
Abstract
Purpose
Circulating tumor DNA (ctDNA) testing has become a promising tool to guide first-line (1L) targeted treatment for advanced non-small cell lung cancer (aNSCLC). This study aims to estimate the clinical validity (CV) and clinical utility (CU) of ctDNA-based next-generation sequencing (NGS) for oncogenic driver mutations to inform 1L treatment decisions in aNSCLC through a systematic literature review and meta-analysis.
Methods
A systematic literature search was conducted in PubMed/MEDLINE and Embase to identify randomized control trials or observational studies reporting CV/CU on ctDNA testing in patients with aNSCLC. Meta-analyses were performed using bivariate random-effects models to estimate pooled sensitivity and specificity. Progression-free/overall survival (PFS/OS) was summarized for CU studies.
Results
A total of 20 studies were identified: 17 CV only, 2 CU only, and 1 both, and 13 studies were included for the meta-analysis on multi-gene detection. The overall sensitivity and specificity for ctDNA detection of any mutation were 0.69 (95% CI 0.63–0.74) and 0.99 (95% CI 0.97–1.00), respectively. However, sensitivity varied greatly by driver gene, ranging from 0.29 (95% CI 0.13–0.53) for ROS1 to 0.77 (95% CI 0.63–0.86) for KRAS. Two studies that compared PFS with ctDNA versus tissue-based testing followed by 1L targeted therapy found no significant differences. One study reported OS curves on ctDNA-matched and tissue-matched therapies but no hazard ratios were provided.
Conclusions
ctDNA testing demonstrated an overall acceptable diagnostic accuracy in patients with aNSCLC, however, sensitivity varied greatly by driver mutation. Further research is needed, especially for uncommon driver mutations, to better understand the CU of ctDNA testing in guiding targeted treatments for aNSCLC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
ctDNA testing demonstrated an overall acceptable diagnostic accuracy in patients with aNSCLC, however, sensitivity varied greatly by driver mutation. |
Further research is needed on ctDNA testing, especially for uncommon driver mutations, to better understand its clinical utility in guiding targeted treatments for aNSCLC. |
1 Introduction
Non-small cell lung cancer (NSCLC) accounts for an estimated 85% of lung cancer cases, with adenocarcinoma, squamous cell carcinoma, and large cell carcinoma being the most common histological subtypes [1]. The estimated 5-year survival is only about 6% in patients with advanced-stage disease [2, 3]. In the past decade, more than 20 targeted therapies have been approved for the treatment of advanced NSCLC (aNSCLC) in patients who harbor EGFR, BRAF, MET, RET, NTRK, KRAS, ALK, or ROS1 alterations [4]. Targeted treatment according to the presence of oncogenic driver mutations has been associated with improved survival outcomes [4,5,6], and current National Comprehensive Cancer Network (NCCN) guidelines specifically recommend that all patients with aNSCLC should get broad genomic profiling with next-generation sequencing (NGS), given its more optimal use of sample availability, reduced procedure time, and favorable testing costs compared with single-gene tests [7,8,9].
In recent years, there has been an increase in the use of circulating tumor DNA (ctDNA) from blood samples as an alternative to tissue biopsy (TB) for identifying oncogenic driver mutations to inform first-line (1L) aNSCLC therapy [10]. Advantages of ctDNA testing include the avoidance of an invasive procedure and possible complications, the ability to identify driver mutations for patients with limited tissue availability for comprehensive NGS, and shorter turnaround time allowing for faster initiation of 1L therapy [11]. Furthermore, solid tumors often exhibit intratumoral heterogeneity, which means a single TB may not fully capture this heterogeneity, as it only represents a small sample from one location within the tumor. In these cases, ctDNA may provide a more comprehensive representation of the tumor’s genetic landscape. Several ctDNA-based NGS tests have been developed and approved for NSCLC, including Guardant360® CDx and FoundationOne®Liquid CDx [12, 13].
However, the diagnostic accuracy of ctDNA testing remains unclear due to variations in technologies and use scenarios [14, 15]. With the broadened adoption of ctDNA-based NGS tests in NSCLC, a better understanding of their clinical validity (CV) and clinical utility (CU) is needed. Clinical validity usually describes the diagnostic accuracy of genetic tests and is generally measured by sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) [16, 17]. Without proper validation of test performance, accurate identification of mutations may be compromised, leading to delays in suitable clinical decisions. Previous systematic literature reviews have examined the CV of ctDNA testing in aNSCLC patients but findings have been inconsistent due to varied review focus and inclusion criteria [18,19,20,21,22,23,24,25,26]. Most of the previous systematic literature reviews and/or meta-analyses [18, 19, 22, 23, 25, 26] did not specify sequencing technologies, which could potentially explain the observed heterogeneity in findings and limit their clinical implications in relation to the currently more recommended NGS tests.
Clinical utility refers to the risks or benefits from test use and in our study is measured as progression-free or overall survival (PFS/OS), considering the therapeutic effectiveness outcomes are the most relevant and commonly reported measure in CU studies and that the focus of our review is 1L targeted treatment informed by ctDNA testing [27,28,29]. CU evidence directly answers if a test is useful in improving patient health outcomes—it is an essential element in value evaluation and affects the acceptance of a test from all parties (patients, healthcare providers, and payers). To our knowledge, no study has systematically reviewed the CU of ctDNA testing for a comprehensive set of biomarkers to inform 1L treatment decisions in aNSCLC.
Given the rapidly evolving field of ctDNA testing technologies and the limitations of previous review studies, an updated synthesis of CV and CU evidence is warranted. The objective of the current study was to estimate the CV and CU of ctDNA-based NGS for oncogenic driver mutations to inform 1L treatment decisions in aNSCLC by means of a systematic literature review and meta-analysis of currently available evidence.
