
Abstract
Objective
We aimed to assess the bioequivalence, safety, and tolerability of Chinese- and French-manufactured Glucophage® immediate-release (GIR) tablets under fasted and fed conditions in healthy volunteers. A bioequivalence study was proposed to support the manufacturing transfer.
Methods
This was an open-label, randomized, two-period, two-sequence, crossover study. Subjects were randomly assigned to receive the test product (one 500 mg GIR tablet manufactured in China) or reference product (one 500 mg GIR tablet manufactured in France). The primary study endpoint was the area under the plasma concentration-time curve from time zero to the last sampling time (AUCt) and maximum observed concentration (Cmax).
Results
In total, 96 subjects were screened and 44 subjects were randomly assigned to treatment (fasted group, 26 subjects; fed group, 18 subjects). All 44 subjects received the study drug, completed the study, and were included in the pharmacokinetic (PK) and safety analysis sets. Under fasted or fed conditions, the mean AUCt and Cmax (primary PK parameters) were comparable between the test and reference products. Point estimates for both parameters were close to 100% and the corresponding 90% confidence intervals were within the specified 80–125% bioequivalence boundary. There were no hypoglycemia-related adverse events (AEs) in either treatment group. All AEs in the present study were mild in severity.
Conclusions
Bioequivalence between the test and reference GIR tablets was demonstrated under fasted and fed conditions and both were safe and well tolerated.
Clinical Trials Registration
This study was registered at ClinicalTrials.gov under the identifying number NCT03393208.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The bioequivalence of French- and Chinese-manufactured Glucophage® 500 mg immediate-release tablets was demonstrated under both fasted and fed conditions in healthy Chinese volunteers. |
1 Introduction
Metformin hydrochloride is an oral glucose-lowering drug that belongs to the class of biguanides. It is considered a foundation therapy for patients with newly diagnosed type 2 diabetes mellitus (T2DM) [1]. Unless contraindicated or not tolerated, metformin is recommended as first-line oral antidiabetic therapy in patients with T2DM by the American Diabetes Association and European Association for the Study of Diabetes [2]. Metformin reduces gluconeogenesis, opposes glucagon-mediated signaling in the liver, and increases glucose uptake in the skeletal muscle [3]. Of note, metformin alone does not cause hypoglycemia because metformin does not directly stimulate insulin secretion [4].
In a previous prospective, open-label pharmacokinetics (PK) study of metformin in adult Chinese patients with T2DM, the mean apparent clearance was 53.0 L/h, apparent volume of distribution was 438 L, absorption rate constant was 1.4 h−1, and lag-time was 0.91 h [5]. Furthermore, the estimated glomerular filtration rate was found to significantly affect the clearance of metformin. In a previous PK study that assessed the bioequivalence of the test and reference metformin hydrochloride tablets in healthy Chinese volunteers under fasting and fed conditions, the median time to reach the maximum observed concentration (tmax) was approximately 2 h for both formulations under fed and fasted conditions [6]. In a randomized phase I study that assessed the bioequivalence and safety of generic metformin hydrochloride (test preparation) and Glucophage® (reference preparation) in healthy Chinese subjects, generic metformin was found to be bioequivalent and as safe as Glucophage® under fed conditions in healthy Chinese subjects [7].
The Glucophage® immediate-release (GIR) tablet is an oral glucose-lowering drug containing metformin hydrochloride as an active ingredient. A bioequivalence study was proposed to support the Generics Quality Consistency Evaluation in China, a campaign started by China’s Health Authority in 2015 to improve the overall quality of generic drug manufacturing by the pharmaceutical industry. This study aimed to investigate the bioequivalence of 500 mg GIR tablets manufactured by Sino-American Shanghai Squibb Pharmaceuticals in China (test product) and 500 mg GIR tablets manufactured by Merck Santé S.A.S in France (reference product) to demonstrate whether the quality of the test product is equal to that of the reference product.
The primary objective of this study was to assess the bioequivalence of the GIR tablets manufactured in China and France following single oral dose administration under fasted and fed conditions in healthy volunteers. The secondary objectives were to compare additional PK parameters and the safety and tolerability of the GIR tablets after single oral dose administrations of the test and reference products.
