
Abstract
Background and Objectives
Islatravir (MK-8591) is a novel nucleoside analogue in development for the treatment and prevention of HIV-1 infection. Doravirine is a non-nucleoside reverse transcriptase inhibitor indicated for the treatment of HIV-1 infection. This study evaluated the pharmacokinetics, safety, and tolerability of islatravir and doravirine coadministration in a double-blind, placebo-controlled, randomized, fixed-sequence study.
Methods
Adult participants without HIV infection were administered oral doravirine 100 mg (n = 10) or placebo (n = 4) once daily (QD) for 5 days, immediately followed by oral islatravir 2.25 mg (n = 10) or placebo QD (n = 4) for 14 days; islatravir 2.25 mg and doravirine 100 mg QD, or placebo QD, were then coadministered for 5 days. Pharmacokinetic and safety data were collected.
Results
Doravirine geometric least-squares mean ratios (90% confidence intervals (CIs)) of (doravirine + islatravir)/doravirine for the area under the plasma drug concentration-time curve over 24 h (AUC0–24h), maximum plasma concentration (Cmax), and plasma concentration at 24 h post-dose (C24h) were not meaningfully impacted. Islatravir geometric least-squares mean ratios (90% CI) of (islatravir + doravirine)/islatravir for AUC0–24h and Cmax were both close to unity, 1.06 (1.01, 1.12) and 1.08 (0.91, 1.27), respectively. All study regimens were generally well tolerated.
Conclusion
These results indicate that coadministration of islatravir and doravirine had no clinically meaningful effect on the pharmacokinetics of either drug, and support further clinical investigation of islatravir in combination with doravirine for the treatment of HIV-1 infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This phase 1 study in adult participants without HIV showed that there was no clinically meaningful drug interaction between islatravir and doravirine when administered together. |
These results support further clinical studies of islatravir in combination with doravirine, which has the potential to provide a simple two-drug regimen for the treatment of HIV-1 infection. |
1 Introduction
HIV-1 infection continues to be prevalent, with an estimated 38 million people living with HIV-1 worldwide as of 2019 [1]. Although current antiretroviral therapy (ART) is effective for the treatment of HIV-1, new agents with improved safety and tolerability, a favorable drug–drug interaction (DDI) profile, a high barrier to the development of resistance, and potent antiviral activity, including against drug-resistant HIV-1 isolates, are needed [2].
Islatravir (MK-8591) is a novel, HIV-1 nucleoside analogue in development for the treatment and prevention of HIV-1 infection [3, 4]. Islatravir uniquely inhibits reverse transcriptase to suppress HIV-1 replication by multiple mechanisms of action, including inhibition of translocation, with potent antiviral activity against wild-type HIV-1 and common nucleotide reverse transcriptase inhibitor (NRTI)-resistant variants [3, 5]. In treatment-naive people living with HIV-1, islatravir administered orally at doses between 0.5 and 30 mg effectively suppressed viral load for at least 7 days and was generally well tolerated [6]. In participants without HIV, single oral doses of up to 400 mg and multiple oral doses of islatravir of up to 100 mg weekly for 3 weeks or up to 5 mg daily for 6 weeks were also generally well tolerated [7].
Islatravir is converted intracellularly by endogenous kinases to its active triphosphate (TP) form, islatravir-TP, which has a long intracellular half-life, allowing the potential for a variety of dosing options and regimens. In participants without HIV, single oral doses of islatravir were rapidly absorbed, with peak plasma concentrations attained at ~ 0.5 h, with islatravir demonstrating a half-life of ~ 49–61 h. Peak intracellular islatravir-TP concentrations were attained 6–24 h after islatravir administration, with a half-life of 118–171 h [8]; similar pharmacokinetics have been observed in people living with HIV-1 [6]. When administered once daily (QD), oral doses of islatravir resulted in steady-state plasma concentrations at approximately Day 14, while intracellular islatravir-TP reached steady state by Day 28 and remained above the concentration threshold associated with antiviral efficacy (0.05 pmol/106 cells) for ~ 30 days following cessation of dosing. In addition, doses as low as 0.25 mg QD produced intracellular islatravir-TP concentrations exceeding the therapeutic concentration threshold on Day 1 [7].