2 Materials and Methods
2.1 Systematic Literature Review
2.1.1 Eligibility Criteria
The study inclusion criteria were defined in terms of the population, interventions, comparisons, outcomes, and study design (PICOS) to guide the identification and selection of relevant studies. Population: adult patients with aNSCLC (at least 80% with stage III or IV, thereby making the assumption that reported results based on the total study population are still applicable to the stage III or IV NSCLC target population of interest), with a subset of treatment-naïve patients for CV studies and all being treatment-naïve for CU studies; interventions: ctDNA-based NGS especially for the detection of clinically relevant driver mutations (i.e., EGFR, BRAF, MET, RET, NTRK, KRAS, ALK, ROS1), and when outcome of interest is CU at least 80% of population receiving matched targeted treatment by driver mutation according to current NCCN guidelines [7]; comparators for CU studies: ctDNA testing versus TB; outcomes in CV studies: sensitivity, specificity, PPV, NPV or any other measures that allow for the calculation of true positive (TP), false positive (FP), false negative (FN), and true negative (TN) rates; outcomes in CU studies: PFS or OS; study design: cohort studies or randomized clinical trials. Studies were excluded if the study population excluded patients with any of the eight clinically relevant driver mutations; published in a non-English language; published before 2012 [1 year before the first Food and Drug Administration (FDA) approval of liquid biopsy test]; and were review articles or conference proceedings.
2.1.2 Study Identification
Relevant studies published between January 2012 and July 2023 were identified by searching MEDLINE and Embase databases with predefined search strategies (Supplementary File, Table S2) through the Embase platform. Furthermore, the official websites of three commonly used ctDNA tests (Guardant360® CDx, InvisionFirst®-Lung, and FoundationOne® Liquid CDx) as well as reference lists of included studies and previous systematic literature reviews [18,19,20,21,22,23,24,25,26] were searched for additional potentially eligible studies.
2.1.3 Study Selection
Two reviewers (C.C. and M.D.) screened the identified abstracts using an open-source, active learning software/platform, ASReview, following recommended approaches for automated screening [30,31,32,33,34]. Studies identified as eligible during abstract screening were subsequently screened at a full-text stage by the same two reviewers according to the eligibility criteria to determine the final set of included studies. Following reconciliation between the two investigators, a third reviewer (J.J.) was included to reach a consensus for any remaining discrepancies. The process of study identification and selection was summarized with a Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram.
2.1.4 Data Extraction
Two reviewers (C.C. and M.D.) extracted data on study characteristics, test and intervention characteristics, patient characteristics, and outcomes for the final list of included studies. Data were stored and managed in a Microsoft Excel workbook, and included the following. Trial characteristics: author name, publication year, country, sample size, oncogenic driver mutations of interest, study duration, and patient in/exclusion criteria; patient characteristics: disease stage, smoking status, race/ethnicity, gender, and age; test and intervention characteristics: ctDNA and NGS technologies and in addition for CU studies, targeted treatment regimen; outcomes in CV studies: overall and oncogenic driver mutation-specific sensitivity, specificity, TP, FP, FN, TN; and outcomes in CU studies: PFS and/or OS.
2.1.5 Quality Assessment
Two authors (C.C. and M.D.) independently assessed the quality of included CV studies on the basis of the revised Quality Assessment of Diagnostic Accuracy Studies-2 (QUADAS-2) criteria [35]. Discrepancies were resolved by consensus.
2.2 Analysis
Study-specific, sample (mutation)-level TP, FP, FN, and TN frequency data for oncogenic driver mutations were used to estimate the overall and mutation-specific sensitivity and specificity for each study as well as across studies by meta-analyses. The overall sensitivity and specificity were estimated on the basis of studies that simultaneously tested at least four driver mutations. Study-specific sensitivity and specificity estimates along with 95% confidence intervals (95% CIs) were obtained according to the Wilson method using continuity corrected cell counts [36, 37]. The bivariate random-effects model proposed by Reitsma et al. (2005) utilizes the standard frequentist approach and was used in our study to obtain pooled estimates for sensitivity and specificity [38,39,40]. Meta-analysis results were also presented with summary receiver operating characteristics (SROC) curves and 95% confidence regions for the pooled sensitivity and specificity estimates. In addition, as a sensitivity analysis, a Bayesian bivariate random-effects meta-analysis was performed to avoid normal approximations of the likelihood and to obtain predictive distributions of the sensitivity and specificity to predict results in a new study. The Bayesian method offers unique advantages in managing uncertainty when dealing with limited or heterogeneous data and allows for a more flexible and informative analysis, providing a range of plausible values for sensitivity and specificity rather than a single point estimate. This is particularly valuable in the context of ctDNA testing, where the performance of assays may vary due to multiple factors. Our dual approach of using both frequentist and Bayesian methods offers complementary insights into the performance of ctDNA testing and helps manage the uncertainty inherent in meta-analyses of diagnostic accuracy studies. Both the frequentist and Bayesian approaches used in our study modeled the sum and differences of true positive and false positive rates as random effects. The Bayesian receiver operating characteristics (BSROC) curves and Bayesian area under the BSROC curve (BAUC) were reported with 95% CI for the ctDNA detection of any and each driver mutation. An AUC of 0.7 or higher is generally considered good accuracy and 0.6–0.7 is considered sufficient; an AUC below 0.5 indicates the test is not useful [41]. There is no standard for good sensitivity/specificity of DNA testing. A ctDNA test might be considered acceptable for coverage if its performance is similar to the FDA-approved Guardant360® [42]. All analyses were conducted using RStudio, version 4.1.2 (©2009–2022 RStudio, PBC) using packages “mada” and “bamdit.” [38, 43] Progression-free/overall survival (PFS/OS) was summarized for CU studies.