2 Materials and Methods
2.1 Study Design
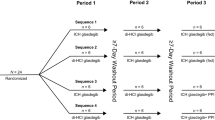
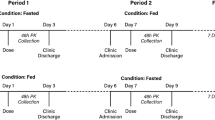
This was an open-label, randomized, two-period, two-sequence, crossover study. The study design is shown in Fig. 1. In each study period, subjects were randomly assigned to receive the test product (one 500 mg GIR tablet manufactured in China) or the reference product (one 500 mg GIR tablet manufactured in France). The study had a screening period of 2 weeks before the first GIR tablet administration, a first dosing/sampling period up to 2 days after dosing, a washout period of at least 7 days after the first GIR tablet administration, a second dosing/sampling period up to 2 days after dosing, and a conditional follow-up examination period (only for subjects with any ongoing adverse events [AEs] at discharge) of up to 7 days following the last study drug administration.

Study design. GIR Glucophage® immediate release
Eligible subjects were assigned to fasted or fed groups first, and subjects in both groups received the allocated treatment according to a computer-generated randomization schedule. In addition, both sexes were to be represented (each sex representing ≥ 25% of the total number) in each study group. In both groups, subjects were required to fast (i.e., refrain from all foods and liquids, except water) for at least 10 h from the previous night to the time of drug administration in the fasted group or the consumption of a high-fat, high-calorie breakfast in the fed group. Additionally, all subjects were required to refrain from drinking water during the first 2 h after drug administration.
This study was approved by the Ethics Committee of Xuanwu Hospital Capital Medical University (No. [2017] 030) and the Chinese regulatory authorities, and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. All study participants provided written informed consent. This study was registered at ClinicalTrials.gov under the identifying number NCT03393208.
2.2 Participants
The main inclusion criteria were Chinese male and female subjects aged 18–55 years with a body mass index of 18–30 kg/m2 who were non-smoking (in the last 3 months), had good physical and mental health status, blood and urine biochemistry and hematology tests within the normal range or showing no clinically relevant deviations, electrocardiogram (ECG) without signs of clinically relevant pathology, and vital signs within the normal range.
The main exclusion criteria were participation in a clinical study within 90 days prior to first drug administration; blood donation (≥500 mL) or significant blood loss within 90 days prior to first drug administration; pregnant or lactating women; any surgical or medical condition that could interfere with the study objectives, conduct, or evaluation; history of surgery of the gastrointestinal tract; history or presence of relevant liver diseases or hepatic dysfunction; hypersensitivity to the active drug substance or excipients; renal failure or renal dysfunction; any contraindication to metformin/GIR; and abnormal and clinically significant chest x-ray finding at screening.
2.3 Study Endpoints
The primary study endpoint was the area under the plasma concentration-time curve from time zero to the last sampling time (AUCt) and maximum observed concentration (Cmax), while the secondary study endpoints were tmax, apparent terminal half-life (t½), AUC from time zero to infinity (AUC∞), AUC from time of the last quantifiable concentration extrapolated to infinity given as a percentage of AUC∞ (AUCextra%), terminal elimination rate constant (λz), apparent total body clearance of drug following extravascular administration (CL/f), and apparent volume of distribution during the terminal phase following extravascular administration (Vz/f). The safety analysis included evaluation of the AEs, vital signs (blood pressure, pulse rate, body temperature, and respiration), clinical laboratory tests (biochemistry, hematology, and urinalysis), ECG, and physical examination. AEs were classified using the Medical Dictionary for Regulatory Activities (MedDRA) version 20.1 classification system by system organ class (SOC) and preferred term (PT), and were rated by severity and potential relationship to the study drug.
2.4 Blood Sampling and Bioanalytical Methods
Blood samples were collected during each treatment period 10 min prior to drug administration (baseline) and at 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 14, 24, 30, 36, and 48 h postdosing. Plasma samples were transferred to the bioanalytical laboratory and were subsequently stored at − 20 °C (± 5 °C) prior to testing. Bioanalytical measurements were conducted using protein precipitation. The lower limit of quantification (LLOQ) of metformin was 10.0 ng/mL. A validated liquid chromatography–mass spectrometry/mass spectrometry method was used to determine metformin in all study samples [6]. The drug concentrations produced by administering the test and reference products were used to derive the PK parameters.
