
Abstract
Aim
The purpose of this study was to conduct and interpret a pooled 12-month analysis of two prospective, multi-center, randomized, double-masked, controlled trials designed to assess the efficacy and safety of the travoprost intracameral implant (slow-eluting [SE] implant in development as a new therapeutic and fast-eluting [FE] implant included for masking purposes) in subjects with open-angle glaucoma (OAG) or ocular hypertension (OHT).
Methods
Subjects with OAG or OHT, on 0–3 intraocular pressure (IOP)-lowering medications, baseline unmedicated mean diurnal IOP of ≥ 21 mmHg, and IOP ≤ 36 mmHg at each baseline diurnal timepoint, received either a travoprost implant and twice-daily (BID) placebo eye drops or BID timolol 0.5% eye drops and a sham procedure. Subjects were followed through 12 months and assessed for IOP, reduction in topical IOP-lowering medications, and safety parameters including treatment-emergent adverse events (TEAEs). IOP at 8AM was prospectively collected at all study visits through 12 months and diurnal IOP, measured at 8AM, 10AM, and 4PM, was prospectively collected at baseline, day 10, week 6, and months 3 and 12.
Results
A total of 1150 subjects were randomized (385 FE implant, 380 SE implant, and 385 sham/timolol) across the two trials. Statistical non-inferiority to timolol and clinically relevant reductions in 8AM IOPs were demonstrated at month 12. In more detail, both implant groups demonstrated statistical non-inferiority to timolol and clinically relevant reductions from baseline in mean diurnal IOP at all visits over the 12-month evaluation period when diurnal IOP was collected. Additionally, both implant groups demonstrated robust treatment effect based on 8AM average IOP from day 10 through the specified visit which ranged from day 10 to month 12 from 6.9 to 8.5 mmHg in the FE implant group; 6.8 to 8.5 mmHg in the SE implant group; and 7.3 to 7.5 mmHg in the sham/timolol group. With regards to reduction in topical pharmacotherapy, at month 12, 77.6% of FE and 81.4% of SE implant eyes were completely free of all topical IOP-lowering medications and a significantly greater proportion of FE and SE implant eyes (89.9% and 93.0%) versus sham/timolol eyes (66.9%) were on the same or fewer topical IOP-lowering medications compared with pre-study (p < 0.0001). Furthermore, of subjects on topical IOP medications at screening, a significantly greater proportion of FE implant (80.2%) and SE implant (85.1%) eyes versus sham/timolol (22.8%) eyes were on fewer topical IOP-lowering medications at month 12 compared with pre-study (p < 0.0001). Lastly, of SE implant eyes on same or fewer topical IOP-lowering medications at month 12, the average through month 12 decreased by 0.9 medications, and of those SE implant eyes on fewer topical IOP-lowering medications compared with pre-study, the average through month 12 decreased by 1.4 medications. The most common TEAEs related to study treatment were hyperemia (conjunctival or ocular), iritis, and IOP increased.
Conclusion
The travoprost intracameral implant demonstrated robust IOP-lowering efficacy that was sustained and statistically non-inferior to timolol over the entire 12 months, resulting in a significant reduction in topical IOP-lowering medication use, with the majority of SE implant eyes remaining completely free of all topical IOP-lowering medications. In addition, the implant demonstrated a favorable safety and tolerability profile based on this pooled 12-month analysis of two pivotal trials.
Trial Registration
ClinicalTrials.gov identifiers NCT03519386 (registered May 09, 2018) and NCT03868124 (registered March 08, 2019).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
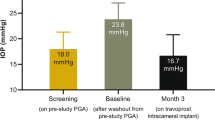
The slow-eluting travoprost intracameral implant demonstrated statistical non-inferiority to timolol 0.5% eye drops and clinically relevant 8AM IOP-lowering efficacy at month 12. |
The slow-eluting travoprost intracameral implant demonstrated statistical non-inferiority to timolol and clinically relevant IOP-lowering efficacy in mean diurnal IOP at all visits through month 12 at which diurnal IOP was prospectively collected. |
Topical IOP-lowering medication burden was significantly reduced in eyes administered a slow-eluting travoprost intracameral implant, with 81.4% of eyes completely free of topical medication at month 12. |
Slow-eluting travoprost intracameral implant demonstrated a favorable safety and tolerability profile. |
1 Introduction
Glaucoma is a chronic progressive optic neuropathy associated with optic nerve damage and visual field changes. Since most glaucoma is associated with elevated intraocular pressure (IOP) and IOP is currently the only modifiable risk factor, treatment strategies focus on lowering IOP. Medical therapy remains the most common initial intervention to lower IOP [1] despite the availability of a variety of laser and incisional surgical techniques, including significant recent advances in microinvasive glaucoma surgery (MIGS) [2].
Adherence to IOP-lowering medical therapy is a primary determinant of glaucoma treatment success, with multiple studies reporting specifically on the association between medication adherence and progression of the visual field loss [3, 4]. Nevertheless, many patients fail to use their medications as prescribed, with more than 90% of all glaucoma patients being non-compliant with refilling their prescriptions for topical IOP-lowering medications [5].
Factors associated with poor medication adherence include the presence of local and/or systemic side effects [6, 7], increased disease severity [8], complicated dosing regimens [9] and comorbidities [10], as well as situational factors such as work schedules or travel [11]. Non-adherence to IOP-lowering medication is also more common in the elderly who have difficulty self-administering eye drops due to decreased manual dexterity or poor vision [12] and who may lack a caregiver to assist them with this task [13, 14]. In addition, glaucoma is chronic and initially asymptomatic in nature; failure to perceive an immediate treatment effect may lead to intentional non-adherence [15].
Given the impact of topical medical burden on glaucoma patients, medication-based therapies designed to reduce the number of topical IOP-lowering medications are of significant interest to patients, providers, and payers.