3 Results
3.1 Study Selection
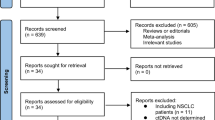
Our initial search generated 1749 potentially relevant publications. After screening titles and abstracts, 58 publications were selected for full-text review. A total of 20 publications corresponding to 20 studies were selected for inclusion (Fig. 1); 17 studies [44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60] reported CV only, 2 study [61] reported CU only, and 1 study [62, 63] provided information on both CV and CU.

PRISMA flowchart of included studies. *ASReview is an open-source, active-learning software for screening titles and abstracts for systematic reviews
3.2 Clinical Validity of ctDNA Testing to Identify Oncogenic Driver Mutations
3.2.1 Characteristics of Included Studies
The majority of the included CV studies [45,46,47, 49,50,51, 53,54,55,56,57,58,59,60, 62] used a prospective cohort design (83.3%, 15/18), and three studies [44, 48, 52] used retrospective medical record data. All studies evaluated ctDNA-based NGS technologies, with 11 studies [44,45,46, 48,49,50,51,52, 58, 60, 62] evaluating branded ctDNA tests and 7 studies [47, 53,54,55,56,57, 59] describing the ctDNA technologies without a specific brand name (Table 1). Guardant360® was the most used ctDNA technology (33.3%, 6/18). TB was the reference standard in all included studies.
In total, seven studies [45, 46, 50, 51, 55, 56, 62] included only untreated patients, five studies [44, 47, 48, 53, 58] included 24–92% of untreated patients, and in six studies [49, 52, 54, 57, 59, 60] the proportion of untreated patients was unclear. Among studies reporting patient racial/ethnic information [46, 51, 52, 62], most patients were Caucasian (Table S3). The type of driver mutations examined differed across studies. No study reported CV on all eight clinically relevant driver mutations. Most of the studies evaluated ctDNA detection of EGFR (77.8%, 14/18) and KRAS (61.1%, 11/18), and 13 studies reported CV information on six or more mutations (Table S4).
Four studies were found to have no risk of bias or applicability concerns and nine studies had two or fewer items of concern according to the QUADAS-2 instrument (Table S16). Eight studies were found to have unclear-risk items, indicating potential issues with the reporting of CV evidence.
3.2.2 Overall Sensitivity and Specificity for Detection of Any Guideline-Recommended Oncogenic Driver Mutation
Overall sensitivity for the detection of any driver mutation varied between 0.52 and 0.81 across 13 studies that simultaneously detected four or more driver mutations (Fig. 2 and Supplementary File Table S4). The overall specificity varied between 0.88 and 1, with 11 studies reporting a specificity value of 0.90 or higher. There was a small degree of between-study heterogeneity regarding sensitivity and specificity (I2 = 20%) [64]. The pooled sensitivity obtained with meta-analysis using the frequentist approach was 0.69 (95% CI 0.63–0.74) and the pooled specificity was 0.99 (95% CI 0.97–1.00). Bayesian meta-analysis generated similar pooled estimates: sensitivity = 0.70 (95% CI 0.60–0.79); specificity = 0.99 (95% CI 0.97–1.00). The SROC curves with both approaches and the 95% confidence regions for the pooled sensitivity and specificity estimates are in Fig. 3. The BAUC was 0.71 (95% CI 0.68–0.73). In general, overall sensitivity was higher in studies that used branded ctDNA tests compared with studies that did not describe tests with a specific brand name, except for the ResBio ctDx_Lung used in the study by Sabari et al. Overall specificity was high regardless of the ctDNA test used. We did not observe a positive relationship between sensitivity/specificity and study quality.

Forest plot of sensitivity and specificity of ctDNA testing for multi-gene detection from bivariate random-effects meta-analyses (n = 13). Sensitivity and specificity of single studies were based on frequentist estimations. I2 = 20% based on the frequentist approach

Summary receiver operating characteristics (SROC) plots based on frequentist and Bayesian bivariate random-effects meta-analyses (n = 13): a each triangle identifies the true positive rate versus the false positive rate (1 − specificity) of each study (observed data); the black circle represents the summary estimate, and the solid contour shows the 95% confidence region around the summary estimate; the dotted contour indicates the 95% prediction region (the region within which a new study will lie); b the panel shows the Bayesian summary receiver operating characteristics (SROC) curve; each blue circle indicates the true positive rate versus the false positive rate (1 − specificity) of each study, and different sizes are used for different sample sizes; the central line corresponds to the posterior median and the upper and lower curves correspond to the quantiles of 2.5% and 97.5%, respectively
3.2.3 Sensitivity and Specificity by Oncogenic Driver Mutation
An overview of meta-analysis results of sensitivity and specificity by oncogenic driver mutation is presented in Table 2. Corresponding SROCs and study-specific estimates are provided in Supplementary File, Tables S6–S12 and Figs. S2–S22. Sensitivity and specificity for detecting EGFR with ctDNA were reported in 14 studies [46, 47, 49,50,51,52,53,54,55,56,57,58, 60, 62]. Study-specific sensitivity estimates varied between 0.56 and 0.83, and specificity estimates varied between 0.68 and 1 (Fig. S2). The pooled sensitivity and specificity as estimated with the meta-analysis were 0.68 and 0.98, respectively, with a BAUC of 0.71 (95% CI 0.68–0.73). Pooled sensitivity for the other driver mutations was generally less than 0.70 (except 0.77 for KRAS), but specificity was close to 1. The pooled sensitivity for each driver mutation detection was generally higher for branded ctDNA tests compared with tests that were not described with a specific brand name; overall pooled specificity was still high regardless of ctDNA tests used. We also conducted an analysis of sensitivity and specificity of ctDNA testing for detecting different mutation classes, including SNVs, indels, and fusions. We found that only SNVs are associated with an acceptable BAUC of 0.72 (95% CI 0.70–0.74), while the other mutation classes demonstrated a BAUC of around 0.52 (Tables S13–S15, Figs. S23–S31).