2.5 Statistical Methods
Bioequivalence was confirmed if the 90% confidence intervals (CIs) for the geometric mean ratios between the test and reference products for both AUCt and Cmax were within the range of 80–125%. Based on the results of previous bioequivalence studies, GIR has shown a relatively low intraindividual coefficient of variation (CV; fasted: assuming a CV of 17.5% for Cmax and 13.8% for AUCt [8] and assuming 5% variation for all ratios [95–105%]; fed: assuming a CV of 7.6% for Cmax and 8.8% for AUCt [Study No. EML056023-H105, data on file] and assuming 5% variation for all ratios [95–105%]). Applying these CVs together with the applicable bioequivalence criteria for AUCt and Cmax (80–125%), and to allow the true treatment ratio of test/reference product to vary within 0.95 and 1.05, we determined 20 evaluable subjects in the fasted group and 12 subjects in the fed group would provide at least 89.9% power and 99.7% power to show bioequivalence in the fasted and fed groups, respectively (90% power for both groups). Assuming a dropout rate of 25%, 26 subjects were to be included in the fasted group. Furthermore, a minimum sample size of 18 subjects was determined for the fed group based on clinical judgment. Thus, the sample size was set to be 44 subjects.
The PK analysis set included all subjects who completed the study with adequate study medication compliance, without any relevant protocol violations or events concerning factors likely to affect the PK results’ comparability, and sufficient evaluable data to determine the primary endpoints (AUCt and Cmax) for both treatments. The safety analysis set included all subjects who received at least one dose of the study drug.
PK parameters were calculated from plasma concentrations of metformin by applying noncompartmental analysis with the linear and logarithmic trapezoidal methods. The primary endpoints, Cmax and AUCt under fasted or fed conditions, were log-transformed and analyzed using a mixed-effects model. The model included fixed effects for sequence, treatment, and period, and subject nested within the sequence as a random effect. Treatment differences on the log scale were estimated for the parameters and their 90% CIs. The least squares means and 95% CIs by treatment were also estimated. Point estimates and CIs were back-transformed to the original scale for presentation, i.e., ratios of geometric means and corresponding 90% CIs for test/reference products, and geometric means and corresponding 95% CIs by product, respectively. Secondary endpoints were summarized using descriptive statistics. Hodges–Lehmann estimates [9] for treatment differences and 90% CIs according to the Tukey method were calculated for tmax. For AUC∞, the mixed-effects model for treatment comparison as described for the primary endpoints was also used. The geometric mean ratios and 90% CIs for the test/reference products were estimated separately for fasted and fed conditions. Safety variables were summarized using descriptive statistics. Statistical analyses were performed using Phoenix® WinNonlin® 6.4 (Certara, LP, Princeton, NJ, USA) and SAS® Windows version 9.4 (SAS Institute Inc., Cary, NC, USA).
3 Results
3.1 Subject Characteristics
A total of 96 subjects were screened and 44 subjects were randomly assigned to treatment (26 subjects to the fasted group and 18 to the fed group). Among the 52 subjects who were not randomized, 43 subjects failed screening and 9 subjects were screened successfully but not randomized. The most common reason for screening failures was ‘eligibility criteria not met’. All 44 randomized subjects received the study drug, completed the study, and were included in the PK and safety analysis sets.
Table 1 shows the baseline demographic and clinical characteristics of the study subjects. More male (68.2%) than female (31.8%) subjects participated in this study, but the sex ratio fulfilled the protocol settings (each sex representing ≥ 25% of the total number per group). Demographic characteristics other than sex were well balanced.
3.2 Pharmacokinetics of Metformin
Figure 2 shows metformin plasma concentration-time profiles for the test and reference products in the fasted and fed groups. Following single oral dose administration of metformin to subjects under fasted conditions, mean metformin plasma concentrations increased and reached peak concentrations at approximately 2 h postdose for both products. Metformin concentrations declined in a multi-exponential manner through 24 h, and mean profiles were superimposable until the 24-h time point between both products. Metformin concentrations were below the LLOQ (10.0 ng/mL) at the scheduled 36- and 48-h time points. After a single oral dose administration of metformin to subjects in the fed group, mean metformin plasma concentrations increased, with peak concentrations reaching between 2.5 and 4 h postdose for both products. Metformin concentrations declined in a multi-exponential manner through 24 h, and mean profiles were essentially superimposable between both products. Metformin concentrations were below the LLOQ (10.0 ng/mL) at the scheduled 36- and 48-h time points.

Arithmetic means ± SD metformin plasma concentration-time profiles for the test and reference products on linear and semi-logarithmic scales in the (a) fasted and (b) fed groups (PK analysis set). PK pharmacokinetics, SD standard deviation
Table 2 shows the PK parameters of metformin for the test and reference products under fasted and fed conditions. Under fasted or fed conditions, the mean AUCt and Cmax (primary PK parameters) and the AUC∞ (secondary PK parameter) were comparable between the test and reference products. For the test and reference products under fasted conditions, the geometric mean t½ was 4.38 and 4.90 h, respectively, and the median tmax was 2.00 h for both treatments. For the test and reference products under fed conditions, the geometric mean t½ was 4.23 and 4.16 h, respectively, and the median tmax was 2.75 h for both treatments.