Coadministration of islatravir and the non-nucleoside reverse transcriptase inhibitor (NNRTI) doravirine has the potential to provide a simple two-drug regimen, with efficacy comparable to that of currently approved regimens for HIV-1 treatment. Doravirine is an NNRTI indicated for the treatment of HIV-1 infection in combination with other antiretroviral agents [9]. Doravirine has potent antiviral activity against wild-type HIV-1 and common NNRTI-resistant variants, and a well-characterized DDI profile [10,11,12]. In treatment-naive adults with HIV-1 infection, doravirine 100 mg administered in combination with two NRTIs demonstrated non-inferior efficacy to ritonavir-boosted darunavir with two NRTIs, and doravirine 100 mg administered as a single-tablet regimen with lamivudine 300 mg and tenofovir disoproxil fumarate 300 mg demonstrated non-inferior efficacy compared with efavirenz 600 mg, emtricitabine 200 mg, and tenofovir disoproxil fumarate 300 mg [13, 14]. Doravirine regimens have been generally well tolerated in clinical trials, with favorable lipid profiles [13,14,15,16,17]. Compared with efavirenz, doravirine demonstrated fewer central nervous system adverse events (AEs) [14], and comparable low effects on weight gain and body mass index through 96 weeks of treatment [18].
Following oral administration, doravirine (6–1200 mg orally) achieved maximum plasma concentrations (Cmax) within 1–5 h with an apparent terminal half-life of 12–21 h [17], while QD dosing (200 mg orally) resulted in attainment of steady state after approximately 5 days [19]. Doravirine pharmacokinetics were similar in people living with and without HIV-1 [9]. Doravirine is metabolized primarily by hepatic cytochrome P450 (CYP) 3A4 and is not an inhibitor or inducer of CYP enzymes or an inhibitor of major drug transporters in vitro [12]. Coadministration of doravirine with the strong CYP3A4 inducer rifampin reduced doravirine exposure by 88%, and coadministration with the moderate CYP3A4 inducer rifabutin reduced doravirine exposure by 50% [20, 21]. The CYP3A4 inhibitors ritonavir and ketoconazole did not have a clinically meaningful effect on doravirine pharmacokinetics when coadministered [22]. Additional studies have demonstrated no clinically meaningful drug–drug interactions after coadministration of doravirine with a range of other drugs [17, 19, 22,23,24].
Islatravir is eliminated primarily by adenosine deaminase-mediated metabolism [25] and is not expected to be a perpetrator of drug–drug interactions against doravirine. Doravirine is not expected to be a perpetrator of drug–drug interactions against islatravir. The effect of doravirine on the adenosine deaminase metabolic pathway is unknown, but very few drugs are known to affect this pathway [26, 27]. To evaluate the potential of islatravir and doravirine as a combination HIV-1 treatment regimen, it is important to characterize the pharmacokinetics and safety profile when coadministered. A DDI study was conducted to assess the safety and pharmacokinetics of islatravir and doravirine coadministration in adult participants without HIV infection.
2 Methods
2.1 Study Design
A double-blind, placebo-controlled, randomized, fixed-sequence, two-period, multiple-dose, DDI study was conducted between June and August 2017 (protocol MK-8591-010-01; see Online Supplementary Material). 14 participants were randomly assigned 5:2 to receive active treatment or placebo. During Period 1, participants received 100 mg doravirine or placebo QD for 5 days. During Period 2, participants in the active treatment arm received islatravir 2.25 mg QD alone for 14 days, followed by doravirine 100 mg coadministered with islatravir 2.25 mg for 5 days. Doravirine dosage was selected based on the therapeutic dose. Islatravir dosage was selected based on pharmacokinetic and viral dynamic simulations predicting daily doses from as low as 0.25 mg to be efficacious, with doses of 0.25 mg, 0.75 mg, and 2.25 mg being assessed in a phase 2 trial [6, 28]. For assessment of drug interactions, 2.25 mg was selected to evaluate a potential effect more thoroughly. Participants who received placebo in Period 1 also received placebo in Period 2. There was no washout period between Periods 1 and 2. All study drug was administered orally with approximately 240 mL water at the same time each day (± 1 h). Participants fasted overnight for ≥ 10 h before dosing on Day 5 of Period 1 and on Days 14 and 19 of Period 2; participants continued to fast for ≥ 4 h post-dose on these days. On other study days, participants fasted 1 h before and 2 h after study drug administration.
Drug allocation was determined by a blinded computerized randomization scheme. The randomization code was available only to clinic pharmacy staff involved in drug preparation; they were not involved in any other aspect of the study, including drug administration. Subjects and members of the staff responsible for the monitoring and evaluation of safety assessments were blinded. The randomization code remained sealed and could have only been broken in the event of a medical emergency requiring drug identification. Placebo tablets of doravirine and placebo capsules of islatravir were matched in appearance to the respective active drug. No subject’s treatment assignment was unblinded by either the Investigator or the clinical monitor during the study. Randomization was revealed after the trial database was officially locked.