3.3 Clinical Utility of ctDNA Testing to Inform 1L Treatment in Patients with aNSCLC
Three studies evaluated the CU of ctDNA testing to inform 1L targeted therapy [61,62,63]. Madison et al. compared PFS among patients on matched 1L therapies following comprehensive genomic profiling of ctDNA (FoundationOne®Liquid or FoundationACT®) and TB (FoundationOne®CDx or FoundationOne®) using data from the Flatiron Health-Foundation Medicine Clinico-Genomic Database [61]. Median PFS in the ctDNA group (n = 33) was 13.8 (95% CI 8.9–NA) months and 10.6 (95% CI 8.7–13.6) months in the TB group (n = 229). The hazard ratio of ctDNA versus TB regarding PFS was 0.68 (95% CI 0.36–1.26) – not statistically significant. OS was only reported according to matched or unmatched 1L treatment but not specifically for TB and ctDNA. Palmero et al. reported PFS for 41 patients treated with 1L targeted therapy informed by ctDNA or TB testing (Guardant360®) (median, 8.6; 95% CI 7.6–11.6 months) [62]. According to the reported Kaplan–Meier (KM) curves, PFS was comparable between these two groups. Jee et al. reported OS for ctDNA-matched (median OS of 39 months) and tissue-matched treatment-naïve patients (29 months), however, they did not provide a relative treatment effect estimate independent of whether driver mutations were detected with ctDNA to infer the clinical utility of ctDNA relative to TB testing [63].
4 Discussion
Our meta-analyses showed that ctDNA testing demonstrated acceptable sensitivity and high specificity for detecting any guideline-recommended oncogenic driver mutation. However, the sensitivity of ctDNA testing varied widely by driver mutation. Pooled sensitivity estimates indicated acceptable performance for KRAS, but estimates were less than 70% for the other driver mutations. Specificity of ctDNA testing was high for all mutations. Evidence regarding the CU of ctDNA testing relative to TB was limited. According to a single study, there was no difference in PFS between ctDNA and TB tests followed by 1L targeted therapy.
To our knowledge, this is the first systematic literature review and meta-analysis specifically focused on the diagnostic performance of ctDNA testing for multi-gene detection with NGS in patients with aNSCLC to inform 1L targeted therapy. In contrast to previous systematic literature reviews/meta-analyses on ctDNA detection of EGFR or KRAS, [18, 22, 23, 25, 26] we assessed the diagnostic accuracy of ctDNA for detecting multiple driver mutations simultaneously rather than focusing on single mutation, thereby providing more relevant information for routine clinical practice where ctDNA-based NGS is rapidly being adopted. Meta-analysis of diagnostic accuracy can be performed on the basis of results reported at the patient level, or the sample (mutation) level. Previous meta-analyses mostly reported ctDNA performance at the patient level [18, 22, 23] or did not specify the data level [19, 20]. We used sample-level data to facilitate diagnostic performance by driver mutation as well as mutation class, which allows for a more detailed assessment of the CV of ctDNA testing and more mutation-specific information.
In our systematic review, we included any study evaluating ctDNA testing for which the study population included at least 80% patients with aNSCLC and any proportion of treatment-naïve patients, which did not strictly align with our target patient population of interest— patients with aNSCLC initiating 1L treatment. The reason to cast a wider net was to ensure we did not miss any study that reported relevant subgroup results. In the actual meta-analyses of CV, we only included data for patients with aNSCLC. Some of these studies included both treatment-naïve and treatment-experienced patients, however, we do not have reason to believe that this has (externally) biased the estimates of diagnostic accuracy focused on the presence of driver mutation for the 1L target population. If we excluded studies comprising not only treatment-naïve patients, the evidence base would have been limited.
The available evidence base regarding the CV and CU of ctDNA was limited. The CV studies had small sample sizes, which made it challenging to reliably estimate sensitivity for rare driver mutations. Regarding the risk of bias, we did not identify any concerns for only four studies. Although we did not observe a difference in overall diagnostic performance between higher and lower quality studies, the limited evidence base makes interpretation of this observation difficult. Similarly, the limited number of studies did not make it feasible to reliably evaluate potential drivers of between-study differences in ctDNA testing performance either. We observed a difference in test performance between branded and non-branded ctDNA tests. The overall sensitivity was higher for branded ctDNA tests, such as Guardant360® CDx (the most common in individual studies), compared with non-branded tests. This difference could be attributed to the fact that branded ctDNA tests have undergone extensive validation and regulatory approval processes, ensuring their reliability and performance. In contrast, non-branded ctDNA tests are often developed in-house by individual laboratories or research institutions and may lack the same level of validation and standardization as their branded counterparts. Additionally, only ten studies reported the time interval between ctDNA and tissue testing, with the number of days varying significantly among these studies (ranging from ≤ 1 day to a median of 207 days). The prolonged interval between testing could potentially expose patients to the emergence of new driver mutations, which may influence false-positive rates.