Boxplots for metformin AUCt and Cmax by group and treatment are shown in Fig. 3. No meaningful differences were observed in the AUCt and Cmax between the test and reference products under fed and fasted conditions.

Boxplot by group and treatment for (a) metformin AUC0−t (upper, fasted; lower, fed) and (b) metformin Cmax (upper, fasted; lower, fed). AUCt area under the plasma concentration–time curve from time zero to the last sampling time, Cmax maximum observed concentration, PK pharmacokinetics
3.3 Bioequivalence
Statistical comparisons of metformin plasma PK parameters between the test and reference products under fasted and fed conditions are shown in Table 3. The statistical assessment of metformin geometric mean primary PK parameters (AUCt and Cmax) and selected secondary parameter (AUC∞) characterizing exposure between the test and reference products was comparable under both fasted and fed conditions. Point estimates for all three parameters were close to 100%, and the corresponding 90% CIs were completely contained within the specified 80–125% bioequivalence boundary. Therefore, the bioequivalence of GIR tablets from China and France was demonstrated under both fed and fasted conditions. The tmax showed no statistically significant difference between the two treatments under fasted or fed conditions, with a median difference of 0.00 and 0.25, respectively.
3.4 Safety Results
Nine of the 44 (20.5%) subjects in the safety analysis set experienced at least one treatment-emergent AE (TEAE) during the study, including 4/26 (15.5%) subjects in the fasted group and 5/18 (27.8%) subjects in the fed group. Overall, 8/44 (18.3%) subjects had TEAEs that were judged as related to the study drug, including 3/26 (11.5%) subjects in the fasted group and 5/18 (27.8%) subjects in the fed group. No serious AE or TEAE leading to withdrawal was reported in this study. There was one non-TEAE (an AE of upper respiratory tract infection was reported and resolved before the first dose of the study drug).
TEAEs by SOC and PT are summarized in Table 4. There were 13 TEAEs reported from two SOCs: 8/44 (18.2%) subjects experienced TEAEs of gastrointestinal disorders, which included diarrhea, abdominal distention, abdominal pain, and frequent bowel movements, and 1/44 (2.3%) subject experienced a TEAE of blood and lymphatic system disorders, namely anemia. The most frequent AEs related to gastrointestinal disorders were diarrhea (6/44 [13.6%] subjects) and abdominal distention (3/44 [6.8%] subjects). There were no hypoglycemia-related AEs in either treatment group. All AEs in the present study were mild in severity.
All TEAEs except anemia were judged to be drug-related. Among all the drug-related TEAEs, diarrhea was reported by subjects in both the fasted group (1/26 [3.8%] subjects after test product administration and 2/26 [7.7%] subjects after reference product administration) and fed group (1/18 [5.6%] subjects after test product administration and 2/18 [11.1%] subjects after reference product administration). Other drug-related TEAEs were reported only in the fed group. In the fed group, abdominal distension was reported in 2/18 (11.1%) subjects after receiving the test product and 1/18 (5.6%) subjects after receiving the reference product. Abdominal pain was reported in 1/18 (5.6%) subjects in both the test and reference product groups, and frequent bowel movements were reported in 1/18 (5.6%) subjects after the reference product administration.
No deaths, serious AEs, or treatment discontinuations due to TEAEs were reported during this study. No clinically relevant abnormalities in vital signs, clinical laboratory tests, ECG, or physical examination were reported during the study.
4 Discussion
Many efforts have been made to reduce the number of in vivo bioavailability and/or bioequivalence studies for drug product approval of generic drug products, namely essential medicines, used for treating common and chronic conditions given the critical implications in healthcare accessibility and cost reduction. The application of biowaivers is based on the Biopharmaceutics Classification System (BCS) class and the drug’s solubility and permeability. Common in vitro testing procedures for biowaivers consists of dissolution tests [10, 11]. According to the Health and Human Services, US FDA, only BCS Class 1 drugs, rapidly dissolving drugs, and highly permeable drugs were applicable for biowaiver testing procedures [10, 11]. As per World Health Organization and European Medicine Agency recommendations, this guidance was later expanded to some BCS Class III drugs [12]. Metformin is a BCS Class III drug with high solubility and low permeability [13]. Nonetheless, the GIR tablets manufactured in China and France do not qualify for an in vivo biowaiver as neither meet the specification of ≥ 85% release within 15 min during the dissolution test in pH 6.8 phosphate buffer (unpublished data). Thus, the present bioequivalence study was conducted to establish bioequivalence of the GIR tablets manufactured in China and France following single oral dose administration under fasted and fed conditions in healthy volunteers and compare the PK parameters and safety of both formulations.
