U, Traore F, Fritsch T, Debanne SM, Friedland RP, Lerner AJ. Height and Alzheimer’s disease: findings from a case-control study. J Alzheimer’s Dis JAD. 2007;11(3):337–41. https://doi.org/10.3233/jad-2007-11310.
Berkhout A, Cools H, Houwelingen HV. Ageing: the relationship between difficulties in feeding oneself and loss of weight in nursing-home patients with dementia. 1998;27(5):637–41. https://doi.org/10.1093/ageing/27.5.637
Atti AR, Palmer K, Volpato S, Winblad B, De Ronchi D, Fratiglioni L. Late-life body mass index and dementia incidence: nine-year follow-up data from the Kungsholmen Project. J Am Geriatrics Soc. 2008;56(1):111–6. https://doi.org/10.1111/j.1532-5415.2007.01458.x.
Visioli F, Burgos-Ramos E. Selected micronutrients in cognitive decline prevention and therapy. Mol Neurobiol. 2016;53(6):4083–93. https://doi.org/10.1007/s12035-015-9349-1.
Morris MC. The role of nutrition in Alzheimer’s disease: epidemiological evidence. Eur J Neurol. 2010;16(1):1–7. https://doi.org/10.1111/j.1468-1331.2009.02735.x.
Loh KW, Vriens MR, Gerritsen A, BorelRinkes IH, van Hillegersberg R, Schippers C, Steenhagen E, Ong TA, Moy FM, Molenaar IQ. Unintentional weight loss is the most important indicator of malnutrition among surgical cancer patients. Netherlands J Med. 2012;70(8):365–9.
Wang C, Fu W, Cao S, Jiang H, Guo Y, Xv H, Liu J, Gan Y, Lu Z. Weight loss and the risk of dementia: a meta-analysis of cohort studies. Curr Alzheimer Res. 2021;18(2):125–35. https://doi.org/10.2174/1567205018666210414112723.
White H, Pieper C, Schmader K. The association of weight change in Alzheimer’s disease with severity of disease and mortality: a longitudinal analysis. J Am Geriatr Soc. 1998;46(10):1223–7. https://doi.org/10.1111/j.1532-5415.1998.tb04537.x.
Takeishi J, Tatewaki Y, Nakase T, Takano Y, Tomita N, Yamamoto S, Mutoh T, Taki Y. Alzheimer’s disease and type 2 diabetes mellitus: the use of MCT oil and a ketogenic diet. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms222212310.
Luca M, Di Mauro M, Di Mauro M, Luca A. Gut microbiota in Alzheimer’s disease, depression, and type 2 diabetes mellitus: the role of oxidative stress. Oxid Med Cell Long. 2019;2019:4730539. https://doi.org/10.1155/2019/4730539.
Tumminia A, Vinciguerra F, Parisi M, Frittitta L. Type 2 diabetes mellitus and Alzheimer’s disease: role of insulin signalling and therapeutic implications. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19113306.
Michailidis M, Moraitou D, Tata DA, Kalinderi K, Papamitsou T, Papaliagkas V. Alzheimer’s disease as type 3 diabetes: common pathophysiological mechanisms between Alzheimer’s disease and type 2 diabetes. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23052687.
Thota RN, Rosato JI, Dias CB, Burrows TL, Martins RN, Garg ML. Dietary supplementation with curcumin reduce circulating levels of glycogen synthase kinase-3β and islet amyloid polypeptide in adults with high risk of type 2 diabetes and Alzheimer’s disease. Nutrients. 2020. https://doi.org/10.3390/nu12041032.
Boccardi V, Murasecco I, Mecocci P. Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res Rev. 2019;54: 100936. https://doi.org/10.1016/j.arr.2019.100936.
Li T, Cao HX, Ke D. Type 2 diabetes mellitus easily develops into Alzheimer’s disease via hyperglycemia and insulin resistance. Curr Med Sci. 2021;41(6):1165–71. https://doi.org/10.1007/s11596-021-2467-2.