Despite the limitations of the available evidence base, the CV findings from our study can have important clinical implications for the role of ctDNA testing in NSCLC. Our results support current guidelines that recommend ctDNA testing as a complementary approach rather than a replacement for TB to inform 1L therapy, unless there is insufficient tissue for NGS or risks of biopsy are excessive [7, 65]. This is particularly relevant for specific driver mutations such as EGFR and KRAS, which had much higher detection rates than ROS1 or ALK. This could indicate the potential benefits of ctDNA testing in clinical practice to help guide the selection of appropriate 1L targeted therapies, particularly for patients with more common driver mutations such as EGFR and KRAS. For example, detecting EGFR mutations through ctDNA testing can prompt the use of EGFR tyrosine kinase inhibitors (TKIs: erlotinib, gefitinib, afatinib, dacomitinib, osimertinib) as 1L treatment, which have been shown to improve outcomes in patients with EGFR-mutant NSCLC. However, the potential drawbacks of low sensitivity (increased risk of false-negative results) in ctDNA tests in the less common driver mutations could lead to missed opportunities for timely and appropriate targeted therapy.
While multiple factors have been implicated in the sensitivity of ctDNA, including tumor burden and the presence of osseous metastases [66], molecular heterogeneity has been relatively unexplored as a predictor of sensitivity for ctDNA. In particular, molecular fusions such as ALK and ROS1 may be less detectable via ctDNA given the number of potential fusion partners, which makes development of a sensitive ctDNA assay challenging [67, 68]. For example, in a patient who has never smoked where risks of biopsy may be high or the initial biopsy may not have sufficient tissue, a negative ctDNA test may not be sufficient to rule out the presence of a ROS1 fusion, which has profound implications for clinical decision-making around repeat biopsies at time of diagnosis and in the future. Another potential factor that may influence the diagnostic accuracy of ctDNA tests compared with tissue biopsies is the availability of DNA enrichment techniques. However, solid tumors often exhibit intratumoral heterogeneity, and a single tumor biopsy may not fully capture this heterogeneity, as it only represents a small sample from one location within the tumor. In these cases, ctDNA testing may be able to provide a more comprehensive representation of the tumor’s genetic landscape.
In addition to informing 1L therapy, ctDNA findings before initiating treatment will also help with the interpretation of ctDNA to monitor disease progression and tumor burden. For example, the absence of a mutation at follow-up that was detected upfront is indicative of effective treatment [69]. Ongoing presence of a particular mutation after initiation of therapy (even in the absence of a targetable mutation) may reflect a higher underlying tumor burden and an independent risk factor for survival [70].
There are only two studies that have evaluated the impact of the CU of ctDNA in terms of PFS in 1L patients with aNSCLC, and no studies evaluating OS. This is a key area of future study, as baseline ctDNA may rarely pick up a mutation that would not be detected with tissue biopsy and influence treatment decisions, ultimately impacting OS. Additionally, we only included PFS and OS in our assessment of CU of ctDNA, and future studies may also want to examine other CU measures such as turn-around time of test results, time to initiation of therapy, number of patients matched to targeted therapy, or treatment decision impacts.
5 Conclusions
On the basis of the currently available evidence, ctDNA testing in patients with aNSCLC has an overall acceptable diagnostic accuracy for detecting any guideline-recommended oncogenic driver mutation when using TB as the reference standard. However, the sensitivity of ctDNA testing varies greatly depending on the specific oncogenic driver mutation. At the time of this review, there were limited studies on less common driver mutations, and this is an essential area of future research. Given the current detection rates, ctDNA cannot be recommended as a replacement for TB, considering the actionable driver mutations of interest, unless there is insufficient tissue for TB or risks associated with the procedure. As the technologies around ctDNA testing and NGS analysis continue to evolve, we anticipate new studies will become available at a rapid pace, necessitating timely updates to this systematic review and meta-analysis.
References
Non-small cell lung cancer treatment (PDQ®)–Health Professional Version—NCI. 2022. https://www.cancer.gov/types/lung/hp/non-small-cell-lung-treatment-pdq. Accessed 4 Oct 2022.
Lung cancer—non-small cell—statistics. Cancer.Net. 2012. https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/statistics. Accessed 9 Sept 2022.
What is lung cancer? | Types of lung cancer. https://www.cancer.org/cancer/lung-cancer/about/what-is.html. Accessed 9 Sept 2022.
Majeed U, Manochakian R, Zhao Y, Lou Y. Targeted therapy in advanced non-small cell lung cancer: current advances and future trends. J Hematol Oncol. 2021;14(1):108. https://doi.org/10.1186/s13045-021-01121-2.
Vestergaard HH, Christensen MR, Lassen UN. A systematic review of targeted agents for non-small cell lung cancer. Acta Oncol. 2018;57(2):176–86. https://doi.org/10.1080/0284186X.2017.1404634.
Yuan M, Huang LL, Chen JH, Wu J, Xu Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Sig Transduct Target Ther. 2019;4(1):1–14. https://doi.org/10.1038/s41392-019-0099-9.
Ettinger DS, Wood DE, Aisner DL, et al. Non-small cell lung cancer, Version 3.2022, NCCN clinical practice guidelines in oncology. J Nat Compr Cancer Netw. 2022;20(5):497–530. https://doi.org/10.6004/jnccn.2022.0025.
De Maglio G, Pasello G, Dono M, et al. The storm of NGS in NSCLC diagnostic-therapeutic pathway: how to sun the real clinical practice. Crit Rev Oncol Hematol. 2022;169: 103561. https://doi.org/10.1016/j.critrevonc.2021.103561.
Pennell NA, Mutebi A, Zhou ZY, et al. Economic impact of next-generation sequencing versus single-gene testing to detect genomic alterations in metastatic non-small-cell lung cancer using a decision analytic model. JCO Precis Oncol. 2019;3:1–9. https://doi.org/10.1200/PO.18.00356.
Aggarwal C, Rolfo CD, Oxnard GR, Gray JE, Sholl LM, Gandara DR. Strategies for the successful implementation of plasma-based NSCLC genotyping in clinical practice. Nat Rev Clin Oncol. 2021;18(1):56–62. https://doi.org/10.1038/s41571-020-0423-x.