Based on the results of primary PK exposure parameters AUCt and Cmax, and secondary parameter AUC∞, GIR metformin exposure was comparable between the test and reference products after single oral dose administration under both fasted and fed conditions. The geometric least square mean ratios for these PK parameters were close to 100% and the corresponding 90% CIs were within the bioequivalence threshold criteria (80–125%). The median tmax and mean t½ for the two products under fasted and fed conditions were also similar. Thus, we consider that the GIR test and reference products met the statistical criteria for bioequivalence under both fasted and fed conditions. The present results are consistent with the results of a randomized, open-label, two-period crossover study conducted in 12 healthy Chinese male volunteers that assessed the bioequivalence of two IR metformin formulations (administered as a single 500 mg dose after an overnight fast), in which the 90% CI of the bioequivalence PK parameters (AUCt and Cmax, and AUC∞,) of both IR products were within the bioequivalence threshold [13]. Furthermore, a recent large-scale study compared the bioequivalence of 42 generic products with 14 orally administered IR reference products, including metformin, using a four-product, four-period, four-sequence, sequence-randomized, crossover bioequivalence study. Among 42 generic-reference comparisons, 100% of AUCt and AUC∞ showed bioequivalence, while only 9.5% of Cmax CIs barely failed to show bioequivalence. Of note, all the generic-reference comparisons for metformin showed bioequivalence based on the abovementioned PK parameters [14]. It has been suggested that providing evidence of the in vivo bioequivalence of different formulations of BCS Class III drugs may help support the extension of biowaivers to this class of drugs. Thus, the present results build on previous research making a case for biowaivers for BCS Class III drugs.
In general, the safety results showed that GIR tablets (both formulations) were safe and well tolerated. Mild TEAEs were reported in fasted and fed conditions, and the events were eventually resolved. The TEAEs found in this study were mostly related to gastrointestinal disorders, which are known AEs encountered with metformin therapy [15]. Subjects presented TEAEs after administration of both the test and reference products. There were no new or unexpected safety findings associated with GIR tablets in this study.
As the effect of food needs to be evaluated for most drugs intended for oral administration [16], and meanwhile the label of GIR indicated that the drug should be taken with food or under fed conditions, the present study evaluated the bioequivalence of GIR tablets under both fed/fasted conditions. The GIR label [15] indicates that food can delay the absorption of metformin and cause a 35-min prolongation of tmax. This is consistent with the slightly longer tmax (2.75 h; mean for both the test and reference groups) in the fed group compared with the tmax (2.00 h for both groups) in the fasted group. As the effect of food on metformin GIR is slight, the product label [15] states that metformin should be administered with a meal as this helps mitigate gastrointestinal AEs. In the present study, the proportion of patients with diarrhea was lower in the fed group (with both the test and reference products) than in the fasted group. However, some events such as bloating and abdominal pain occurred only in the fed group. A limitation of the present study was that no statistical analysis was conducted regarding the food effect.
5 Conclusion
Bioequivalence was demonstrated between the test (manufactured by Sino-American Shanghai Squibb Pharmaceuticals, China) and reference (manufactured by Merck Santé S.A.S, France) GIR tablets under fasted and fed conditions, and both the test and reference GIR tablets were considered safe and well tolerated.
References
Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60:1586–93. https://doi.org/10.1007/s00125-017-4336-x.
Davies MJ, D’Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669–701. https://doi.org/10.2337/dci18-0033.
Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–56. https://doi.org/10.1038/nrendo.2013.256.
Bodmer M, Meier C, Krähenbühl S, et al. Metformin, sulfonylureas, or other antidiabetes drugs and the risk of lactic acidosis or hypoglycemia: a nested case-control analysis. Diabetes Care. 2008;31:2086–91. https://doi.org/10.2337/dc08-1171.
Li L, Guan Z, Li R, et al. Population pharmacokinetics and dosing optimization of metformin in Chinese patients with type 2 diabetes mellitus. Medicine (Baltimore). 2020;99: e23212. https://doi.org/10.1097/MD.0000000000023212.