2.2 Participants
Male and female adults without HIV infection aged 19–55 years whose body mass index ranged from 19.0 to 32.0 kg/m2 at screening were eligible to enroll. Participants with a presence or history of a clinically significant medical or psychiatric condition were excluded. Participants were also excluded if they had used nicotine-containing products within 3 months or had a history of alcoholism or drug abuse within 2 years before dosing. Participants were prohibited from taking any other drugs, herbal products, or vitamin supplements within 14 days of the study and were prohibited from taking any products that could significantly interact with CYP enzymes or P-glycoprotein (such as St. John’s wort) for 28 days before the study.
2.3 Pharmacokinetic Sampling
Serial blood samples were collected pre-dose and at 0.5, 1, 2, 3, 4, 8, 12, 16, 20, and 24 h post-dose on Day 5 of Period 1 for measurement of doravirine plasma concentrations and at 0.25, 0.5, 1, 2, 4, 8, 20, and 24 h post-dose on Day 14 of Period 2 for measurement of islatravir plasma concentrations. Blood samples were collected pre-dose and at 0.5, 1, 2, 3, 4, 8, 12, 16, 20, 24, 120, 192, and 336 h post-dose on Day 19 of Period 2 for measurement of islatravir plasma concentrations and at 0.25, 0.5, 1, 2, 4, 8, 20, 24, 120, 192, and 336 h post-dose on Day 19 for measurement of doravirine plasma concentrations.
Blood (4 mL) was drawn into collection tubes containing anticoagulant (K2EDTA) at the protocol-specified time points, gently inverted six times (for islatravir plasma concentration determination) or eight to ten times (for doravirine) and placed immediately in an ice water bath. Within 30 min of collection, the blood samples were centrifuged at 4 °C and plasma from each sample was aliquoted in a labeled cryotube and stored at − 20 °C within 60 min of collection. The plasma samples were packed on dry ice and sent to inVentiv Health Clinique (now Syneos, Quebec, Canada) for assay.
2.4 Pharmacokinetics
Plasma concentrations of islatravir and doravirine were determined by inVentiv Health Clinique (Québec City, QC, Canada) using validated high-performance liquid chromatography coupled to tandem mass spectrometry methods [29]. Doravirine was extracted by an automated liquid-liquid extraction method using [13C215N3-d3]doravirine as internal standard. Extracted samples were assessed on a system equipped with an Atlantis T3, 50 × 2.1 mm, 3 µm column with a water/acetonitrile with formic acid mobile phase. Over the calibration range, intraday precision and accuracy ranged from 0.70 to 1.51% CV and 99.93 to 105.0%, respectively. Interday precision and accuracy ranged from 0.85 to 1.81% CV and 99.72 to 100.87%, respectively. The lower limit of quantitation for doravirine was 1.00 ng/mL, with an analytical range of 1.00–1000.00 ng/mL.
Islatravir was extracted using an automated protein precipitation extraction method using [13C15N3]islatravir as internal standard. Extracted samples were assessed on a system equipped with an X Select HSS T3, 50 × 2.1 mm, 2.5 µm column with a water/acetonitrile with formic acid mobile phase. Over the calibration range, intraday precision and accuracy ranged from 1.28 to 3.00% CV and 99.72 to 104.37%, respectively. Interday precision and accuracy ranged from 1.39 to 2.82% CV and 99.22 to 100.31%, respectively. The lower limit of quantitation for islatravir was 0.10 ng/mL, with an analytical range of 0.10–100.00 ng/mL. Values below the limit of quantitation for each drug were treated as zero.
Cmax plasma concentration at 24 h post-dose (C24h), and time to maximum concentration (Tmax) were all obtained directly from plasma concentration-time data. C24h for islatravir was not reported, because islatravir-TP is the relevant moiety for efficacy and was not evaluated in this trial for purposes of the DDI evaluation. AUC0–24h was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations over time (linear-up/log-down).
2.5 Safety Assessments
Safety and tolerability were evaluated based on AE monitoring, physical examinations, vital signs, electrocardiography, and laboratory safety tests conducted or collected throughout the study. All clinical AEs reported by the participant or observed by the investigator were graded as mild, moderate, or severe in intensity.
AEs are defined as any untoward medical occurrence in a clinical investigation subject administered a pharmaceutical product, with or without a causal relationship with this treatment. A serious AE (SAE) is any AE occurring at any dose that (1) resulted in death, (2) was life-threatening in the view of the investigator, (3) resulted in persistent or significant disability/incapacity, (4) resulted in or prolonged an existing inpatient hospitalization, or (5) resulted in a congenital anomaly/birth defect in a subject’s offspring.