Sugimoto T, Tokuda H, Miura H, Kawashima S, Ando T, Kuroda Y, Matsumoto N, Fujita K, Uchida K, Kishino Y, et al. Cross-sectional association of metrics derived from continuous glucose monitoring with cognitive performance in older adults with type 2 diabetes. Diabetes Obes Metab. 2023;25(1):222–8. https://doi.org/10.1111/dom.14866.
Cummings J, Ortiz A, Castellino J, Kinney J. Diabetes: Risk factor and translational therapeutic implications for Alzheimer's disease. 2022;56(9):5727–57. https://doi.org/10.1111/ejn.15619.
Shieh JC, Huang PT, Lin YF. Alzheimer’s disease and diabetes: insulin signaling as the bridge linking two pathologies. Mol Neurobiol. 2020;57(4):1966–77. https://doi.org/10.1007/s12035-019-01858-5.
Haraguchi Y, Mizoguchi Y, Noguchi T, Arai T, Fukuyama J, Kato TA, Kawashima T, Monji A. A patient with Alzheimer’s disease complicated by elderly-onset Cushing’s syndrome who had undergone surgical treatment for adrenocorticotropic hormone-independent macronodular adrenal hyperplasia. Psychogeriatrics. 2016;16(4):274–6. https://doi.org/10.1111/psyg.12146.
Tan B, Babur E, Toy N, Günaydın B, Dursun N, Süer C. Tau protein is differentially phosphorylated in young- and old-aged rats with experimentally induced hyperthyroidism. Int J Dev Neurosci. 2022;82(7):654–63. https://doi.org/10.1002/jdn.10220.
Bitiktas S, Kandemir B, Tan B, Kavraal S, Liman N, Dursun N, Dönmez-Altuntas H, Aksan-Kurnaz I, Suer C. Adult-onset hyperthyroidism impairs spatial learning: possible involvement of mitogen-activated protein kinase signaling pathways. 2016;27(11):802–8. https://doi.org/10.1097/wnr.0000000000000612.
Peeters RP. Thyroid hormones and aging. Hormones (Athens). 2008;7(1):28–35. https://doi.org/10.14310/horm.2002.1111035.
Tan ZS, Vasan RS. Thyroid function and Alzheimer’s disease. J Alzheimer’s Dis JAD. 2009;16(3):503–7. https://doi.org/10.3233/jad-2009-0991.
Liu C, Lu Y, Chen J, Qiu W, Zhan Y, Liu Z. Basal metabolic rate and risk of multiple sclerosis: a Mendelian randomization study. Metab Brain Dis. 2022;37(6):1855–61. https://doi.org/10.1007/s11011-022-00973-y.
Blakemore S-J, Burnett S, Dahl RE. The role of puberty in the developing adolescent brain. 2010;31(6):926–33. https://doi.org/10.1002/hbm.21052
Hyder F, Rothman DL, Bennett MR. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci USA. 2013;110(9):3549–54. https://doi.org/10.1073/pnas.1214912110.
Watts ME, Pocock R, Claudianos C. Brain energy and oxygen metabolism: emerging role in normal function and disease. Front Mol Neurosci. 2018;11:216. https://doi.org/10.3389/fnmol.2018.00216.
Vanni S, Baldeschi AC, Zattoni M, Legname G. Brain aging: a Ianus-faced player between health and neurodegeneration. J Neurosci Res. 2019. https://doi.org/10.1002/jnr.24379.
Sun Y, Ma C, Sun H, Wang H, Peng W, Zhou Z, Wang H, Pi C, Shi Y, He X. Metabolism: a novel shared link between diabetes mellitus and Alzheimer’s disease. J Diabetes Res. 2020;2020:4981814. https://doi.org/10.1155/2020/4981814.
McDonald TS, Lerskiatiphanich T, Woodruff TM, McCombe PA, Lee JD. Potential mechanisms to modify impaired glucose metabolism in neurodegenerative disorders. J Cerebral Blood Flow Metab. 2023;43(1):26–43. https://doi.org/10.1177/0271678x221135061.
Cunnane S, Nugent S, Roy M, Courchesne-Loyer A, Croteau E, Tremblay S, Castellano A, Pifferi F, Bocti C, Paquet N, et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition. 2011;27(1):3–20. https://doi.org/10.1016/j.nut.2010.07.021.