Huang XM, Wang GZ, He BB, et al. Bioequivalence and pharmacokinetic evaluation of two metformin hydrochloride tablets under fasting and fed conditions in healthy Chinese volunteers. Clin Pharmacol Drug Dev. 2020;9:910–7. https://doi.org/10.1002/cpdd.849.
Sun ML, Qi L, Luo XD, et al. Pharmacokinetic bioequivalence and safety assessment of two metformin hydrochloride tablet formulations using a phase I, randomized, open, two-period, two cross-over, single-dose, fed study in healthy Chinese adult subjects. Int J Clin Pharmacol Ther. 2021;59:630–8. https://doi.org/10.5414/CP204002.
Valizadeh H, Nayyeri-Maleki P, Ghanbarzadeh S, et al. Pharmacokinetics and bioequivalence of two brands of metformin 500 mg tablets in Iranian healthy volunteers. J Pharm Investig. 2014;44:61–8. https://doi.org/10.1007/s40005-013-0102-3.
Lehmann EL. Nonparametrics: statistical methods based on ranks (Reprinting of 1988 revision of 1975 Holden-Day ed.). New York: Springer; 2006. p. xvi+463 (ISBN978-0-387-35212-1).
World Health Organization Expert Committee on Specifications for Pharmaceutical Preparations. Annex 12: WHO “Biowaiver List”: proposal to waive in vivo bioequivalence requirements for WHO Model List of Essential Medicines immediate-release, solid oral dosage forms. https://www.who.int/publications/m/item/trs-1025-annex-12-who-biowaiver-eml
World Health Organization. Annex 8. Proposal to waive in vivo bioequivalence requirements for WHO Model List Essential Medicines immediate-release, solid oral dosage forms. WHO Technical Report Series, No. 937. Geneva: World Health Organization; 2006.
Davit BM, Kanfer I, Tsang YC, et al. BCS biowaivers: similarities and differences among EMA, FDA, and WHO requirements. AAPS J. 2016;18(3):612–8.
Cheng CL, Yu LX, Lee HL, et al. Biowaiver extension potential to BCS Class III high solubility-low permeability drugs: bridging evidence for metformin immediate-release tablet. Eur J Pharm Sci. 2004;22(4):297–304. https://doi.org/10.1016/j.ejps.2004.03.016.
Hammami MM, De Padua SJS, Hussein R, et al. Generic-reference and generic-generic bioequivalence of forty-two, randomly-selected, on-market generic products of fourteen immediate-release oral drugs. BMC Pharmacol Toxicol. 2017;18(1):78. https://doi.org/10.1186/s40360-017-0182-1.
Metformin investigational brochure. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/020357s037s039,021202s021s023lbl.pdf. Accessed 19 Jun 2022.
US Food and Drug Administration. Guidance for industry: food-effect bioavailability and fed bioequivalence studies. 2002. https://www.fda.gov/media/70945/download. Accessed 19 Jun 2022.
Acknowledgements
The authors thank Michelle Belanger, MD, of Edanz (www.edanz.com) for providing medical writing support, which was funded by Merck Serono Co., Ltd, China, an affiliate of Merck KGaA, Darmstadt, Germany, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Merck Serono Co., Ltd, China, an affiliate of Merck KGaA, Darmstadt, Germany.
Conflicts of interest/Competing interest
Dandan Li and Dongli Zhou are employees of Merck Serono (Beijing) Pharmaceutical R&D Co., Ltd, China, an affiliate of Merck KGaA. Chaoying Hu, Dan Gao, and Lan Zhang report no conflicts of interest.
Ethics approval
The protocol and all documents were reviewed and approved by the Ethics Committee of Xuanwu Hospital Capital Medical University (No. [2017]030).
Consent to participate
All study participants provided written informed consent.
Consent for publication
All researchers, participants, institutions, and sponsors consented to the submission of this report to the journal.
Author contributions
LZ was the principal investigator and contributed to all aspects of this work. CH and DG contributed to the study design and conduct. DL and DZ interpreted the data and drafted the manuscript. All authors approved the final version of the article for submission and agree to be accountable for all aspects of this work.
Data and code availability
The datasets used in the current analysis are available from Dandan Li upon reasonable request.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hu, C., Gao, D., Li, D. et al. Chinese- and French-Manufactured Immediate-Release Glucophage® Bioequivalence: A Randomized, Open-Label, Crossover Study. Drugs R D 22, 301–309 (2022). https://doi.org/10.1007/s40268-022-00405-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-022-00405-3