2.6 Data Analysis
Pharmacokinetic analyses were based on the per-protocol population, which included the subset of participants who complied with the protocol up to the relevant pharmacokinetic sampling period. One participant was discontinued on Day 18 of Period 2 by the investigator due to a positive urine drug screen. This participant had completed all PK assessments on Day 5 of Period 1 (doravirine alone) and Day 14 of Period 2 (islatravir alone). Therefore, data from all ten participants who received doravirine alone in Period 1 and islatravir alone in Period 2, and the nine participants who received the combination treatment in Period 2, were included in the PK analyses. Safety and tolerability analyses included all participants who received one or more dose of study drug or placebo.
Pharmacokinetic parameter values were calculated using Phoenix WinNonlin version 6.3 (Certara). Individual values for AUC0–24h, Cmax, and C24h were natural-log (ln)-transformed and evaluated separately with a linear mixed-effects model with a fixed effect for treatment. An unstructured covariance matrix was used to allow for unequal treatment variances and to model the correlation between treatment measurements for each participant with the use of REPEATED statement in PROC MIXED (SAS Institute, Cary, NC, USA). The Kenward-Roger adjustment was used to calculate the denominator degrees of freedom for the fixed effects. A two-sided 90% confidence interval (CI) was computed for the true mean difference for each doravirine parameter on the ln-scale and exponentiated to obtain the 90% CI for the true geometric mean ratio (GMR) of (doravirine + islatravir)/doravirine area under the plasma drug concentration-time curve over 24 h (AUC0–24h), Cmax, and C24h. Similar analyses were conducted for islatravir AUC0–24h and Cmax. Similar analyses for islatravir and doravirine Tmax were not performed.
Sample size was determined based on available doravirine pharmacokinetic variability data. Power calculations for AUC0–24h, C24h, and Cmax of doravirine were based on assumed true within-subject standard deviations of 0.266, 0.410, and 0.198, respectively, for ln-AUC0–24h, ln-C24h, and ln-Cmax. With ten subjects, there was at least 99%, 93%, and 99% probability for AUC0–24h, C24, and Cmax, respectively, of obtaining a 90% CI for the true GMR of 1 to lie within a 0.5–2.0 interval.
3 Results
3.1 Study Population
Fourteen participants (12 male, two female) were enrolled in the trial (Fig. 1). Ten participants were randomly assigned to receive active treatment and four were randomly assigned to receive placebo. Demographics are listed in Table 1. The mean (range) age was 33.5 (23–55) years and mean body mass index (range) was 27.58 (24.25–31.75) kg/m2.

Participant disposition. QD once daily
Thirteen participants completed the study per protocol. One participant in the active treatment arm discontinued on Day 18 of Period 2 because of a positive result on urine drug screening and was not included in the Period 2 coadministration analysis.
3.2 Effect of Islatravir Co-Administration on Doravirine Pharmacokinetics
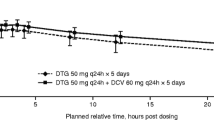
Statistical analysis of doravirine pharmacokinetic parameter values demonstrated comparable results with and without islatravir coadministration (Table 2 and Fig. 2A). Doravirine geometric least-squares mean ratios (GMR) and 90% CIs of (doravirine + islatravir)/doravirine for the AUC0–24h, Cmax, and C24h were not meaningfully changed. Doravirine median time to Tmax did not differ significantly with or without islatravir. The mean concentration–time profile of doravirine over 24 h demonstrated a mono-exponential decline after administration alone and after coadministration with islatravir.

Mean plasma concentration-time profiles of A doravirine following multiple doses of 100 mg doravirine with and without 2.25 mg islatravir, and B islatravir following multiple doses of 2.25 mg islatravir with and without 100 mg doravirine (inset = semi-log scale)
3.3 Effect of Doravirine on Islatravir Pharmacokinetics
The pharmacokinetic profile of islatravir was comparable when administered alone and with doravirine (Table 3 and Fig. 2B). Islatravir GMR (90% CI) of (islatravir + doravirine)/islatravir for AUC0–24h and Cmax were both close to unity and islatravir Tmax was comparable with and without doravirine.
3.4 Safety and Tolerability
Doravirine and islatravir administered alone or in combination were generally well tolerated. AEs were reported by eight of 14 participants (57%) who received study treatment. All AEs were mild and resolved or were resolving by the end of the study. Of the eight AEs reported, seven were considered to be drug related. The most common drug-related AE was mild somnolence, which was reported by two participants during treatment with doravirine alone, one of whom also reported somnolence during treatment with coadministered doravirine and islatravir (Table 4). No SAEs, deaths, discontinuations due to an AE, or clinically meaningful changes in laboratory values, vital signs, or electrocardiographic parameter values were reported in the study.