A variety of ocular drug delivery systems are being developed to reduce the burden of administering topical IOP-lowering medications, the challenges associated with patients’ adherence to these medications, and poor drug bioavailability [16]. One of these systems is the travoprost intracameral implant (Glaukos Corporation, Aliso Viejo, CA, USA), a micro-scale implant (approximately 0.5 mm in diameter and 1.8 mm in length) that is pre-loaded into a sterile, single-dose inserter for administration into the anterior chamber via a micro-invasive ab interno surgical approach. The implant is actively anchored through the trabecular meshwork into the sclera at the iridocorneal angle via scleral anchor. A proprietary formulation of travoprost is released through a drug-eluting membrane with the fast-eluting implant (FE implant) primarily used for clinical trial masking purposes as required by the US FDA and the slow-eluting implant (SE implant) designated for development as a sustained-release IOP-lowering pharmacotherapy.
A phase II clinical trial evaluating the FE implant and SE implant versus twice-daily timolol 0.5% eye drops found that IOP was reduced significantly for up to 3 years after a single implant administration, and that a greater proportion of FE and SE implant patients versus timolol patients had a reduced topical medication burden compared with their pre-study regimen. In addition, the safety profile of the implants was favorable over the 36-month study [17].
Two phase III trials were conducted to evaluate the safety and IOP-lowering efficacy of the SE and FE implant compared with twice-daily timolol 0.5% eye drops in patients with open-angle glaucoma (OAG) or ocular hypertension (OHT) [18, 19]. In both trials, preplanned primary efficacy analyses of data through month 3 have demonstrated non-inferiority of both implant models to timolol for IOP-lowering efficacy, as well as an acceptable safety profile through month 12. The company obtained marketing approval from the United States Food and Drug Administration (U.S. FDA) for the SE implant (iDose® TR, travoprost intracameral implant 75 μg) based on its favorable benefit-to-risk profile. The trials remain underway to collect safety data through 3 years post-administration.
Herein we report on the 12-month safety, IOP-lowering efficacy, and topical medication burden based on the pooled analysis of the combined phase III trials evaluating the FE implant and SE implant versus twice-daily timolol 0.5% eye drops.
2 Methods
2.1 Study Design
Both phase III trials were prospective, multi-center (93 sites in the U.S., one in Armenia, and one in the Philippines), randomized, double-masked, sham- and active-controlled trials to evaluate the efficacy and safety of the FE and SE travoprost intracameral implant versus timolol eye drops in patients with OAG or OHT. The trials were registered at ClinicalTrials.gov (NCT03519386 and NCT03868124) and were conducted in accordance with current good clinical practices, the tenets of the Declaration of Helsinki, and applicable regulatory requirements. Institutional Review Board/Ethics Committee approval was obtained for the trials for each site, with each participant providing written informed consent prior to undergoing any study-related procedures.
2.2 Participants
Key eligibility criteria for the prospective study eye at the screening visit included the following: diagnosis of OAG or OHT with cup-to-disc (C/D) ratio no greater than 0.8 and visual field mean deviation no worse than −12 dB; on no IOP-lowering medication, or on one to three topical IOP-lowering medications and able to safely undergo a washout period; best-corrected visual acuity (BCVA) of 20/80 Snellen or better; open anterior chamber angle (Shaffer grade ≥ 3) with absence of structural abnormalities that could interfere with placement of the implant; and central corneal thickness ≥ 440 but ≤ 620 microns.
Key eligibility criteria for the study eye at the baseline visit following a washout period, if applicable, included the following: mean diurnal IOP ≥ 21 mmHg and IOP at each of the individual diurnal timepoints (8AM, 10AM, and 4PM) ≤ 36 mmHg.
Key exclusion criteria for the study eye included the following: prior glaucoma surgery or argon laser trabeculoplasty; iridotomy, selective laser trabeculoplasty, or micropulse laser trabeculoplasty in the past 90 days; corneal inflammation, infection, clinically significant dystrophy or guttata, opacities or disorders that could affect the reliability of applanation tonometry or hamper the ability to visualize the nasal iridocorneal angle; visually significant cataract or prior complicated cataract surgery; retinal or choroidal disorders or anomalies that could impact the conduct of the study; and ocular infection, inflammation, or other pathology.
In addition, a subject would be excluded if the non-study eye had a BCVA of worse than 20/80 Snellen; if the subject was < 18 years of age; was pregnant or planning a pregnancy; had uncontrolled systemic disease or an immunodeficiency disorder; was using systemic steroids (other than inhaled steroids or those applied to the skin within ¼ inch of the eye); had any allergy or contraindication to the use of an ophthalmic prostaglandin analog (PGA) or β-blocker, or to benzalkonium chloride (BAK)-preserved eye drops; or was currently participating in another clinical trial or had done so within the past 30 days.
Both eyes could be evaluated for study inclusion/exclusion at the screening and baseline visits; however, only one eye was subsequently randomized and treated with study medication. If both eyes qualified at the baseline visit, the right was selected as the study eye. The non-study eye was treated in accordance with the investigators’ judgement.
2.3 Procedures
At the screening visit, subjects underwent the informed consent process. Medical/ophthalmic history and concomitant medications were documented, and manifest refraction, Snellen BCVA measurement, standard automated perimetry (Humphrey 24-2 SITA standard, Octopus 24-2 Dynamic, or Compass 24-2 ZEST), slit lamp biomicroscopy, applanation tonometry, ultrasound pachymetry, gonioscopy, and dilated ophthalmoscopy (including assessment of C/D ratio) were performed.
Subjects on IOP-lowering medication(s) at the screening visit but who otherwise qualified underwent a washout period (56 days for rho-kinase inhibitors, 28 days for PGAs and β-blockers, 21 days for α-adrenergic agonists, 7 days for carbonic anhydrase inhibitors [CAIs], and 5 days for miotics). To mitigate the risk of a long washout period and its possible effect on the health of the eye, the subject could be placed on an IOP-lowering medication with a shorter washout period (i.e., a CAI) provided the appropriate duration washout from that medication was undertaken prior to the baseline visit. Subjects on no IOP-lowering medication at the screening visit could proceed directly to the baseline visit.