Rolfo C, Mack PC, Scagliotti GV, et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): a statement paper from the IASLC. J Thorac Oncol. 2018;13(9):1248–68. https://doi.org/10.1016/j.jtho.2018.05.030.
Commissioner O of the. FDA approves first liquid biopsy next-generation sequencing companion diagnostic test. FDA. 2020. https://www.fda.gov/news-events/press-announcements/fda-approves-first-liquid-biopsy-next-generation-sequencing-companion-diagnostic-test. Accessed 4 Oct 2022.
U.S. Food and Drug Administration (FDA) Approves FoundationOne®LiquidCDx as a companion diagnostic for Pfizer’s BRAFTOVI® (encorafenib) in combination with cetuximab to identify patients with BRAF V600E alterations in metastatic colorectal cancer | Foundation Medicine. https://www.foundationmedicine.com/press-releases/f9b285eb-db6d-4f61-856c-3f1edb803937. Accessed 16 Oct 2023.
Chae YK, Davis AA, Carneiro BA, et al. Concordance between genomic alterations assessed by next-generation sequencing in tumor tissue or circulating cell-free DNA. Oncotarget. 2016;7(40):65364–73. https://doi.org/10.18632/oncotarget.11692.
Weber S, Spiegl B, Perakis SO, et al. Technical evaluation of commercial mutation analysis platforms and reference materials for liquid biopsy profiling. Cancers (Basel). 2020;12(6):1588. https://doi.org/10.3390/cancers12061588.
Burke W. Genetic tests: clinical validity and clinical utility. Curr Protoc Hum Genet. 2014;81:9.15.1–9.15.8. https://doi.org/10.1002/0471142905.hg0915s81.
Promoting Safe and Effective Genetic testing in the United States. Genome.gov. https://www.genome.gov/10001733/genetic-testing-report. Accessed 4 Nov 2022.
Passiglia F, Rizzo S, Di Maio M, et al. The diagnostic accuracy of circulating tumor DNA for the detection of EGFR-T790M mutation in NSCLC: a systematic review and meta-analysis. Sci Rep. 2018;8:13379. https://doi.org/10.1038/s41598-018-30780-4.
van Delft F, Koffijberg H, Retèl V, van den Heuvel M, IJzerman M. The validity and predictive value of blood-based biomarkers in prediction of response in the treatment of metastatic non-small cell lung cancer: a systematic review. Cancers (Basel). 2020;12(5):1120. https://doi.org/10.3390/cancers12051120.
Sebastião MM, Ho RS, de Carvalho JPV, Nussbaum M. Diagnostic accuracy of next generation sequencing panel using circulating tumor DNA in patients with advanced non-small cell lung cancer: a systematic review and meta-analysis. J Health Econ Outcomes Res. 2020;7(2):158–63. https://doi.org/10.36469/jheor.2020.17088.
Esagian SM, Grigoriadou GΙ, Nikas IP, et al. Comparison of liquid-based to tissue-based biopsy analysis by targeted next generation sequencing in advanced non-small cell lung cancer: a comprehensive systematic review. J Cancer Res Clin Oncol. 2020;146(8):2051–66. https://doi.org/10.1007/s00432-020-03267-x.
Luo J, Shen L, Zheng D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: a systematic review and meta-analysis. Sci Rep. 2014;4:6269. https://doi.org/10.1038/srep06269.
Qiu M, Wang J, Xu Y, et al. Circulating tumor DNA is effective for the detection of EGFR mutation in non-small cell lung cancer: a meta-analysis. Cancer Epidemiol Biomark Prev. 2015;24(1):206–12. https://doi.org/10.1158/1055-9965.EPI-14-0895.
Saarenheimo J, Andersen H, Eigeliene N, Jekunen A. Gene-guided treatment decision-making in non-small cell lung cancer—a systematic review. Front Oncol. 2021;11: 754427. https://doi.org/10.3389/fonc.2021.754427.
Chen Z, Miao H, Zeng Q, Xu S, Chen Z, Liu K. Circulating cell-free DNA as a diagnostic and prognostic biomarker for non-small-cell lung cancer: a systematic review and meta-analysis. Biomark Med. 2020;14(7):587–97. https://doi.org/10.2217/bmm-2018-0093.
Wang N, Zhang X, Wang F, et al. The diagnostic accuracy of liquid biopsy in EGFR-mutated NSCLC: a systematic review and meta-analysis of 40 studies. SLAS Technol. 2021;26(1):42–54. https://doi.org/10.1177/2472630320939565.
Dang DK, Park BH. Circulating tumor DNA: current challenges for clinical utility. J Clin Investig. 132(12):e154941. https://doi.org/10.1172/JCI154941.
Stadler JC, Belloum Y, Deitert B, et al. Current and future clinical applications of ctDNA in immuno-oncology. Cancer Res. 2022;82(3):349–58. https://doi.org/10.1158/0008-5472.CAN-21-1718.
Walcott SE, Miller FA, Dunsmore K, Lazor T, Feldman BM, Hayeems RZ. Measuring clinical utility in the context of genetic testing: a scoping review. Eur J Hum Genet. 2021;29(3):378–86. https://doi.org/10.1038/s41431-020-00744-2.
ASReview LAB: Active learning for systematic reviews—ASReview LAB documentation. https://asreview.readthedocs.io/en/latest/index.html. Accessed 23 Aug 2022.
van de Schoot R, de Bruin J, Schram R, et al. An open source machine learning framework for efficient and transparent systematic reviews. Nat Mach Intell. 2021;3(2):125–33. https://doi.org/10.1038/s42256-020-00287-7.
Ferdinands G, Schram R, de Bruin J, et al. Active learning for screening prioritization in systematic reviews—a simulation study. Open Science Framework. 2020. 10.31219/osf.io/w6qbg.