4 Discussion
Islatravir, a novel, potent nucleoside analogue in development for the treatment and prevention of HIV-1 infection, and doravirine, a novel approved NNRTI, have the potential to be coadministered as ART. This study evaluated whether the coadministration of islatravir and doravirine has any effect on the pharmacokinetic or safety profile of either drug to support the continued evaluation of this combination as a potential treatment regimen for HIV-1 infection.
Results from this DDI study demonstrate that coadministration of multiple oral doses of islatravir 2.25 mg QD and doravirine 100 mg QD does not have a clinically meaningful effect on the pharmacokinetics of either drug in adult participants without HIV. Islatravir 2.25 mg QD administered alone or with doravirine 100 mg QD was generally well tolerated, and no clear AE trends were observed in this phase 1 trial with 10 participants.
Extensive DDI studies of doravirine demonstrated interactions only with CYP3A4 inhibitors and inducers [20, 21]. Islatravir was not expected to affect the pharmacokinetics of doravirine. The 90% CIs for doravirine AUC0–24h, Cmax, and C24h GMR of (doravirine + islatravir)/doravirine spanned from 0.99 to 1.32. The etiology of the minor increases seen is unknown and could be secondary to variability. Based on collective data from the doravirine clinical program, variations of up to threefold increases are not expected to significantly impact safety as doses of up to 1,200 mg have been generally well tolerated in clinical studies [30]. With respect to antiviral efficacy, only decreases in C24h GMR would be expected to impact efficacy, and a decrease in C24h was not observed in this study [17, 30, 31].
Overall, multiple QD doses of islatravir 2.25 mg had no clinically meaningful effect on plasma doravirine AUC0–24h, Cmax, or C24h. Consistent with previous observations, the plasma concentration of doravirine declined in a mono-exponential manner over 24 h regardless of whether it was coadministered with islatravir [17]. Median Tmax values of 2.5 h for doravirine alone and 2 h for doravirine coadministered with islatravir were within the range of 1–4 h, as previously reported [20, 32].
The elimination of islatravir is expected to be balanced between renal excretion and adenosine deaminase-mediated metabolism [25]. Islatravir is not metabolized by CYP enzymes nor is it a substrate of major transporters [12, 24]. Doravirine has not been tested as an inhibitor or inducer of adenosine deaminase. Adenosine deaminase is a critical enzyme in nucleotide metabolism, and a genetic lack of adenosine deaminase results in severe combined immunodeficiency syndrome, a condition that, if untreated, is often fatal in early childhood [33]. Doravirine treatment was not associated with AEs that would be expected from impairment of adenosine deaminase, such as neutropenia, thrombocytopenia, anemia, vasculitis, inflammation, or skin rash, suggesting that doravirine is unlikely to interact with this metabolic pathway [14, 34]. Mean islatravir AUC0–24h and Cmax GMR of (islatravir + doravirine)/islatravir were close to unity, indicating doravirine had no clinically meaningful effect on plasma islatravir AUC0–24h or Cmax.
Doravirine is not anticipated to influence intracellular islatravir-TP levels. Uptake of NRTIs occurs by diffusion or by a combination of diffusion and active transport, and the subsequent phosphorylation to the active form is catalyzed by endogenous kinases, with multiple enzymes capable of catalyzing each phosphorylation step [35]. Thus, the potential for doravirine to completely inhibit islatravir-TP formation is low. Because of this underlying physiology, DDIs affecting only the phosphorylated forms of nucleoside analogues are considered unlikely. Supporting this, previous DDI trials of nucleoside analogs either do not assess active forms [36], or do not demonstrate a difference in nucleotide concentrations [37].
As a result of the fixed-sequence design, potential period effects cannot be ruled out, which may be considered a possible limitation of this study. However, a randomized crossover design was not implemented due to the complexities associated with the long apparent terminal half-life of islatravir-TP, which exceeded 7 days when dosed daily to steady state [38]. Only two of the 14 participants enrolled were female; however, studies conducted thus far have not demonstrated any difference in safety, potency, or pharmacokinetics after doravirine or islatravir administration in male and female participants, and this will be investigated in future clinical studies [14]. Based on the similar pharmacokinetic properties of both islatravir and doravirine in people living with and without HIV-1, the findings from this trial can be reasonably bridged between the populations.
The findings from this study supported the evaluation of islatravir doses of up to 2.25 mg coadministered with doravirine 100 mg QD in a phase 2b study in treatment-naive adults with HIV-1 infection. The combination of islatravir 0.75 mg and doravirine 100 mg QD demonstrated sustained viral suppression through 48 weeks of treatment and was generally well tolerated in treatment-naïve adults with HIV-1 infection who switched following initiation on a three-drug regimen of oral islatravir and doravirine in combination with lamivudine for 24 weeks [28]. A comprehensive phase 3 clinical trial program will investigate the treatment potential of islatravir 0.75 mg and doravirine 100 mg across populations of people living with HIV-1 including treatment-naïve individuals (NCT04233879); individuals who are virologically suppressed on an ART regimen and switch to islatravir and doravirine (NCT04223778, NCT04223791); and heavily treatment-experienced individuals whose current ART regimen is failing, in whom islatravir and doravirine will be coadministered in combination with optimized background ART (NCT04233216).