At the baseline visit, any changes in medical/ophthalmic history and concomitant medications were documented, and a pregnancy test (for women of childbearing potential), manifest refraction, Early Treatment Diabetic Retinopathy Study (ETDRS) BCVA measurement, slit lamp biomicroscopy (including assessment of conjunctival hyperemia, iris color, and grading of lens opacities using the Age-Related Eye Disease Study [AREDS] Clinical Lens Grading System [ARLNS] in phakic eyes) [20], specular microscopy (at selected sites), and applanation tonometry were performed.
Eligible subjects were scheduled for their day 0 operative visit and dispensed a topical antibiotic to be used for at least 1 day pre-operatively.
At day 0, subjects were randomized in a 1:1:1 treatment allocation to the FE implant, SE implant or sham procedure. Both implant models were evaluated in order to maintain masking of the implants. The randomization was also stratified by baseline mean diurnal IOP (average of the 8AM, 10AM, and 4PM IOP ≤ 25 mmHg and average of the 8AM, 10AM, and 4PM IOP > 25 mmHg).
Procedures were performed under aseptic conditions at ambulatory surgical centers, at hospital outpatient facilities, or in surgical suites in office-based settings. Administration of the travoprost intracameral implant was composed of the following steps: a drop of antibiotic was administered 30 minutes pre-operatively; the eye was anesthetized using general, retrobulbar, peribulbar, or topical anesthesia; a temporal clear corneal incision was made peripherally and a cohesive viscoelastic was added to the anterior chamber to form the chamber and improve visualization of the angle. The implant injector was then advanced nasally (to the approximately 2 o’clock position for the right eye; approximately 10 o’clock position for the left eye) under direct gonioscopy to the trabecular meshwork where the implant was securely anchored through the meshwork into the sclera (Fig. 1).

Gonioscopic view of travoprost intracameral implant anchored into the sclera at the iridocorneal angle
The sham procedure consisted of administering a drop of antibiotic and a topical anesthetic prior to pressing a sterile tuberculin syringe against the conjunctiva of the anesthetized eye.
Following administration of the implant or sham procedure, subjects in the implant groups were dispensed masked kits containing placebo eye drops (a BAK-preserved artificial tear solution) and those who received a sham procedure were dispensed masked kits containing timolol maleate ophthalmic solution, 0.5%. All subjects were instructed to administer their eye drops twice daily to the study eye, in the morning at approximately 8AM ± 1 hour and in the evening at approximately 8PM ± 1 hour. On the day of study visits, the investigator or their designee administered the masked eye drop after the 8AM IOP assessment. Masking of the eye drops was accomplished by having identical-appearing dropper bottles, cap color, and labeling.
Follow-up visits through the month-12 timepoint included day 1–2, day 10, week 4, week 6, month 3, 6, 9, and 12. An assessment of concomitant medications and adverse events was undertaken at all follow-up visits, as was measurement of IOP, and slit lamp biomicroscopy (including assessment of conjunctival hyperemia). Snellen pinhole visual acuity was measured at day 10, and ETDRS visual acuity was measured at all subsequent visits. Specular microscopy was performed at months 3 and 12, standard automated perimetry at months 6 and 12, pachymetry at month 12, gonioscopy at day 10, week 4, week 6, months 3, 6, 9, and 12, and ophthalmoscopy at months 3, 6, 9, and 12 (under dilation at month 12, and including C/D assessment at months 6 and 12). Additional masked kits of eye drops (timolol or placebo) were dispensed to subjects as needed at the follow-up visits.
IOP was measured via Goldmann applanation tonometry at all visits using a 2-examiner technique in which the examiner who viewed the mires on the cornea and turned the dial of the tonometer was unmasked, and the individual who read and recorded the measurement was masked. At the screening visit, day 1–2, week 4, and at months 6 and 9, IOP was assessed at a single timepoint (8AM, 10AM, or 4PM). At the baseline visit, day 10, week 6, month 3, and month 12, diurnal IOP (8AM, 10AM, and 4PM) was assessed. At each evaluation timepoint, two readings of the IOP were taken. If these readings differed by > 2 mmHg, a third reading was taken. The IOP for each timepoint was the mean of the two readings or the median of the three readings.
If the IOP in the study eye was > 22 mmHg at day 3 or later, IOP was to be rechecked within 7 days. Additional IOP-lowering medication, preferably a topical CAI, was to be prescribed if the rechecked IOP was > 22 mmHg and ≤ 25 mmHg, and the IOP reduction from baseline was < 20%, or if the rechecked IOP was > 25 mmHg regardless of the percent reduction from baseline.
2.4 Outcomes
Efficacy outcome measures for this analysis included change from baseline in mean diurnal IOP for all visits at which diurnal IOP was prospectively collected, 8AM IOP change from baseline by study visit, the proportion of subjects without additional topical IOP-lowering medication, the proportion of subjects on the same or fewer topical IOP-lowering medications at month 12 compared with pre-study and their number of medications pre-study and average through month 12, the proportion of subjects on fewer topical IOP-lowering medications at month 12 compared with pre-study and their number of medications pre-study and average through month 12.
Treatment-emergent adverse events (TEAEs) (solicited and observations from safety assessments), BCVA, conjunctival hyperemia, endothelial cell density (from specular microscopy images analyzed by a masked centralized reading center), visual fields, and central corneal thickness were evaluated as safety outcomes.
2.5 Statistical Analysis
The sample size for each of the two phase III trials was based on the primary efficacy endpoint for regulatory approval in the US described previously [19].