ASReview. Active learning explained. ASReview. 2022. https://asreview.nl/blog/active-learning-explained/. Accessed 23 Aug 2022.
ASReview—screen papers for literature review using machine learning. Singapore Management University (SMU). https://library.smu.edu.sg/topics-insights/asreview-screen-papers-literature-review-using-machine-learning. Accessed 23 Aug 2022.
Whiting PF, Rutjes AWS, Westwood ME, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med. 2011;155(8):529–36. https://doi.org/10.7326/0003-4819-155-8-201110180-00009.
Wilson EB. Probable inference, the law of succession, and statistical inference. J Am Stat Assoc. 1927;22(158):209–12. https://doi.org/10.1080/01621459.1927.10502953.
Newcombe RG. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat Med. 1998 (Published online).
Sousa-Pinto PD with contributions from B. mada: Meta-analysis of diagnostic accuracy. 2022. https://CRAN.R-project.org/package=mada. Accessed 13 Sept 2022.
Holling H, Böhning W, Masoudi E, Böhning D, Sangnawakij P. Evaluation of a new version of I2 with emphasis on diagnostic problems. Commun Stat Sim Comp. 2020;49(4):942–72. https://doi.org/10.1080/03610918.2018.1489553.
Reitsma JB, Glas AS, Rutjes AWS, Scholten RJPM, Bossuyt PM, Zwinderman AH. Bivariate analysis of sensitivity and specificity produces informative summary measures in diagnostic reviews. J Clin Epidemiol. 2005;58(10):982–90. https://doi.org/10.1016/j.jclinepi.2005.02.022.
Šimundić AM. Measures of diagnostic accuracy: basic definitions. EJIFCC. 2009;19(4):203–11.
LCD - MolDX: Plasma-based genomic profiling in solid tumors (L38065). https://www.cms.gov/medicare-coverage-database/view/lcd.aspx?lcdid=38065&ver=9&keyword=guardant360&keywordType=starts&areaId=all&docType=NCA,CAL,NCD,MEDCAC,TA,MCD,6,3,5,1,F,P&contractOption=all&sortBy=relevance&bc=1. Accessed 9 Dec 2022.
Verde PE. bamdit: Bayesian meta-analysis of diagnostic test data. 2022. https://CRAN.R-project.org/package=bamdit. Accessed 13 Sept 2022.
Bustamante Alvarez JG, Janse S, Owen DH, et al. Treatment of non-small-cell lung cancer based on circulating cell-free DNA and impact of variation allele frequency. Clin Lung Cancer. 2021;22(4):e519–27. https://doi.org/10.1016/j.cllc.2020.11.007.
Fernandes MGO, Cruz-Martins N, Souto Moura C, et al. Clinical application of next-generation sequencing of plasma cell-free DNA for genotyping untreated advanced non-small cell lung cancer. Cancers (Basel). 2021;13(11):2707. https://doi.org/10.3390/cancers13112707.
Leighl NB, Page RD, Raymond VM, et al. Clinical utility of comprehensive cell-free DNA analysis to identify genomic biomarkers in patients with newly diagnosed metastatic non-small cell lung cancer. Clin Cancer Res. 2019;25(15):4691–700. https://doi.org/10.1158/1078-0432.CCR-19-0624.
Li BT, Janku F, Jung B, et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: results from the Actionable Genome Consortium. Ann Oncol. 2019;30(4):597–603. https://doi.org/10.1093/annonc/mdz046.
Lin LH, Allison DHR, Feng Y, et al. Comparison of solid tissue sequencing and liquid biopsy accuracy in identification of clinically relevant gene mutations and rearrangements in lung adenocarcinomas. Mod Pathol. 2021;34(12):2168–74. https://doi.org/10.1038/s41379-021-00880-0.
Müller JN, Falk M, Talwar J, et al. Concordance between comprehensive cancer genome profiling in plasma and tumor specimens. J Thorac Oncol. 2017;12(10):1503–11. https://doi.org/10.1016/j.jtho.2017.07.014.
Park S, Olsen S, Ku BM, et al. High concordance of actionable genomic alterations identified between circulating tumor DNA-based and tissue-based next-generation sequencing testing in advanced non-small cell lung cancer: the Korean Lung Liquid versus Invasive Biopsy Program. Cancer. 2021;127(16):3019–28. https://doi.org/10.1002/cncr.33571.
Pritchett MA, Camidge DR, Patel M, et al. Prospective clinical validation of the InVisionFirst-lung circulating tumor DNA assay for molecular profiling of patients with advanced nonsquamous non–small-cell lung cancer. JCO Precis Oncol. 2019;3:1–15. https://doi.org/10.1200/PO.18.00299.
Schwaederlé MC, Patel SP, Husain H, et al. Utility of genomic assessment of blood-derived circulating tumor DNA (ctDNA) in patients with advanced lung adenocarcinoma. Clin Cancer Res. 2017;23(17):5101–11. https://doi.org/10.1158/1078-0432.CCR-16-2497.
Yao Y, Liu J, Li L, et al. Detection of circulating tumor DNA in patients with advanced non-small cell lung cancer. Oncotarget. 2016;8(2):2130–40. https://doi.org/10.18632/oncotarget.12883.
Xu S, Lou F, Wu Y, et al. Circulating tumor DNA identified by targeted sequencing in advanced-stage non-small cell lung cancer patients. Cancer Lett. 2016;370(2):324–31. https://doi.org/10.1016/j.canlet.2015.11.005.
Liu L, Liu H, Shao D, et al. Development and clinical validation of a circulating tumor DNA test for the identification of clinically actionable mutations in nonsmall cell lung cancer. Genes Chromosomes Cancer. 2018;57(4):211–20. https://doi.org/10.1002/gcc.22522.