Wang Q, Duan L, Li X, Wang Y, Guo W, Guan F, Ma S. Glucose metabolism, neural cell senescence and Alzheimer’s disease. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23084351.
Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20(3):148–60. https://doi.org/10.1038/s41583-019-0132-6.
Huo Z, Yu L, Yang J, Zhu Y, Bennett DA, Zhao J. Brain and blood metabolome for Alzheimer’s dementia: findings from a targeted metabolomics analysis. Neurobiol Aging. 2020;86:123–33. https://doi.org/10.1016/j.neurobiolaging.2019.10.014.
Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14(6):724–38. https://doi.org/10.1016/j.cmet.2011.08.016.
Cuperlovic-Culf M, Badhwar A. Recent advances from metabolomics and lipidomics application in Alzheimer’s disease inspiring drug discovery. Expert Opin Drug Discov. 2020;15(3):319–31. https://doi.org/10.1080/17460441.2020.1674808.
Devi L, Anandatheerthavarada HK. Mitochondrial trafficking of APP and alpha synuclein: relevance to mitochondrial dysfunction in Alzheimer’s and Parkinson’s diseases. Biochimica et Biophysica Acta (BBA) – Mol Basis Dis. 2010;1802(1):11–9. https://doi.org/10.1016/j.bbadis.2009.07.007.
Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. 2008;105(35):13145–50. https://doi.org/10.1073/pnas.0806192105.
Lopez Sanchez MIG, Waugh HS, Tsatsanis A, Wong BX, Crowston JG, Duce JA, Trounce IA. Amyloid precursor protein drives down-regulation of mitochondrial oxidative phosphorylation independent of amyloid beta. Sci Rep. 2017;7(1):9835. https://doi.org/10.1038/s41598-017-10233-0.
Pera M, Larrea D, Guardia-Laguarta C, Montesinos J, Velasco KR, Agrawal RR, Xu YM, Chan RB, Di Paolo G, Mehler MF, et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. Embo J. 2017;36(22):3356–71. https://doi.org/10.15252/embj.201796797.
Reddy PH, Oliver DM. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells. 2019;8(5):488. https://doi.org/10.3390/cells8050488.
Bavarsad K, Hosseini M, Hadjzadeh MAR, Sahebkar A. The effects of thyroid hormones on memory impairment and Alzheimer's disease. 2019;234:14633–40.
Acknowledgements
We thank the participants and researchers from the UK Biobank cohort as well as the participants and surveyors from the MR-BASE platform. We thank IGAP for providing summary statistics for AD.
Funding
This research was financially supported by the Key R&D Plan of the Science and Technology Plan of Tibet Autonomous Region (XZ202201ZY0026G) and Science and Technology Cooperation Project of Shandong Provincial Department of Science and Technology for Counterpart Assistance to Tibet (YDZX2021083). This study was supported the Natural Science Foundation of Shandong Province, China (ZR2021QH271). Xiaorui Cheng funded the journal’s Rapid Service Fee.
Author contributions
Conception and design: Yuexiao Zou. Analysis and interpretation: Yuexiao Zou, Qingxian Wang. Data collection: Yuexiao Zou. Critically revised the manuscript: Xiaorui Cheng. Obtained funding: Qingxian Wang, Xiaorui Cheng. Overall responsibility: Yuexiao Zou, Qingxian Wang, Xiaorui Cheng.
Disclosures
Yuexiao Zou, Qingxian Wang and Xiaorui Cheng have nothing to disclose.
Compliance with ethics guidelines
Ethics approval and informed consent were not required for the present study as they were obtained in the original studies. The original studies were conducted in compliance with the Declaration of Helsinki.
Data availability
The datasets generated during and/or analyzed during the current study are available in the Github repository, https://github.com/zoudasheng/MR-BMRandAD.git. Some data generated or analyzed during this study are included in this published article/as supplementary information files.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zou, Y., Wang, Q. & Cheng, X. Causal Relationship Between Basal Metabolic Rate and Alzheimer’s Disease: A Bidirectional Two-sample Mendelian Randomization Study. Neurol Ther 12, 763–776 (2023). https://doi.org/10.1007/s40120-023-00458-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-023-00458-9