The sample size of approximately 380 patients per treatment group in this combined analysis of the phase III trials provides > 99% power to reject the null hypothesis that the difference (implant group minus sham/timolol group) in mean change from baseline in mean diurnal IOP ≥ 1.5 mmHg (i.e., using a non-inferiority margin of 1.5 mmHg) at each visit over the first 3 months. (Note: the study was not powered to demonstrate a treatment effect with statistical significance in individual subgroups.)
Non-inferiority was declared for a visit (day 10, week 6, month 3, and month 12 visits) if the upper limit of the two-sided 95% confidence interval (CI) around the difference (implant group minus sham/timolol group) in mean change from baseline in mean diurnal IOP was < 1.5 mmHg. Note, the day 10, week 6, month 3, and month 12 visits were the visits at which diurnal IOP was prospectively collected.
Data analyses were performed using SAS version 9.4 (SAS Institute, Cary, North Carolina, USA). Within each treatment group, the change from baseline in IOP at each visit was analyzed using a one-sample t-test. Two-sample t-tests were used to test for differences between the treatment groups for continuous variables (including 8AM IOP reductions in average IOP from day 10 through the specified visit), and chi-square or Fisher’s exact tests were used to test categorical variables. A p-value of < 0.05 indicated statistical significance.
Treatment group differences with 95% confidence intervals (CIs) for mean diurnal IOP were based on an analysis of covariance model with treatment group as a factor and using time-matched baseline IOP as the covariate at each visit. Multiple imputation techniques were used to impute missing data.
3 Results
3.1 Disposition
The first subject was screened in May 2018, and the last subject completed their month 12 visit in June 2022.
A total of 1973 subjects were screened, of whom 1150 were randomized and comprised the intent-to-treat (ITT) analysis set: 385 FE implant subjects, 380 SE implant subjects, and 385 sham/timolol subjects. The safety analysis set comprised 1149 subjects: 385 FE implant subjects, 378 SE implant subjects, and 386 sham/timolol subjects.
Of the 1150 subjects randomized at the day 0 operative visit, 96.0% completed the 12-month evaluation period.
3.2 Demographics and Baseline Characteristics
Subject demographics and study eye disease characteristics were well balanced across the three treatment groups (Tables 1 and 2, respectively).
Most subjects had a diagnosis of OAG in the study eye (83.0%), were on one or more topical IOP-lowering medications at screening (70.4%), and had a post-washout mean diurnal IOP (average of 8AM, 10AM, and 4PM) of 25 mmHg or less (71.0%). The mean number of pre-study topical IOP-lowering medications was 0.9, 0.9, and 1.0 in the FE implant, SE implant, and sham/timolol groups, respectively.
3.3 Efficacy
3.3.1 Intraocular Pressure (IOP) Reductions Through Month 12
Mean (SD) baseline diurnal IOP (average of the 8AM, 10AM, and 4PM timepoints) was 24.2 (2.88) mmHg for the overall study population. Reductions from baseline in mean diurnal IOP (i.e., the minimum and maximum across the day 10, week 6, month 3, and month 12 visits) were clinically relevant and statistically significant ranging from 5.4 to 8.2 mmHg for the FE implant group, 5.4 to 8.4 mmHg for the SE implant group, and from 6.1 to 7.2 mmHg for the sham/timolol group across the four visits at which diurnal IOP was prospectively collected. In addition, both the SE and FE implant groups demonstrated statistical non-inferiority to the sham/timolol group in reduction from baseline in mean diurnal IOP across all study visits over the entire 12 months when diurnal IOP was prospectively collected. The upper limit of the two-sided 95% confidence interval on the difference between the implant groups and the sham/timolol group was < 1.5 mmHg at all four visits (Table 3).
Mean (SD) baseline 8AM IOP was 24.6 (3.55) mmHg for the overall study population. The 8AM IOP reductions in average IOP from day 10 through the specified visit ranged from 6.9 to 8.5 mmHg in the FE implant group, from 6.8 to 8.5 mmHg in the SE implant group, and from 7.3 to 7.5 mmHg in the sham/timolol group (Fig. 2). The 8AM IOP reductions in average IOP from day 10 through the specified visit were statistically significant (p < 0.0001) and clinically relevant at all visits for all three treatment groups. In the same analysis, these 8AM IOP reductions in both the SE and FE implant groups demonstrated statistical non-inferiority to the sham/timolol group over the entire 12 months.

Mean (± standard error) 8AM IOP change from baseline for the FE implant, SE implant, and sham/timolol averaged from day 10 through each specified visit. IOP change from baseline was statistically significant (p < 0.0001; one-sample t tests) for all treatment groups at all visits. IOP change from baseline was significantly greater in the FE implant and SE implant groups versus sham/timolol group at day 10 (p = 0.0011 and p = 0.0005, respectively). FE Implant fast-eluting travoprost intracameral implant, IOP intraocular pressure, SE Implant slow-eluting travoprost intracameral implant
3.3.2 Topical IOP-Lowering Medications
Pre-study, subjects in both the FE and SE implant groups were on a mean (SD) of 0.9 (0.8) medications per study eye, and 21.6% and 23.1% of FE and SE implant eyes, respectively, were on two or three pre-study topical IOP-lowering medications.
Over the 12-month evaluation period, the proportion of subjects without additional IOP-lowering medications in the study eye decreased in all three treatment groups (Table 4). However, at month 12, the proportion of implant eyes that remained completely free of topical IOP-lowering medications was high at 77.6% of FE implant eyes and 81.4% of SE implant eyes despite the fairly high treatment burden pre-study.
In addition, at month 12, a significantly greater proportion of implant eyes (89.9% of FE implant eyes, 93.0% of SE implant eyes) compared with sham/timolol eyes (66.9%) were on the same or fewer topical IOP-lowering medications compared with pre-study (p < 0.0001), and of eyes that were on IOP-lowering medication at screening, a significantly greater proportion of implant eyes (80.2% of FE implant eyes, 85.1% of SE implant eyes) compared with sham/timolol eyes (22.8%) were on fewer topical IOP-lowering medications compared with pre-study (p < 0.0001).