Couraud S, Vaca-Paniagua F, Villar S, et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: a proof-of-concept study from BioCAST/IFCT-1002. Clin Cancer Res. 2014;20(17):4613–24. https://doi.org/10.1158/1078-0432.CCR-13-3063.
Chen Y, Han T, Zhou Y, Mao B, Zhuang W. Comparing the efficacy of targeted next-generation sequencing in the identification of somatic mutations in circulating tumor DNA from different stages of lung cancer. Neoplasma. 2019;66(4):652–60. https://doi.org/10.4149/neo_2018_181130N910.
Remon J, Lacroix L, Jovelet C, et al. Real-world utility of an amplicon-based next-generation sequencing liquid biopsy for broad molecular profiling in patients with advanced non-small-cell lung cancer. JCO Precis Oncol. 2019;3:PO.18.00211. https://doi.org/10.1200/PO.18.00211.
Guo N, Lou F, Ma Y, et al. Circulating tumor DNA detection in lung cancer patients before and after surgery. Sci Rep. 2016;6(1):33519. https://doi.org/10.1038/srep33519.
Sabari JK, Offin M, Stephens D, et al. A prospective study of circulating tumor DNA to guide matched targeted therapy in lung cancers. J Natl Cancer Inst. 2018;111(6):575–83. https://doi.org/10.1093/jnci/djy156.
Madison R, Schrock AB, Castellanos E, et al. Retrospective analysis of real-world data to determine clinical outcomes of patients with advanced non-small cell lung cancer following cell-free circulating tumor DNA genomic profiling. Lung Cancer. 2020;148:69–78. https://doi.org/10.1016/j.lungcan.2020.07.033.
Palmero R, Taus A, Viteri S, et al. Biomarker discovery and outcomes for comprehensive cell-free circulating tumor DNA versus standard-of-care tissue testing in advanced non–small-cell lung cancer. JCO Precis Oncol. 2021;5:93–102. https://doi.org/10.1200/PO.20.00241.
Jee J, Lebow ES, Yeh R, et al. Overall survival with circulating tumor DNA-guided therapy in advanced non-small-cell lung cancer. Nat Med. 2022;28(11):2353–63. https://doi.org/10.1038/s41591-022-02047-z.
Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines: 7. Rating the quality of evidence—inconsistency. J Clin Epidemiol. 2011;64(12):1294–302. https://doi.org/10.1016/j.jclinepi.2011.03.017.
Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J Thorac Oncol. 2018;13(3):323–58. https://doi.org/10.1016/j.jtho.2017.12.001.
Lee Y, Park S, Kim WS, et al. Correlation between progression-free survival, tumor burden, and circulating tumor DNA in the initial diagnosis of advanced-stage EGFR-mutated non-small cell lung cancer. Thorac Cancer. 2018;9(9):1104–10. https://doi.org/10.1111/1759-7714.12793.
Pascual J, Attard G, Bidard FC, et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2022;33(8):750–68. https://doi.org/10.1016/j.annonc.2022.05.520.
Zografos E, Dimitrakopoulos FI, Koutras A. Prognostic value of circulating tumor DNA (ctDNA) in oncogene-driven NSCLC: current knowledge and future perspectives. Cancers (Basel). 2022;14(19):4954. https://doi.org/10.3390/cancers14194954.
Singh AP, Li S, Cheng H. Circulating DNA in EGFR-mutated lung cancer. Ann Transl Med. 2017;5(18):379. https://doi.org/10.21037/atm.2017.07.10.
Zou W, Yaung SJ, Fuhlbrück F, et al. ctDNA predicts overall survival in patients with NSCLC treated with PD-L1 blockade or with chemotherapy. JCO Precis Oncol. 2021;5:827–38. https://doi.org/10.1200/PO.21.00057.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Author contributions
Concept and design: Jansen and Chen; acquisition, analysis, or interpretation of data: all authors; drafting of the manuscript: Chen, Jansen, and Ragavan; critical revision of the manuscript for important intellectual content: all authors; statistical analysis: Chen and Jansen; administrative, technical, or material support: Douglas and Phillips; and supervision: Jansen and Phillips.
Funding
This work was supported by grants from the National Cancer Institute (R01 CA221870) and the National Human Genome Research Institute (U01 HG009599 and HG011792) to Dr. Phillips.
Disclaimer
The National Human Genome Research Institute and the National Cancer Institute had no role in the preparation, review, or approval of the manuscript or decision to submit the manuscript for publication.
Conflict of interest
Mr. Douglas has disclosed that he receives consulting fees from Illumina, Inc. Dr. Phillips has disclosed that she receives consulting fees from Illumina, Inc; serves on an evidence review panel (the California Technology Assessment Forum), which is an independent appraisal committee for the Institute for Clinical and Economic Review (ICER); served as an advisor on two panels for Roche Diagnostics on tumor sequencing; and served on the Association of Community Cancer Centers Policy Advisory Committee in 2021. Dr. Jansen is a part-time salaried employee of the Precision Medicine Group, where he provides methodological expertise for health economics and outcomes research studies unrelated to this paper. Dr. Ragavan serves as a consultant/advisor to Trial Library and received royalties from Pharmacy Times. C.C. has nothing to disclose.
Availability of data and codes
The data and R codes used in the study are available from the corresponding author on reasonable request.
Ethics approval
Not applicable.
Consent
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Chen, C., Douglas, M.P., Ragavan, M.V. et al. Clinical Validity and Utility of Circulating Tumor DNA (ctDNA) Testing in Advanced Non-small Cell Lung Cancer (aNSCLC): A Systematic Literature Review and Meta-analysis. Mol Diagn Ther 28, 525–536 (2024). https://doi.org/10.1007/s40291-024-00725-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-024-00725-x