5 Conclusion
Coadministration of islatravir and doravirine was generally well tolerated in participants without HIV and demonstrated no clinically meaningful effects on the pharmacokinetic profiles of either doravirine or islatravir. The lack of drug–drug interactions observed indicates that islatravir in combination with doravirine for the treatment of HIV-1 infection is suitable for further clinical investigation.
References
UNAIDS. Fact Sheet—Global HIV & AIDS Statistics—2020 Fact Sheet. 2021. https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf. Accessed Mar 2021.
DHHS. Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services. 2021. https://clinicalinfo.hiv.gov/sites/default/files/guidelines/documents/AdultandAdolescentGL.pdf. Accessed Mar 2021.
Markowitz M, Sarafianos SG. 4’-Ethynyl-2-fluoro-2’-deoxyadenosine, MK-8591: a novel HIV-1 reverse transcriptase translocation inhibitor. Curr Opin HIV AIDS. 2018;13(4):294–9. https://doi.org/10.1097/COH.0000000000000467.
Markowitz M, Grobler JA. Islatravir for the treatment and prevention of infection with the human immunodeficiency virus type 1. Curr Opin HIV AIDS. 2020;15(1):27–32. https://doi.org/10.1097/COH.0000000000000599.
Grobler K, Huang Q, Hazuda D, Lai M. Efficacy of MK-8591 against diverse HIV-1 subtypes and NRTI-resistant clinical isolates. J Int AIDS Soc. Abstract #0343; HIV Glasgow, 28–31 October 2018, Glasgow, UK.
Schurmann D, Rudd DJ, Zhang S, De Lepeleire I, Robberechts M, Friedman E, et al. Safety, pharmacokinetics, and antiretroviral activity of islatravir (ISL, MK-8591), a novel nucleoside reverse transcriptase translocation inhibitor, following single-dose administration to treatment-naive adults infected with HIV-1: an open-label, phase 1b, consecutive-panel trial. Lancet HIV. 2020;7(3):e164–72. https://doi.org/10.1016/S2352-3018(19)30372-8.
Matthews RP, Jackson Rudd D, Levine V, Zhang S, Sterling L, Grobler J, et al. Multiple daily doses of MK-8591 as low as 0.25 mg are expected to suppress HIV. In: Conference on Retroviruses and Opportunistic Infections (CROI); Abstract #26; March 4–7, 2018; Boston, MA, USA.
Matthews R, Ankrom W, Friedman E, Rudd DJ, Liu Y, Zhang S, et al. Safety, tolerability and pharmacokinetics of single- and multiple-dose administration of islatravir (MK-8591) in adults without HIV. Clin Transl Sci. 2021.
PIFELTRO™. (doravirine) [prescribing information]. Whitehouse Station, NJ, USA: Merck Sharp & Dohme Corp. Revised 09 2019.
Feng M, Sachs NA, Xu M, Grobler J, Blair W, Hazuda DJ, et al. Doravirine suppresses common nonnucleoside reverse transcriptase inhibitor-associated mutants at clinically relevant concentrations. Antimicrob Agents Chemother. 2016;60(4):2241–7. https://doi.org/10.1128/AAC.02650-15.
Lai MT, Feng M, Falgueyret JP, Tawa P, Witmer M, DiStefano D, et al. In vitro characterization of MK-1439, a novel HIV-1 nonnucleoside reverse transcriptase inhibitor. Antimicrob Agents Chemother. 2014;58(3):1652–63. https://doi.org/10.1128/AAC.02403-13.
Bleasby K, Fillgrove KL, Houle R, Lu B, Palamanda J, Newton DJ, et al. In vitro evaluation of the drug interaction potential of doravirine. Antimicrob Agents Chemother. 2019;63:4. https://doi.org/10.1128/AAC.02492-18.
Molina JM, Squires K, Sax PE, Cahn P, Lombaard J, DeJesus E, et al. Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. Lancet HIV. 2018;5(5):e211–20. https://doi.org/10.1016/S2352-3018(18)30021-3.
Orkin C, Squires KE, Molina JM, Sax PE, Wong WW, Sussmann O, et al. Doravirine/lamivudine/tenofovir disoproxil fumarate is non-inferior to efavirenz/emtricitabine/tenofovir disoproxil fumarate in treatment-naive adults with human immunodeficiency virus-1 infection: week 48 results of the DRIVE-AHEAD Trial. Clin Infect Dis. 2019;68(4):535–44. https://doi.org/10.1093/cid/ciy540.