Of the 93.0% of subjects in the SE implant group who were on the same or fewer topical IOP-lowering medications at month 12 compared with pre-study, the average through month 12 decreased to a mean (SD) of 0.1 (0.2) medications per eye from 1.0 (0.8) medications per eye, and of the 85.1% subjects in the SE implant group who were on fewer topical IOP-lowering medications compared with pre-study, the average through month 12 decreased to a mean (SD) of 0.0 (0.1) medications per eye from 1.4 (0.6) medications per eye.
3.4 Safety
3.4.1 Study Eye Treatment-Emergent Adverse Events (TEAEs)
TEAEs were reported in 35.8% and 35.7% of FE implant (138/385) and SE implant (135/378) study eyes, respectively, and in 18.9% of sham/timolol (72/386) eyes (Table 5).
Serious TEAEs were reported in 0.3% and 0.8% of FE implant (1/385) and SE implant (3/378) study eyes, respectively, and in no sham/timolol study eyes. These serious TEAEs included IOP increased (one FE implant eye and one SE implant eye), retinal detachment (one SE implant eye), and endophthalmitis (one SE implant eye). Both TEAEs of IOP increased and the TEAE of endophthalmitis were related to study treatment. The TEAEs of IOP increased were reported at study day 19 and day 42 and resolved with the use of topical IOP-lowering medication. The TEAE of retinal detachment, which was unrelated to study treatment, was reported at study day 189 and resolved with surgical intervention (pars plana vitrectomy, laser, and perfluropropane gas). The TEAE of endophthalmitis was reported at study day 8 and resolved following treatment with intravitreal ceftazidime and vancomycin.
TEAEs leading to study discontinuation were reported in a low proportion of study eyes in each treatment group; 1.8%, 1.4%, and 0.8% in the FE implant (7/385), SE implant (4/378) and sham/timolol (3/386) eyes, respectively. One study eye TEAE leading to study discontinuation was characterized as a serious TEAE (endophthalmitis, in an SE implant eye).
Ocular TEAEs in the study eyes were mild or moderate in severity, with only 1.8%, 1.6%, and 0.5% of FE implant (7/385), SE implant (6/378), and sham/timolol (2/386) study eyes, respectively, having severe TEAEs.
Treatment-related TEAEs were reported in 19.2% and 18.5% of FE implant (74/385) and SE implant (70/378) study eyes, and in 2.3% of sham/timolol (9/386) study eyes. The most common treatment-related TEAEs (reported at an incidence of > 2% in any treatment group) included intraocular pressure increased, iritis, and conjunctival/ocular hyperemia.
As shown in Table 6, treatment-related TEAEs of intraocular pressure increased were reported in 5.2% of FE implant eyes (20/385), 2.9% of SE implant eyes (11/378), and 0.3% of sham/timolol eyes (1/386); however, the vast majority of these events were mild or moderate, and transient in nature. In six of the FE implant eyes (1.6%) and in five of the SE implant eyes (1.3%) with a TEAE of increased IOP, the event was observed 1 or 2 days post-operatively and rapidly resolved. In the remaining 14 FE implant (3.6%) and 6 SE implant eyes (1.3%), reports of increased IOP occurred mostly after month 6 and were treated with the addition of topical IOP-lowering medication.
Treatment-related TEAEs of iritis were reported in 2.1% of FE implant eyes (8/385), 4.8% of SE implant eyes (18/378), and no sham/timolol eyes. Most of these events were mild or moderate, and transient in nature.
Treatment-related TEAEs of conjunctival or ocular hyperemia were reported in 3.9% of FE implant eyes (15/385), 1.9% of SE implant eyes (7/378), and 0.3% of sham/timolol eyes 1/386), with only one study eye (an SE implant eye) having severe conjunctival hyperemia that was transient in nature and coincided with an occurrence of moderate iritis.
Notably, there were no serious corneal TEAEs, no TEAEs related to corneal endothelial cell loss, and no reports of iris hyperpigmentation or periorbital fat atrophy. An adverse event of device dislocation was reported in < 2% of eyes that received an FE or SE implant.
3.4.2 Non-Ocular or Non-Study Eye TEAEs
Non-ocular or non-study eye TEAEs were reported in similar proportions of subjects in the three treatment groups (Table 5). Similarly, serious non-ocular or non-study eye TEAEs were reported in similar proportions of subjects in the three treatment groups, as were severe non-ocular or non-study eye TEAEs. Deaths were reported in 0.8%, 0.3%, and 1.6% of subjects in the FE implant (3/385), SE implant (1/378), and sham/timolol (6/386) groups, respectively. None of the deaths were related to study treatment. Study discontinuation due to non-ocular or non-study eye TEAE was reported in 0.5% of FE implant subjects (2/385) and 0.8% of sham/timolol subjects (3/386). Few subjects had non-ocular or non-study eye TEAEs that were considered related to study treatment; 0.3% and 0.5% of subjects in the FE implant (1/385) and SE implant (2/378) groups, respectively.
3.4.3 Visual Acuity
At baseline, mean (SD) BCVA was 83.0 (6.0), 83.4 (5.4), and 82.4 (5.9) ETDRS letters in the study eyes of subjects in the FE implant, SE implant, and sham/timolol groups, respectively. At month 12, mean (SD) BCVA was 82.5 (5.7), 82.9 (6.0), and 83.0 (6.4) in the FE implant eyes, SE implant eyes, and sham/timolol eyes. This less than one ETDRS letter mean difference between all three treatment groups at month 12 was not clinically meaningful.
Investigators were provided guidance to report a TEAE for any clinically relevant loss of 10 or more ETDRS letters relative to baseline. Less than 2% of subjects in any treatment group had a treatment-related TEAE of reduced visual acuity.
3.4.4 Conjunctival Hyperemia
At baseline, the mean (SD) conjunctival hyperemia score (on a scale of 0 to 3) was 0.26 (0.34), 0.25 (0.36), and 0.27 (0.36) in the FE implant, SE implant, and sham/timolol groups. Post-operatively, hyperemia increased in the implant groups, peaking at day 10 with a mean (SD) of 0.45 (0.56) in the FE implant and 0.44 (0.48) in the SE implant group before decreasing to 0.31 (0.39) and 0.32 (0.41), respectively, at month 12. These low mean values indicate that the vast majority of study eyes experienced no to mild hyperemia.
3.4.5 Endothelial Cell Density
Evaluation of central corneal endothelial cell density via specular microscopy was performed on a subset of subjects (55/385 FE implant subjects [14.3%]; 45/378 SE implant subjects [11.9%]; 53/386 sham/timolol subjects [13.7%]). At baseline, mean (SD) values were 2367.6 (423.34), 2453.4 (406.13), and 2437.1 (358.35) cells/mm2 in the study eyes of subjects in the FE implant, SE implant, and sham/timolol groups, respectively. At month 12, mean (SD) endothelial cell density values were 2308.9 (464.92), 2382.5 (428.92), and 2436.6 (368.49) cells/mm2. There were no TEAEs of corneal endothelial cell loss in any group based on a pre-defined threshold of a confirmed ≥ 30% reduction from baseline.
3.4.6 Visual Fields
At baseline, evaluation of visual fields by automated perimetry indicated that the mean (SD) mean deviation (MD) score in study eyes was −2.14 (2.88), −1.93 (2.86), and −2.26 (3.05) dB in the FE implant, SE implant, and sham/timolol groups, respectively. At month 12, the mean change from baseline in MD was −0.23 (2.86), −0.33 (2.73), and −0.24 (3.00) dB in the FE implant, SE implant, and sham/timolol groups, respectively. There was no clinically meaningful difference in visual field MD between the FE implant and sham/timolol groups or between the SE implant and sham/timolol groups.
3.4.7 Central Corneal Thickness
At baseline, mean (SD) central corneal thickness as measured by ultrasonic pachymetry was 554.9 (35.9), 554.5 (35.4), and 554.5 (34.8) µm in the study eyes of the FE implant, SE implant, and sham/timolol groups. At month 12, the change from baseline was −5.5 (18.2), −4.3 (15.7), and +0.7 (13.4) µm in the FE implant, SE implant, and sham/timolol groups, respectively, with differences in change from baseline considered to be not clinically relevant.
3.4.8 Lens Status
At baseline, 12.1%, 10.1%, and 12.6% of subjects in the FE implant, SE implant, and sham/timolol groups had Grade 2 or greater nuclear lens opacities (on a scale ranging from < 1 to > 3). At month 12, this percentage had increased in all treatment groups with the highest level observed in the sham/timolol group (15.6%, 12.8%, and 16.1% of subjects in the FE implant, SE implant, and sham/timolol groups, respectively). Treatment-related TEAEs of cataract were reported in < 2% of phakic eyes that received an FE or SE implant.
4 Discussion
This study in a large population of OAG and OHT subjects demonstrated that both the fast-eluting and slow-eluting models of the travoprost intracameral implant delivered statistically significant and clinically relevant 8AM IOP reductions through month 12. In addition, both implant models delivered clinically relevant reductions from baseline in mean diurnal IOP, that were statistically non-inferior to timolol, at all visits through month 12 when diurnal IOP was measured. The ability of the implants to produce clinically relevant IOP-lowering reductions through month 12 is corroborated by a human pharmacokinetic study which demonstrated that aqueous humor concentrations of travoprost free acid at month 12 were above the established efficacious concentration, and that 50% of the travoprost remained in SE implants explanted 12 months post-administration [21].
Another demonstrated benefit of the travoprost intracameral implant was its ability to reduce subjects’ topical IOP-lowering medication burden. Adequate IOP control without the need for additional topical IOP-lowering medication was observed in up to 77.6% and 81.4% of FE and SE implant eyes, respectively, through month 12. These eyes had been on an average of 0.9 medications per eye pre-study. In addition, at month 12, a significantly greater proportion of implant eyes versus sham/timolol eyes were on the same or fewer topical medications compared with pre-study. Moreover, of eyes that were on topical IOP-lowering medication(s) at screening, a significantly greater proportion of implant eyes versus sham/timolol eyes were actually on fewer topical IOP-lowering medications at month 12 compared with pre-study. Subjects in the implant groups received a single administration of an FE or SE implant in the study eye whereas subjects in the sham/timolol group were required to administer their glaucoma therapy twice daily to their study eye, equating to 730 administrations in a 12-month period. This finding has a direct impact on the quality of life of glaucoma patients since a reduction in topical medications has the potential to reduce local and/or systemic side effects including ocular surface disease [22, 23].
The safety and tolerability of the travoprost intracameral implants was favorable. Despite a greater proportion of FE and SE implant eyes versus sham/timolol eyes having one or more TEAEs, most TEAEs were of mild or moderate severity and transient in nature, with few severe TEAEs being related to study treatment. The relatively low incidence of serious TEAEs or severe treatment-related TEAEs is outweighed by the guarantee of the anchored implant eluting IOP-lowering medication 24/7 directly into the anterior chamber, thereby circumventing the issue of patient non-adherence with topical medications.
Not unexpectedly, given the administration procedure, a greater incidence of treatment-related conjunctival or ocular hyperemia was reported in the implant groups versus the sham/timolol group. However, the investigator-graded mean conjunctival hyperemia score was low in the implant groups, indicative of the small number of eyes with moderate or severe hyperemia. There were no reports of the most common side effects associated with topical PGAs; specifically, there were no reports of prostaglandin-associated periorbitopathy, iris or periocular skin hyperpigmentation, nor lengthening/darkening of the eyelashes during the 12-month evaluation period. Bilateral macular edema was reported in a diabetic subject who had an FE implant in the study eye and was receiving bimatoprost eye drops in the fellow eye; hence the edema was not considered treatment related.