Wong A, Goldstein D, Mallolas J, DeJesus E, Johnson M, Molina JM, et al. Efficacy and safety of doravirine/lamivudine/tenofovir disoproxil fumarate (DOR/3TC/TDF) in treatment-naive HIV-1 infected adults with transmitted non-nucleoside reverse transcriptase inhibitor (NNRTI) resistance mutations. J Acquir Immune Defic Syndr. 2019;82(4):e47–9. https://doi.org/10.1097/QAI.0000000000002153.
Johnson M, Kumar P, Molina JM, Rizzardini G, Cahn P, Bickel M, et al. Switching to doravirine/lamivudine/tenofovir disoproxil fumarate (DOR/3TC/TDF) maintains HIV-1 virologic suppression through 48 weeks: results of the DRIVE-SHIFT trial. J Acquir Immune Defic Syndr. 2019;81(4):463–72. https://doi.org/10.1097/QAI.0000000000002056.
Anderson MS, Gilmartin J, Cilissen C, De Lepeleire I, Van Bortel L, Dockendorf MF, et al. Safety, tolerability and pharmacokinetics of doravirine, a novel HIV non-nucleoside reverse transcriptase inhibitor, after single and multiple doses in healthy subjects. Antivir Ther. 2015;20(4):397–405. https://doi.org/10.3851/IMP2920.
Orkin C, Elion R, Thompson M, Rockstroh J, Xu ZJ, Martin EA, et al. Effect of doravirine on body weight and body mass index in treatment naive adults with HIV-1. Abstract # PS3/2; Oral presentation at European AIDS Conference (EACS); November 6–9, 2019; Basel, Switzerland.
Anderson MS, Khalilieh S, Yee KL, Liu R, Fan L, Rizk ML, et al. A two-way steady-state pharmacokinetic interaction study of doravirine (MK-1439) and dolutegravir. Clin Pharmacokinet. 2017;56(6):661–9. https://doi.org/10.1007/s40262-016-0458-4.
Yee KL, Khalilieh SG, Sanchez RI, Liu R, Anderson MS, Manthos H, et al. The effect of single and multiple doses of rifampin on the pharmacokinetics of doravirine in healthy subjects. Clin Drug Investig. 2017;37(7):659–67. https://doi.org/10.1007/s40261-017-0513-4.
Khalilieh SG, Yee KL, Sanchez RI, Liu R, Fan L, Martell M, et al. Multiple doses of rifabutin reduce exposure of doravirine in healthy subjects. J Clin Pharmacol. 2018;58(8):1044–52. https://doi.org/10.1002/jcph.1103.
Khalilieh SG, Yee KL, Sanchez RI, Fan L, Anderson MS, Sura M, et al. Doravirine and the potential for CYP3A-mediated drug-drug interactions. Antimicrob Agents Chemother. 2019;63:5. https://doi.org/10.1128/AAC.02016-18.
Khalilieh S, Yee KL, Sanchez RI, Triantafyllou I, Fan L, Maklad N, et al. Results of a doravirine-atorvastatin drug-drug interaction study. Antimicrob Agents Chemother. 2017;61:2. https://doi.org/10.1128/AAC.01364-16.
Sanchez RI, Yee KL, Fan L, Cislak D, Martell M, Jordan HR, et al. Evaluation of the pharmacokinetics of metformin following coadministration with doravirine in healthy volunteers. Clin Pharmacol Drug Dev. 2019;9(1):107–14. https://doi.org/10.1002/cpdd.685.
Kirby KA, Michailidis E, Fetterly TL, Steinbach MA, Singh K, Marchand B, et al. Effects of substitutions at the 4’ and 2 positions on the bioactivity of 4’-ethynyl-2-fluoro-2’-deoxyadenosine. Antimicrob Agents Chemother. 2013;57(12):6254–64. https://doi.org/10.1128/AAC.01703-13.
Amanlou M, Saboury AA, Bazl R, Ganjali MR, Sheibani S. Adenosine deaminase activity modulation by some street drug: molecular docking simulation and experimental investigation. Daru. 2014;22:42. https://doi.org/10.1186/2008-2231-22-42.
Kutryb-Zajac B, Mierzejewska P, Slominska EM, Smolenski RT. Therapeutic perspectives of adenosine deaminase inhibition in cardiovascular diseases. Molecules. 2020;25:20. https://doi.org/10.3390/molecules25204652.