There were several limitations to this study. First, treatment of the fellow (non-study) eye was not standardized, which may have confounded safety results and/or had an impact on IOP in the study eye if timolol was used [24]. Second, study staff may have become unmasked to study treatment via visualization of the implant at the iridocorneal angle. To mitigate the risk of unmasking, subjects were randomized to one of two models of the identically appearing implant despite the desire to commercialize only the SE implant based on its better benefit-to-risk profile in the phase II trial [17] and its enhanced manufacturability relative to the FE implant. Further steps to prevent unmasking included the use of a sham surgical procedure for subjects randomized to timolol, and the use of placebo eye drops for subjects randomized to an implant. Third, timolol 0.5% eye drops served as the active comparator rather than PGA eye drops. Despite PGA eye drops being the most commonly used IOP-lowering medication in clinical practice, timolol 0.5% eye drops remain the most commonly used comparator in glaucoma trials conducted for registration of new IOP-lowering medications in the U.S. and were also used as the comparator in phase III trials with the bimatoprost intracameral implant [25, 26].
However, the study had several noteworthy strengths including its replication of 8AM IOP reductions from baseline compared with those observed through month 12 in the prior phase II trial with the travoprost intracameral implants [17]. In addition, both the current study and the phase II trial showed similar high proportions of implant subjects on the same or fewer IOP-lowering medications at month 12 compared with pre-study [17].
5 Conclusion
This analysis demonstrates that the SE travoprost intracameral implant (iDose® TR [travoprost intracameral implant] 75 μg) provided sustained, clinically relevant IOP lowering through month 12, with a substantial reduction in topical IOP-lowering medication burden.
References
Gedde SJ, Vinod K, Wright MM, American Academy of Ophthalmology Preferred Practice Pattern Glaucoma Panel, et al. Primary open-angle glaucoma preferred practice pattern®. Ophthalmology. 2021;128(1):71–150.
Fellman RK, Mattox C, Singh K, et al. American Glaucoma Society position paper: microinvasive glaucoma surgery. Ophthalmology Glaucoma. 2020;3(1):1–6.
Newman-Casey PA, Niziol LM, Gillespie BW, et al. The association between medication adherence in visual field progression in the collaborative initial glaucoma treatment study. Ophthalmology. 2020;127(4):477–83.
Shu YH, Wu J, Luong T, et al. Topical medication adherence and visual field progression in open-angle glaucoma: analysis of a large US health care system. J Glaucoma. 2021;30:1047–55.
Nordstrom BL, Friedman DS, Mozaffari E, Quigley HA, Walker AM. Persistence and adherence with topical glaucoma therapy. Am J Ophthalmol. 2005;140:598–606.
Slota C, Sayner R, Vitko M, et al. Glaucoma patient expression of medical problems and nonadherence. Optom Vis Sci. 2015;92(5):537–43.
Wolfram C, Stahlberg E, Pfeiffer N. Patient-reported nonadherence with glaucoma therapy. J Ocular Pharmacol Therapeut. 2019;35(4):223–8.
Sleath B, Blalock S, Covert D, et al. The relationship between glaucoma medication adherence, eye drop technique, and visual field defect severity. Ophthalmology. 2011;118(12):2398–402.
Schmier JK, Hulme-Lowe CK, Covert DW. Adjunctive therapy patterns in glaucoma patients using prostaglandin analogs. Clin Ophthalmol. 2014;8:1097–104.
Serbin M, Devine B, Campbell J, Basu A. Assessing health care burden in glaucoma patients with and without physical and mental comorbidities. J Manag Care Spec Pharm. 2020;26(3):325–31.
European Glaucoma Society Terminology and Guidelines for Glaucoma. 5th Edition. Br J Ophthalmol. 2021;105(Suppl 1):1–169.
Tanito M, Mochiji M, Tsutsui A, et al. Factors associated with topical medical instillation failure in glaucoma: VRAMS-QPiG Study. Adv Ther. 2023;40:4907–18.
Tsai T, Robin AL, Smith JP 3rd. An evaluation of how glaucoma patients use topical medications: a pilot study. Trans Am Ophthalmol Soc. 2007;105:29–33 (discussion 33-5).
The Advanced Glaucoma Intervention Study (AGIS). 3. Baseline characteristics of black and white patients. Ophthalmology. 1998;105(7):1137–45.
Rees G, Leong O, Crowston JG, et al. Intentional and unintentional nonadherence to ocular hypotensive treatment in patients with glaucoma. Ophthalmology. 2010;117(5):903–8.
Agrawal AK, Das M, Jain S. In situ gel systems as “smart” carriers for sustained ocular drug delivery. Expert Opin Drug Deliv. 2012;9:383–402.
Berdahl JP, Sarkisian SR Jr, Ang RE, Doan LV, Kothe AC, Usner DW, Katz LJ, Navratil T, Travoprost Intraocular Implant Study Group. Efficacy and safety of the travoprost intraocular implant in reducing topical IOP-lowering medication burden in patients with open-angle glaucoma or ocular hypertension. Drugs. 2024;84(1):83–97.
Katz LJ, Sarkisian SR, Voskanyan LA, et al. Results of the prospective, randomized, controlled, multicenter phase 3 trials of the travoprost intraocular implant versus topical timolol. Invest Ophthalmol Vis Sci. 2023;64:4296.
Sarkisian SR, Ang RE, Lee AM, et al. Phase 3 randomized clinical trial of the safety and efficacy of travoprost intraocular implant in patients with open-angle glaucoma or ocular hypertension. Ophthalmology. 2024. https://doi.org/10.1016/j.ophtha.2024.02.022.