Molina JM, Yazdanpanah Y, Afani Saud A, Bettacchi C, Chahin Anania C, DeJesus E, et al. MK-8591 at doses of 0.25 to 2.25 mg QD, in combination with doravirine establishes and maintains viral suppression through 48 weeks in treatment-naïve adults with HIV-1 infection. Abstract #4789. Presented at 10th Internationa AIDS Society Conference on HIV Science (IAS); July 21–24, 2019; Mexico City, Mexico.
Taylor PJ, Tai CH, Franklin ME, Pillans PI. The current role of liquid chromatography-tandem mass spectrometry in therapeutic drug monitoring of immunosuppressant and antiretroviral drugs. Clin Biochem. 2011;44(1):14–20. https://doi.org/10.1016/j.clinbiochem.2010.06.012.
Khalilieh S, Yee KL, Sanchez R, Stoch SA, Wenning L, Iwamoto M. Clinical pharmacokinetics of the novel HIV-1 non-nucleoside reverse transcriptase inhibitor doravirine: an assessment of the effect of patient characteristics and drug-drug interactions. Clin Drug Investig. 2020;40(10):927–46. https://doi.org/10.1007/s40261-020-00934-2.
Yee KL, Ouerdani A, Claussen A, de Greef R, Wenning L. Population pharmacokinetics of doravirine and exposure-response analysis in individuals with HIV-1. Antimicrob Agents Chemother. 2019;63:4. https://doi.org/10.1128/AAC.02502-18.
Ankrom W, Yee KL, Sanchez RI, Adedoyin A, Fan L, Marbury T, et al. Severe renal impairment has minimal impact on doravirine pharmacokinetics. Antimicrob Agents Chemother. 2018;62:8. https://doi.org/10.1128/AAC.00326-18.
Flinn AM, Gennery AR. Adenosine deaminase deficiency: a review. Orphanet J Rare Dis. 2018;13(1):65. https://doi.org/10.1186/s13023-018-0807-5.
Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. 2018;38(5):569–78. https://doi.org/10.1007/s10875-018-0525-8.
Holec AD, Mandal S, Prathipati PK, Destache CJ. Nucleotide reverse transcriptase inhibitors: a thorough review, present status and future perspective as HIV therapeutics. Curr HIV Res. 2017;15(6):411–21. https://doi.org/10.2174/1570162X15666171120110145.
Begley R, Das M, Zhong L, Ling J, Kearney BP, Custodio JM. Pharmacokinetics of tenofovir alafenamide when coadministered with other HIV antiretrovirals. JAIDS J Acquir Immune Defic Syndromes. 2018;78(4):465–72. https://doi.org/10.1097/qai.0000000000001699.
Lahiri CD, Tao S, Jiang Y, Sheth AN, Acosta EP, Marconi VC, et al. Impact of protease inhibitors on intracellular concentration of tenofovir-diphosphate among HIV-1 infected patients. AIDS. 2015;29(9):1113–5. https://doi.org/10.1097/QAD.0000000000000659.
Jackson Rudd D, Cao Y, Vaddady P, Grobler J, Asante-Appiah E, Diamond T, et al. Modeling-supported islatravir dose selection for phase 3. Abstract # 462. Presented at the Conference on Retroviruses and Opportunistic Infections (CROI); March 8–11, 2020; Boston, MA, USA.
Acknowledgements
The authors thank the participants and clinical research staff who participated in this study, and thank Sabrina Fox-Bosetti (Merck & Co., Inc., Kenilworth, NJ, USA) for outstanding operational support. Medical writing and editorial assistance, under the direction of the authors, were provided by Yee-Man Ching, PhD, of ApotheCom (UK) in accordance with Good Publication Practice (GPP3) guidelines. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD), who was also the sponsor of the open access fee.
Conflict of interest
RPM, DJR, KLF, SZ, SAS, and MI are current or former employees of MSD when the study was conducted. CM has nothing to disclose.
Ethics approval
The study was approved by the Chesapeake Research Review, Inc. institutional review board and was conducted in accordance with the Declaration of Helsinki following Good Clinical Practice Guidelines.
Consent to participate
Study participants provided written informed consent.
Consent to publish
Not applicable.
Data availability
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA’s data sharing policy, including restrictions, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.
Code availability
Not applicable.
Author contributions
All authors wrote the manuscript. RPM, DJR, KLF, SAS, and MI designed the trials. CT performed the research. SZ performed the statistical analysis. RPM, DJR, KLF, SZ, SAS, and MI analyzed the data.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Matthews, R.P., Jackson Rudd, D., Fillgrove, K.L. et al. A Phase 1 Study to Evaluate the Drug Interaction Between Islatravir (MK-8591) and Doravirine in Adults Without HIV. Clin Drug Investig 41, 629–638 (2021). https://doi.org/10.1007/s40261-021-01046-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-021-01046-1