Chew EY, Kim J, Sperduto RD, et al. Evaluation of the Age-Related Eye Disease Study clinical lens grading system AREDS Report No. 31. Ophthalmology. 2010;117(11):2112–9.
Szekely G, Katz LJ, Voskanyan LA, et al. Travoprost intraocular implant (iDoseTR®) delivers therapeutically relevant and durable aqueous humor drug concentration levels at 12 months and demonstrates safety of implant repeat dosing. Invest Ophthalmol Vis Sci. 2023;64:4301 (presented at the 2023 ARVO Annual Meeting, held in New Orleans, LA, April 23-27, 2023).
Leung EW, Medeiros FA, Weinreb RN. Prevalence of ocular surface disease in glaucoma patients. J Glaucoma. 2008;17(5):350–5.
Fechtner RD, Godfrey DG, Budenz D, et al. Prevalence of ocular surface complaints in patients with glaucoma using topical intraocular pressure-lowering medications. Cornea. 2010;29(6):618–21.
Piltz J, Gross R, Shin DH, et al. Contralateral effect of topical beta-adrenergic antagonists in initial one-eyed trials in the ocular hypertension treatment study. Am J Ophthalmol. 2000;130(4):441–53.
Medeiros FA, Walters TR, Kolko M, ARTEMIS 1 Study Group, et al. Phase 3, randomized, 20-month study of bimatoprost implant in open-angle glaucoma and ocular hypertension (ARTEMIS 1). Ophthalmology. 2020;127(12):1627–41.
Bacharach J, Tatham A, Ferguson G, ARTEMIS 2 Study Group, et al. Phase 3, randomized, 20-month study of the efficacy and safety of bimatoprost implant in patients with open-angle glaucoma and ocular hypertension (ARTEMIS 2). Drugs. 2021;81(17):2017–33.
Acknowledgements
The authors acknowledge Sara Heedy, PhD of Glaukos Corporation for her assistance with preparation of the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was sponsored by Glaukos Corporation (Aliso Viejo, CA, USA). Glaukos participated in the design of the study, data management, analysis and interpretations, and the preparation, review, and approval of the manuscript.
Conflict of Interest
I. Paul Singh has received consulting fees from Allergan, Alcon, Bausch + Lomb, Glaukos, iStar Medical, New World Medical, Nova Eye, and Sight Sciences; and lecture fees from Allergan, Alcon, Bausch + Lomb, Glaukos, iStar Medical, New World Medical, Nova Eye, Sight Sciences, Thea and Viatris. John P. Berdahl has received consulting fees, has been a paid advisory board member or received fees for attending a meeting for AbbVie, Aerpio, Alcon, Aldeyra, Aurea Medical, Aurion Biotech/CorneaGen, Balance Ophthalmics, Bausch and Lomb, Dakota Lions Eye Bank, Elios Vision Inc., Equinox, Expert Opinion, Glaukos, Gore, Imprimis, Interfeen, iRenix, Iacta Pharmaceuticals, IVERIC bio, Inc., JNJ, Kala, Kedalion, MELT Pharmaceuticals, MicroOptx, New World Medical, Ocular Surgical Data, Ocular Therapeutix, Omega Ophthalmic, Orasis, Oyster Point, RxSight, Santen, Sight Sciences, Surface Inc., Tarsus, Tear Clear, Vertex Ventures, ViaLase, Vittamed, Vance Thompson Vision, Versea Biologics, Visionary Ventures, Visus and Zeiss; has received lecture fees (honoraria), travel fees or reimbursements when speaking for AbbVie, Alcon and Glaukos; has equity ownership/stock options of Aurion Biotech/CorneaGen, Balance Ophthalmics, Equinox, Expert Opinion, Interfeen, Ocular Surgical Data, Omega Ophthalmic, Surface Inc, Vance Thompson Vision, Verana Health and Zeiss; owns stock of Glaukos; has patents and/or royalties with Imprimis. Steven R. Sarkisian Jr is a consultant/advisor for Alcon, Allergan, Bausch + Lomb, Beaver-Visitec International, Glaukos, Icare USA, Katena Products, MicroSurgical Technology, Ocular Science, Santen, and Sight Sciences; is on the speaker’s bureau for Alcon, Allergan, and Bausch + Lomb; has received grant support from Alcon, Allergan, Allysta Pharmaceuticals, Elios, Glaukos, iSTAR Medical, Ocular Science, Ocular Therapeutix, and Sight Sciences; and holds stock or stock options in Ocular Science and Sight Sciences. Lilit A. Voskanyan has received research support from Glaukos. Robert E. Ang has been a speaker for and has received research support from Glaukos. Long V. Doan, David Applegate, Yannan Shen, L. Jay Katz, Angela C. Kothe, and Tomas Navratil are employees of Glaukos Corporation and may have Glaukos stock and/or stock options.
Ethics Approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval for the majority of sites was granted by WCG IRB (formerly known as Western Institutional Review Board); approval for single sites each was granted by Wills Eye Hospital Institutional Review Board, St. Cabrini Medical Center-Asian Eye Institute Ethics Review Committee, and the Ethics Committee of Ophthalmological Center named after S.V. Malayan.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Authors’ Contributions
The study was designed by LVD, LJK and DA; data collection was performed by IPS, JPB, SRS, LAV, and REA; statistical analyses were performed by YS; data interpretation was performed by DA, YS, ACK, and TN. Authoring, revisions, and approval of the manuscript: IPS, JPB, SRS, LAV, REA, LVD, DA, YS, LJK, ACK, and TN.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Singh, I.P., Berdahl, J.P., Sarkisian, S.R. et al. Long-Term Safety and Efficacy Evaluation of Travoprost Intracameral Implant Based on Pooled Analyses from Two Phase III Trials. Drugs (2024). https://doi.org/10.1007/s40265-024-02074-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s40265-024-02074-9